

Abstract
RNase H2 is a susceptibility gene for the Aicardi–Goutières syndrome (AGS), a genetic auto‐inflammatory disease. Mackenzie and colleagues now report a tractable mouse model for the disease, implicating the cGAS‐STING pathway.
Subject Categories: Immunology
The auto‐inflammatory genetic disease termed Aicardi–Goutières syndrome (AGS) is characterized by an increased expression of interferon‐stimulated genes (ISG) and is now considered an interferonopathy (Crow & Manel, 2015). Mackenzie and colleagues report the generation of a new mouse model for AGS mutations in Rnaseh2b, and leverage it to provide mechanistic insights into the disease (Fig 1) (Mackenzie et al, 2016).
Figure 1. AGS mutation in RNase H2 activates the cGAS‐STING pathway.
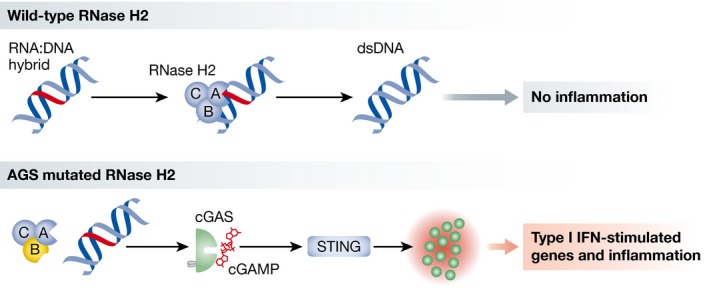
Top: RNA:DNA hybrids are produced during transcription and DNA replication. RNase H2, composed of 3 subunits A, B, and C, excises ribonucleotides, leading to DNA repair. Bottom: When RNase H2 carries a mutation found in patients with Aicardi–Goutières syndrome (AGS), RNA:DNA hybrids are not processed properly and lead to the activation of the nucleic acid sensor cGAS. cGAS produces the dinucleotide cyclic GMP‐AMP (cGAMP), which activates STING, leading to the expression of type I IFN‐stimulated genes and inflammation, as observed in AGS.
Diseases of the immune system remain a considerable challenge. Effective immune responses are impaired in genetic disorders such as SCIDs, and ineffective immune responses prevent the control of microbes such as HIV. On the flipside, unchecked immune responses are responsible for pathologies such as lupus. A fascinating aspect of the immune system is that it is—by essence—shaped through interactions between genes, cells, environment, and microbes. In AGS, genetic mutations cause an auto‐inflammation that is associated with an increased production of type I interferon. This is indicative of an elevated immune response belonging to the innate (or non‐clonal) arm of the immune system. A distinctive feature of innate immunity is that, unlike adaptive immune responses that are mediated by specialized hematopoietic cell types, all cells of the organisms have the capacity to contribute. Immune cells still, such as dendritic cells, are nonetheless key players in innate immunity since they uniquely link innate to adaptive immune activation. Consequently, diseases due to unchecked innate immunity range from auto‐inflammation to autoimmunity (McGonagle & McDermott, 2006).
Remarkably in AGS, all the genes mapped so far—RNASEH2A, RNASEH2B, and RNASEH2C, SAMHD1, TREX1, ADAR1, and IFIH1—code for proteins with enzymatic activities on nucleic acids or nucleotides. This points to the idea that the products of these genes would normally prevent the accumulation of misplaced or misprocessed nucleic acids, as the latter would otherwise trigger downstream nucleic acid sensing pathways, leading to auto‐inflammation and AGS. For some AGS mutations in TREX1, a pathogenic function independent of the activity on nucleic acid has also been reported, extending the activities of AGS genes (Hasan et al, 2015).
Here, the authors focused on RNase H2. RNase H2 cleaves RNA in DNA:RNA hybrids, which are known to occur during genomic DNA replication, in R‐loops formed during transcription and during retro‐transcription. However, the endogenous substrates of RNase H2 are not fully resolved.
While mutation in RNase H2 is viable and causes AGS in humans, homozygote knock‐out mice developed prior to the work of Mackenzie and colleagues are embryonically lethal. Based on the human AGS missense mutation RNASEH2B‐A177T, they generated the corresponding murine missense A174T mutation in Rnaseh2b. Strikingly, the authors found that Rnaseh2b A174T/A174T mice are viable. The mice have no overt phenotype, but the absence of AGS‐like clinical features was not a surprise, since it was also not observed in mice deficient for Samhd1 and Trex1.
In heart and kidney, but not brain, a clear increased expression of ISGs was detected. To dissect the underlying mechanism of innate immune activation, the authors moved to in vitro cell cultures. They cleverly bred Rnaseh2b −/− with p53 −/− to generate viable mouse embryonic fibroblasts. These cells phenocopied in vitro the ISG induction seen in Rnaseh2b A174T/A174T mice, constituting a relevant in vitro model.
They hypothesized that an innate immune sensing pathway was active in these cells. After considering the various pathways that have been previously implicated in sensing RNA:DNA hybrids, they focused on the cGAS‐STING pathway. Cyclic GMP‐AMP synthase (cGAS) is an essential sensor of cytosolic dsDNA that can also respond to RNA:DNA hybrids and signal through the cyclic GMP‐AMP receptor STING to induce a type I interferon response. With multiple genetic approaches to manipulate the cGAS‐STING pathway, they collectively provide strong evidences that this cytosolic nucleic acid sensing axis is required for the ISG induction in the Rnaseh2b −/− and Rnaseh2b A174T/A174T models.
What is the immunostimulatory pathogenic molecular product in RNase H2 AGS? cGAS responds to RNA:DNA hybrids, but its response to dsDNA is more potent. The authors thus reasoned that cGAS could respond either to accumulated RNA:DNA hybrids or to cytosolic dsDNA released as a result of genome instability due to unexcised ribonucleotide in genomic DNA. To approach this question, they rescued Rnaseh2b −/− p53 −/− cells with either RNase H1, which restores cellular RNase H activity, but lacks the ability of RNase H2 to excise ribonucleotides from DNA (Sparks et al, 2012), or wild‐type RNase H2B. Only reconstitution with RNase H2B reduced DNA damage and repressed the expression of ISGs, reinforcing their idea that the ISG response is associated with the instability of DNA species presumably containing unexcised ribonucleotides. Reflecting on the idea that cGAS is required for the ISG induction, it will be important to examine at the biochemical level the cGAS substrate requirements and response to dsDNA containing ribonucleotides and other damages, and conversely, to precisely identify the relevant substrates of cGAS in these cellular models of RNase H2B AGS. It will also be important to understand how these dsDNA products would gain access to the cytosol to reach cGAS.
Of note, Mackenzie and colleagues do not report on the detection of the ISG signature in in vitro cultured cells from Rnaseh2b A174T/A174T mice and RNASEH2‐A177T patients. If the ISG signature is not systematically observed in cells carrying AGS mutations, this would indicate that factors additional to the mutation are required, opening an exciting area of research tractable in cell culture.
Overall, Rnaseh2b A174T/A174T mouse and Rnaseh2b −/− p53 −/− cells constitute novel validated models to further dissect the pathogenic process of AGS. At the molecular level, the models should enable to identify the critical steps that lead to cGAS activation. Worthy of mention, a distinct study simultaneously reported a mouse model for a different RNase H2 AGS mutation, also implicating the cGAS‐STING pathway and identifying retrotransposable elements as possible substrates for the DNA sensor (Pokatayev et al, 2016). The identification of endogenous substrates for cGAS remains an open and interesting question that AGS mouse models can help to pinpoint. Critically, none of the mouse model of AGS reported so far recapitulates the clinical feature of AGS—understanding this shortcoming remains a major outstanding question.
There is important additional physiopathological relevance of AGS genes beyond interferonopathies. For example, SAMHD1 and TREX1 have been found to dampen the innate immune sensing of HIV‐1 in lymphocytes, dendritic cells and macrophages, possibly pointing to the desirable immune responses that need to be restored in infected patients or in a vaccine (Manel et al, 2010; Yan et al, 2010). The Rnaseh2b models described by Mackenzie and colleagues thus constitute fertile grounds not only to unravel the molecular mechanisms of AGS, but also to shed a broad light on immune defense mechanisms.
Acknowledgements
This work is supported by an ERC Grant HIVINNATE 309848 to NM.
See also: KJ Mackenzie et al (April 2016)
References
- Crow YJ, Manel N (2015) Aicardi‐Goutieres syndrome and the type I interferonopathies. Nat Rev Immunol 15: 429–440 [DOI] [PubMed] [Google Scholar]
- Hasan M, Fermaintt CS, Gao N, Sakai T, Miyazaki T, Jiang S, Li QZ, Atkinson JP, Morse HC 3rd, Lehrman MA, Yan N (2015) Cytosolic nuclease TREX1 regulates oligosaccharyltransferase activity independent of nuclease activity to suppress immune activation. Immunity 43: 463–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Lettice L, Tarnauskaite Z, Reddy K, Dix F, Revuelta A, Abbondati E, Rigby RE, Rabe B, Kilanowski F, Grimes G, Fluteau A, Devenney PS, Hill RE, Reijns MA, Jackson AP (2016) Ribonuclease H2 mutations induce a cGAS/STING‐dependent innate immune response. EMBO J 35: 831–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manel N, Hogstad B, Wang Y, Levy DE, Unutmaz D, Littman DR (2010) A cryptic sensor for HIV‐1 activates antiviral innate immunity in dendritic cells. Nature 467: 214–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGonagle D, McDermott MF (2006) A proposed classification of the immunological diseases. PLoS Med 3: e297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokatayev V, Hasin N, Chon H, Cerritelli SM, Sakhuja K, Ward JM, Morris HD, Yan N, Crouch RJ (2016) RNase H2 catalytic core Aicardi‐Goutieres syndrome‐related mutant invokes cGAS‐STING innate immune‐sensing pathway in mice. J Exp Med 213: 329–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks JL, Chon H, Cerritelli SM, Kunkel TA, Johansson E, Crouch RJ, Burgers PM (2012) RNase H2‐initiated ribonucleotide excision repair. Mol Cell 47: 980–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan N, Regalado‐Magdos AD, Stiggelbout B, Lee‐Kirsch MA, Lieberman J (2010) The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat Immunol 11: 1005–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]