

Abstract
Autophagy, mediated by a number of autophagy‐related (ATG) proteins, plays an important role in the bulk degradation of cellular constituents. Beclin‐1 (also known as Atg6 in yeast) is a core protein essential for autophagic initiation and other biological processes. The activity of Beclin‐1 is tightly regulated by multiple post‐translational modifications, including ubiquitination, yet the molecular mechanism underpinning its reversible deubiquitination remains poorly defined. Here, we identified ubiquitin‐specific protease 19 (USP19) as a positive regulator of autophagy, but a negative regulator of type I interferon (IFN) signaling. USP19 stabilizes Beclin‐1 by removing the K11‐linked ubiquitin chains of Beclin‐1 at lysine 437. Moreover, we found that USP19 negatively regulates type I IFN signaling pathway, by blocking RIG‐I‐MAVS interaction in a Beclin‐1‐dependent manner. Depletion of either USP19 or Beclin‐1 inhibits autophagic flux and promotes type I IFN signaling as well as cellular antiviral immunity. Our findings reveal novel dual functions of the USP19‐Beclin‐1 axis by balancing autophagy and the production of type I IFNs.
Keywords: autophagy, Beclin‐1, cross talk, type I interferon, ubiquitination
Subject Categories: Autophagy & Cell Death; Immunology; Post-translational Modifications, Proteolysis & Proteomics
Introduction
Autophagy is a homeostatic process that takes place in all eukaryotic cells and involves the sequestration of cytoplasmic components in double‐membraned autophagosomes (Choi et al, 2013). The autophagosomes fuse with the endosomal–lysosomal system to form the autolysosome, mediating the clearance of cellular components through lysosomal degradation (Deretic et al, 2013; Lamb et al, 2013). Autophagy is a critical process to maintain cellular homeostasis, development, tumorigenesis, and infection in mammalian cells. Nevertheless, the cross talk between autophagy and antiviral innate immunity remains a heated topic that warrants further investigation (Kuballa et al, 2012).
Autophagy is governed by a series of autophagy‐related proteins (ATGs). Beclin‐1, a mammalian homolog of ATG6, plays an important role in autophagy initiation and progression. It participates in VPS34 complex formation and recruits additional proteins, such as ATG14L, AMBRA1, UVRAG, Bcl‐2, and Rubicon, to modulate the activity of VPS34 (Nazio et al, 2013; Russell et al, 2013). Recently, Beclin‐1 has been shown to interact with cyclic GMP‐AMP (cGAMP) synthetase (cGAS) and suppress cGAMP synthesis to halt type I interferon (IFN) production upon recognition of microbial DNA (Liang et al, 2014). Given the complexity of mammals, it is not surprising that Beclin‐1 undergoes multiple layers of regulation to fine‐tune its function (Levine et al, 2015). AMPK activates the pro‐autophagic Beclin‐1 complex by phosphorylating serine (Ser) 91 and Ser94 on Beclin‐1 to induce autophagy. ULK1 phosphorylates Beclin‐1 on Ser14, thereby enhancing the activity of the ATG14L‐containing VPS34 complexes (Kim et al, 2013; Russell et al, 2013).
Ubiquitin and ubiquitin‐like modifications participate in most of the cellular signaling pathways (Husnjak & Dikic, 2012). Ubiquitination, as well as its reversal process, deubiquitination, plays critical roles in the regulation of autophagy (Nazio et al, 2013; Simicek et al, 2013; Rogov et al, 2014; Xia et al, 2014). The E3 ligase TNF receptor‐associated factor 6 (TRAF6) mediates the K63‐linked ubiquitination of Beclin‐1 to disrupt the interaction between Beclin‐1 and Bcl‐2, while the deubiquitinating enzyme A20 can reverse this process (Shi & Kehrl, 2010). A recent report suggests that many tripartite motif (TRIM) proteins serve as the platforms for assembly of Beclin‐1 into a function complex to initiate autophagy (Mandell et al, 2014). Furthermore, ubiquitination is associated with proteasomal degradation of the members of Beclin‐1 complex. Ring finger protein 216 (RNF216) promotes the ubiquitination of Beclin‐1, leading to the degradation of Beclin‐1 in a proteasome‐dependent manner (Xu et al, 2014). Two ubiquitin‐specific peptidases, USP10 and USP13, maintain the stability of Beclin‐1 by mediating the deubiquitination of Beclin‐1 in VPS34 complex (Liu et al, 2011). Despite of these progresses, the mechanism underlying the precise regulation of Beclin‐1 by ubiquitination in autophagy remains largely unknown.
USP19 is a tail‐anchored ubiquitin‐specific protease, which has been reported to localize to the endoplasmic reticulum (ER) and function as a target of the unfolded protein response (UPR) (Hassink et al, 2009). USP19 deubiquitinates and thereby stabilizes the KPC1 ligase for p27Kip1, c‐IAP1, and c‐IAP2 (Mei et al, 2011). USP19 can also interact with several components of the hypoxia pathway including HIF‐1α and inhibit its degradation in a deubiquitinase‐independent manner (Hassink et al, 2009; Sundaram et al, 2009). However, whether USP19 is involved in the regulation of autophagy or innate immune response is unknown. In this study, we identified USP19 as a positive regulator of autophagy. USP19 stabilizes Beclin‐1 by removing the K11‐linked ubiquitin chains of Beclin‐1 at lysine 437. Moreover, we found that USP19 negatively regulates type I IFN signaling pathway by blocking RIG‐I‐MAVS interaction in a Beclin‐1‐dependent manner. USP19 or Beclin‐1 deficiency led to enhanced type I IFN signaling as well as cellular antiviral immunity upon RNA viral infection. Our findings demonstrate that USP19 acts as a mediator of autophagy and antiviral immunity by balancing autophagic flux and type I IFN signaling.
Results
USP19 positively regulates autophagy
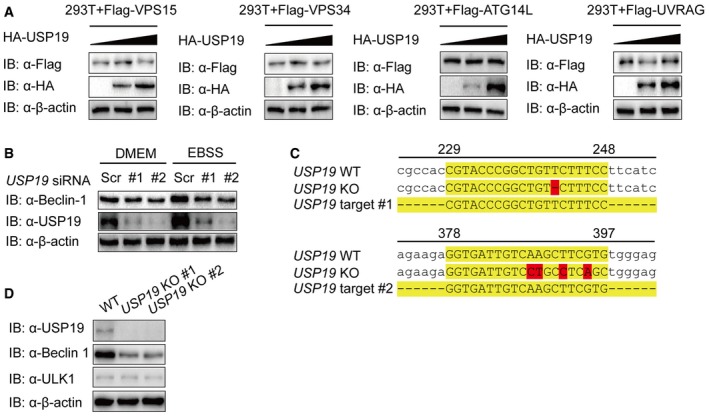
To gain insight into the functions of ubiquitin modification in autophagy, we performed a screen by using small interfering RNAs (siRNAs) targeting 84 human deubiquitinating enzyme (DUB) genes and identified USP19 as a positive regulator of autophagy (Appendix Fig S1A). We further confirmed the knockdown efficiency of siRNAs targeting two different regions of USP19. Both of the two siRNAs efficiently inhibited the expression of transfected and endogenous USP19 in 293T cells and HeLa cells (Fig 1A). Under both basal conditions and autophagy‐induced conditions with Earle's balanced salt solution (EBSS) treatment, depletion of USP19 remarkably reduced the formation of GFP‐LC3B puncta (Fig 1B and C). We then monitored the autophagic flux by detecting the autophagy marker light chain 3 (LC3) using immunoblotting. We found that USP19 knockdown inhibited the accumulation of LC3 II under both basal conditions and rapamycin‐treated conditions (Fig 1D). Consistent with the inhibition of autophagy, the level of p62/SQSTM1 (an autophagy substrate) was higher in USP19 knockdown cells than that in control cells (Fig 1E). In human peripheral blood mononuclear cells (PBMCs), we also found that USP19 depletion decreased the accumulation of LC3 II and consumption of p62 (Fig 1F).
Figure 1. USP19 positively regulates autophagy.
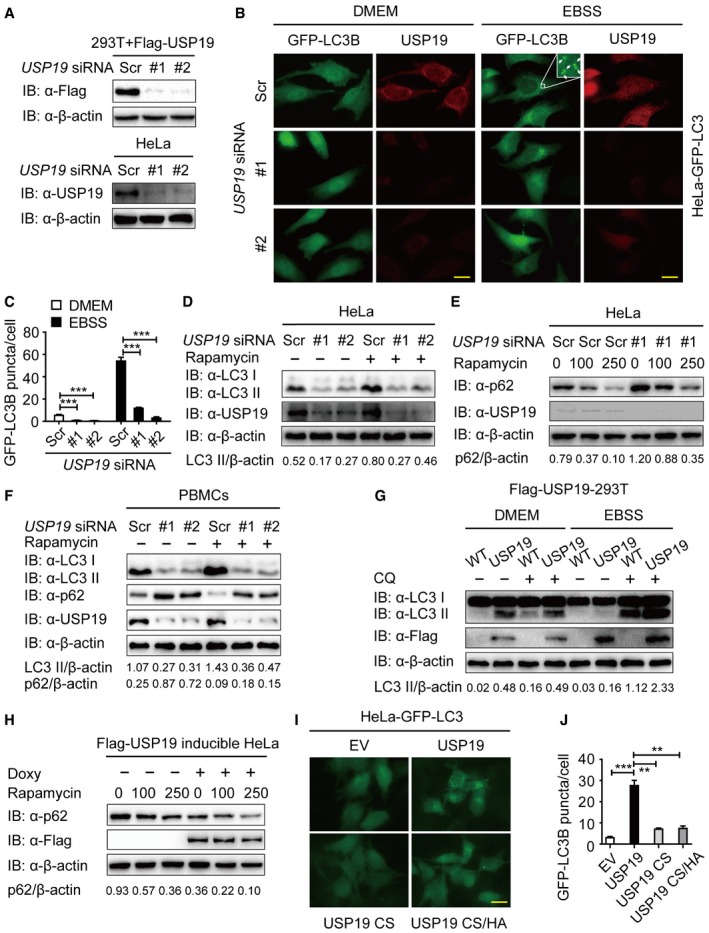
-
AImmunoblot analysis of the knockdown of exogenous USP19 in 293T cells expressing Flag‐USP19 (top) or endogenous USP19 in HeLa cells (bottom) treated with USP19‐specific siRNA or scrambled (Scr) siRNA.
-
B, CRepresentative images of GFP‐LC3B puncta in HeLa‐GFP‐LC3B cells transfected with USP19‐specific siRNA or scrambled siRNA during growth in normal medium (DMEM) or EBSS medium for 3 h. Arrows denote representative autophagosomes. Pictures (B) were taken using Leica DMI3000 B microscopy with a × 100 oil‐immersion objective. Scale bar, 200 μm. Quantification of GFP‐LC3B puncta in HeLa‐GFP‐LC3B cells transfected with USP19‐specific siRNA or scrambled siRNA (C). Bars represent mean ± standard error of the mean (SEM) of triplicate samples (20 cells per sample). ***P < 0.001 (two‐tailed Student's t‐test).
-
DImmunoblot detection of the relative accumulation of LC3‐I and LC3‐II in HeLa cells transfected with USP19‐specific siRNA or scrambled siRNA with rapamycin (250 nM) treatment for 12 h.
-
EHeLa cells were transfected with USP19‐specific siRNA or scrambled siRNA and treated with the indicated concentration of rapamycin for 12 h. p62 levels were detected by immunoblot.
-
FHuman peripheral blood mononuclear cells (PBMCs) transfected with control or USP19‐specific siRNA were treated with rapamycin (250 nM) for 18 h, and the lysates were analyzed with the indicated antibodies.
-
G293T cells stably expressing Flag‐USP19 were incubated with DMEM or EBSS medium for 3 h in the absence or presence of 50 μM of chloroquine (CQ). Cells were then assayed for the relative accumulation of LC3‐I and LC3‐II by immunoblot analysis using anti‐LC3 antibody.
-
HFlag‐USP19‐inducible HeLa cells were treated with 200 ng/ml doxycycline (Doxy) overnight to induce the expression of Flag‐USP19. The protein levels of p62 were analyzed after rapamycin treatment for 12 h at the indicated concentrations.
-
I, JHeLa cells stably expressing GFP‐LC3B were transfected with pcDNA3.1 empty vector or plasmid expressing the wild‐type and the mutant forms of USP19 (CS or CS/HA). Pictures were taken using Leica DMI3000 B microscopy with a × 100 oil‐immersion objective. Scale bar, 200 μm. Average GFP‐LC3B puncta per cell were calculated (J). Bars represent mean ± SEM of triplicate samples (20 cells per sample). **P < 0.01 and ***P < 0.001 (two‐tailed Student's t‐test).
To further confirm the role of USP19 in autophagy, we generated a HEK293T cell line stably expressing Flag‐tagged USP19 and found that overexpression of USP19 significantly enhanced the amount of LC3 II in both basal conditions and EBSS‐treated conditions (Fig 1G). We also generated an USP19‐inducible HeLa cell line to investigate the role of USP19 in autophagy induction (Appendix Fig S1B and C) and observed that ectopic expression of USP19 consumed more p62 by rapamycin treatment (Fig 1H). These results suggest that USP19 is a positive regulator of autophagic flux. Since USP19 is a member of ubiquitin‐specific protease family, we next examined whether the catalytic activity of USP19 is essential for its function in autophagy. We generated catalytically inactive USP19 mutant CS (C506S) and CS/HA (C506S/H1157A) (Appendix Fig S1D) and transfected HeLa‐GFP‐LC3B cells with plasmids encoding wild‐type (WT), CS, or CS/HA mutant of USP19. As shown in Fig 1I and J, cells expressing WT USP19 displayed an enhanced accumulation of GFP‐LC3B puncta even in the absence of nutrient limitation. By contrast, ectopic expression of CS and CS/HA mutants USP19 failed to promote autophagy. Taken together, our findings suggest that USP19 positively regulates autophagy in a deubiquitinase‐dependent manner.
Identification of USP19 as a new member of the Beclin‐1 complex
To identify the potential molecular targets of USP19 in autophagy, we co‐transfected 293T cells with expression vectors containing Flag‐tagged ATG proteins together with HA‐tagged USP19. Coimmunoprecipitation (Co‐IP) and immunoblot analysis showed that Flag‐tagged Beclin‐1, VPS15, VPS34, ATG14L, UVRAG, and AMBRA1 interacted with HA‐USP19 (Fig 2A). However, we did not observe any apparent interaction between the ATG8 subfamily members (LC3A, LC3B, LC3C, GABRAPL1, or GABRAPL1) or cargo receptors (p62, OPTN, NIX, Tollip, or NBR1) with USP19 (Fig EV1A and B). We further show that the interaction between USP19 and Beclin‐1 complex component proteins was not dependent on Beclin‐1, because the interaction between USP19 and those proteins was not affected in BECN1 knockout (KO) cells (Fig EV1C). Our results also showed that USP19 was not a binding partner to regulate the protein–protein interaction of Beclin‐1 complex components (Fig EV1D–F). To determine the physiological relevance of those findings, we treated HeLa cells, A549 cells, or human PBMCs with EBSS medium and collected cell lysates at varying time points. We used a specific antibody against Beclin‐1 (anti‐Beclin‐1) to immunoprecipitate Beclin‐1‐associated protein complexes, and found that the endogenous association between Beclin‐1 and USP19 increased considerably at 1–2 h upon starvation treatment (Figs 2B and C, and EV1G). Confocal imaging further demonstrated that Beclin‐1 colocalized with USP19 after starvation (Fig 2D and E). These results suggest that USP19 specifically interacts with Beclin‐1 complex during autophagy.
Figure 2. USP19 interacts with Beclin‐1 complex member.
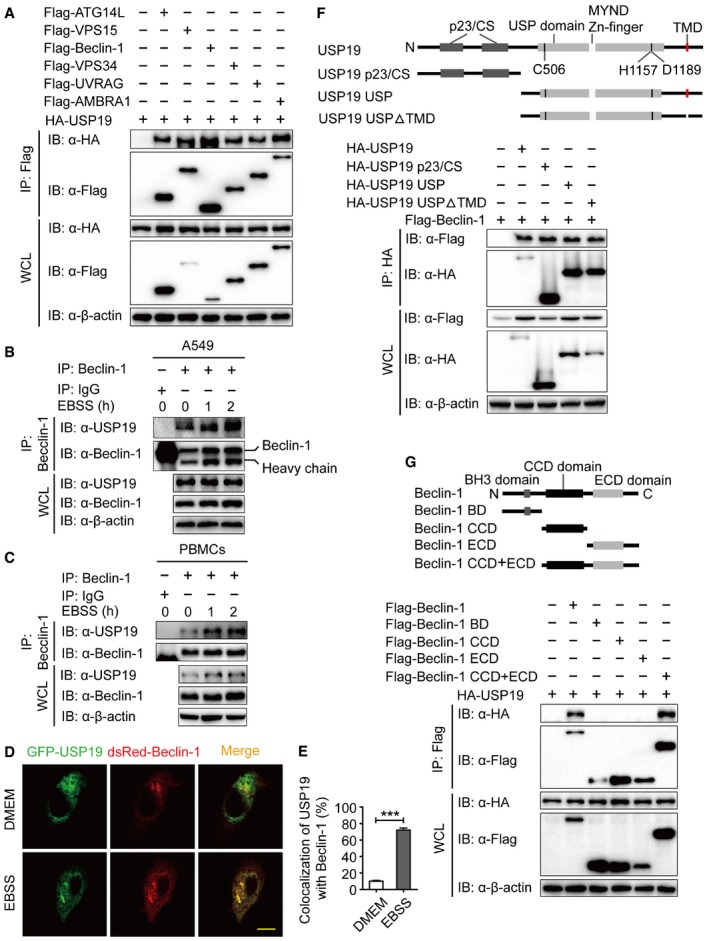
-
A293T cells were transfected with plasmids encoding HA‐USP19 and Flag‐tagged Beclin‐1 complex members (Flag‐ATG14L, Flag‐VPS15, Flag‐Beclin‐1, Flag‐VPS34, Flag‐UVRAG, and Flag‐AMBRA1), followed by immunoprecipitation (IP) with anti‐Flag beads and immunoblot analysis with anti‐HA. Throughout was the immunoblot analysis of whole‐cell lysates (WCL) without immunoprecipitation.
-
B, CExtracts of A549 cells (B) or human PBMCs (C) incubated with EBSS for various time points (above lanes) were subjected to immunoprecipitation with anti‐Beclin‐1 and immunoblot analysis with the indicated antibodies (shown on the left).
-
D, EHeLa cells were incubated in EBSS medium for 1 h after co‐transfection eGFP‐USP19 and dsRed‐Beclin‐1 for 48 h. Cells were imaged for eGFP and dsRed. Scale bar, 200 μm. Quantitative data are mean ± SEM of triplicate samples (20 cells per sample). ***P < 0.001 (two‐tailed Student's t‐test).
-
FCoimmunoprecipitation and immunoblot analysis of 293T cells transfected with deletion mutants of USP19 plasmid along with vector encoding Flag‐Beclin‐1.
-
GCoimmunoprecipitation and immunoblot analysis of 293T cells transfected with deletion mutants of Beclin‐1 plasmid along with vector encoding HA‐USP19.
Figure EV1. USP19 interacts with Beclin‐1 complex.
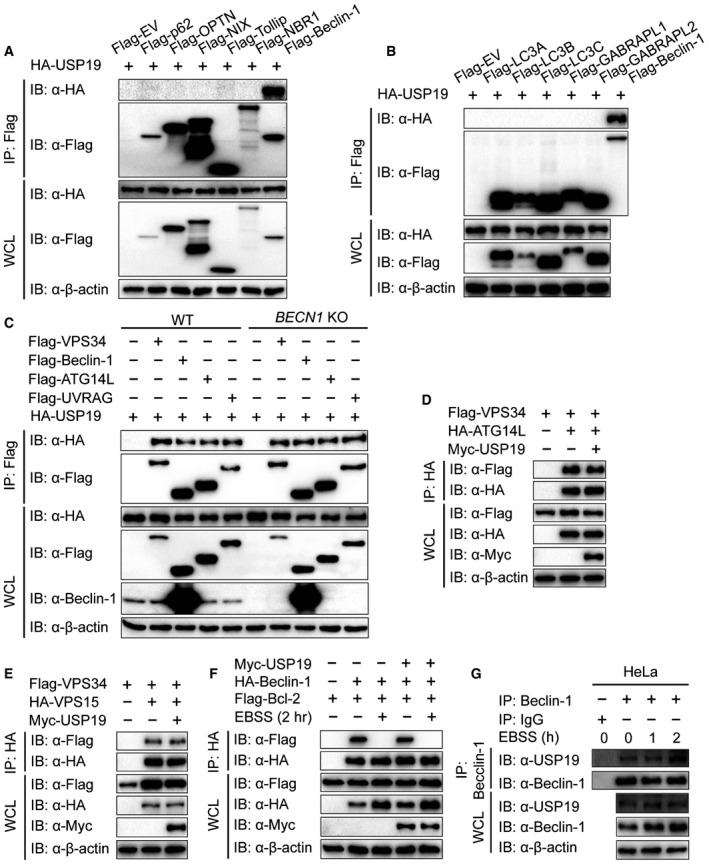
- 293T cells were transfected with vectors encoding HA‐USP19 and Flag‐tagged cargo receptors (Flag‐p62, Flag‐OPTN, Flag‐NIX, Flag‐VPS34, Flag‐Tollip, or Flag‐NBR1), followed by immunoprecipitation with anti‐Flag beads and immunoblot analysis with anti‐HA.
- 293T cells were transfected with vectors encoding HA‐USP19 and Flag‐tagged LC3 superfamily members (Flag‐LC3A, Flag‐LC3B, Flag‐LC3C, Flag‐GABRAPL1, or Flag‐GABRAPL2), followed by immunoprecipitation with anti‐Flag beads and immunoblot analysis with anti‐HA.
- WT and BECN1 KO 293T cells were transfected with vectors encoding HA‐USP19 and Flag‐tagged Beclin‐1 complex members (Flag‐VPS34, Flag‐Beclin‐1, Flag‐ATG14L, and Flag‐UVRAG), followed by immunoprecipitation and immunoblot analysis.
- 293T cells were transfected with vectors encoding HA‐ATG14L and Flag‐VPS34 together with or without Myc‐USP19, followed by immunoprecipitation with anti‐HA beads and immunoblot analysis with anti‐Flag.
- 293T cells were transfected with vectors encoding HA‐VPS15 and Flag‐VPS34 together with or without Myc‐USP19, followed by immunoprecipitation with anti‐HA beads and immunoblot analysis with anti‐Flag.
- 293T cells were transfected with vectors encoding HA‐Beclin‐1 and Flag‐Bcl‐2 together with or without Myc‐USP19, followed by immunoprecipitation with anti‐HA beads and immunoblot analysis with anti‐Flag.
- Extracts of HeLa cells incubated with EBSS for various time points were subjected to immunoprecipitation with anti‐Beclin‐1 and immunoblot analysis with the indicated antibodies.
To map the essential domains of USP19 that mediate its association with Beclin‐1, we generated three deletion constructs: USP19 p23/CS (aa 1–496), USP19 USP (aa 497–1318), and USP19 USPΔTMD (aa 497–1291 plus 1313–1318). We found that all the three USP19 deletion proteins could interact with the full‐length Beclin‐1 protein. Moreover, USP19 interacted with Beclin‐1 in an ER‐independent manner, as the interaction was not affected by the deletion of USP19 transmembrane domain (TMD) (Fig 2F). In order to determine which domain of Beclin‐1 was needed for the interaction between USP19 and Beclin‐1, we generated Beclin‐1 truncation mutants and performed Co‐IP experiment. As shown in Fig 2G, USP19 could only interact with the CCD plus ECD (aa 144–450) domain of Beclin‐1, but not BD (aa 1–143), CCD (aa 144–268), or ECD (aa 269–450) domain of Beclin‐1 alone. This result indicates that USP19 might bind Beclin‐1 through the linkage region between the CCD and ECD domains.
USP19 stabilizes Beclin‐1
We next sought to determine how USP19 prompts autophagy by interacting with Beclin‐1 complex. When we transfected 293T cells with plasmid encoding Flag‐Beclin‐1 and HA‐USP19 together, we found that the expression of Beclin‐1 protein positively correlated with USP19 expression (Fig 3A), thus pointing to a potential stabilizing role of USP19 toward Beclin‐1. To determine the specificity of the USP19‐mediated stabilization of Beclin‐1, we carried out similar experiments in cells expressing VPS15, VPS34, ATG14L, and UVRAG with increasing USP19 expression, and found that USP19 did not affect the concentration of other members of Beclin‐1 complex (Fig EV2A).
Figure 3. USP19 stabilizes Beclin‐1.
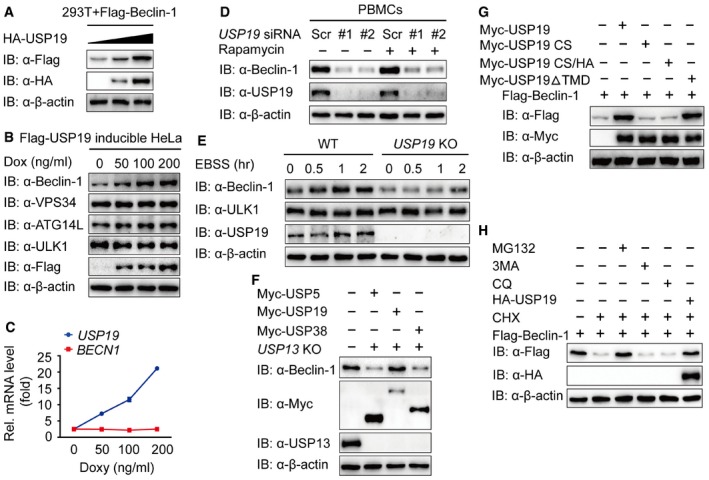
- Immunoblot analysis of extracts of 293T cells transfected with expression vector for Flag‐Beclin‐1 and increasing doses of expression vector for HA‐USP19 (wedge).
- HeLa cell line harboring inducible Flag‐USP19 was induced with indicated concentration of Doxy overnight, and the protein was harvested for immunoblot analysis.
- BECN1 mRNA levels of the same sample as in (B) were detected by real‐time PCR. Bars represent mean ± SEM of triplicate samples.
- Human PBMCs were transfected with USP19‐specific or scramble siRNA and treated with rapamycin (250 nM) for 18 h. The extracts were analyzed by immunoblot with the indicated antibodies.
- Wild‐type (WT) and USP19 KO A549 cells were cultured in EBSS for the indicated time points, and the protein expression levels of Beclin‐1 and ULK1 were detected by immunoblot.
- USP13 KO HEK293T cells were transfected with plasmids encoding Myc‐USP5, Myc‐USP19, and Myc‐USP38, and the cellular extracts were analyzed by immunoblot.
- 293T cells transfected with plasmid expressing Flag‐Beclin‐1 and empty vector or Myc‐tagged USP19 (WT, CS, CS/HA, or ΔTMD), and the lysates were analyzed by immunoblot.
- 293T cells were transfected with plasmid encoding Flag‐Beclin‐1 and treated with CHX (100 μg/ml) alone or with MG132 (10 μM), 3MA (10 mM), CQ (50 μM) as combination for 6 h or transfected with the HA‐USP19 plasmid. The cell lysates were analyzed by immunoblot.
Figure EV2. USP19 specifically stabilizes Beclin‐1.
- Immunoblot analysis of extracts of 293T cells transfected with plasmid for Flag‐VPS15, Flag‐VPS34, Flag‐ATG14L, or Flag‐UVRAG and increasing doses of plasmid for HA‐USP19 (wedge).
- HeLa cells were transfected with USP19‐specific or scramble siRNA and incubated in DMEM or EBSS medium for 2 h, and the extracts were analyzed by immunoblot with the indicated antibodies.
- Sequence alignment of USP19 KO A549 cells with WT.
- Immunoblot analysis of extracts of USP19 KO A549 cells.
To assess whether USP19 is able to mediate the stability of endogenous Beclin‐1, we treated Flag‐USP19‐inducible HeLa cells with increasing amount of doxycycline (Doxy), and found that endogenous Beclin‐1 was upregulated upon increasing expression of USP19, whereas the abundance of endogenous VPS34, ATG14L, and ULK1 remained largely unaltered (Fig 3B). To exclude the possibility that the upregulation of Beclin‐1 protein was caused by higher transcription of the gene (BECN1), we used real‐time PCR to analyze the same cell line and found that the abundance of BECN1 mRNA did not change with increased expression of USP19 (Fig 3C). We then knocked down the expression of USP19 in HeLa cells or human PBMCs and found that the protein levels of endogenous Beclin‐1 were downregulated under both normal and autophagy‐induced conditions (Figs 3D and EV2B). We also generated USP19 KO A549 cell lines by using the CRISPR/Cas9 genome‐editing tool (Fig EV2C) and observed that the expression of Beclin‐1 was downregulated in USP19 KO cells, while the ULK1 levels remained unaffected (Figs 3E and EV2D). As previous research has reported that USP13 can stabilize Beclin‐1 (Liu et al, 2011), we transfected the plasmids encoding Myc‐USP5, Myc‐USP19, or Myc‐USP38 in USP13 KO cells and found that only USP19 overexpression rescued the degradation of Beclin‐1 in USP13 KO cells (Fig 3F), implying that USP19 stabilized Beclin‐1 in a USP13‐independent manner. Moreover, we found that the catalytically inactive USP19 mutants CS and CS/HA could not stabilize Beclin‐1, but USP19ΔTMD can still stabilize Beclin‐1 (Fig 3G). These results suggest that USP19‐mediated Beclin‐1 stabilization depends on its enzyme catalytic activity but not endoplasmic reticulum localization of USP19. Two major systems which are associated with the proteasome or lysosome, respectively, exist in eukaryotic cells to control protein degradation (Sahtoe & Sixma, 2015). In order to reveal which degradation system dominantly mediates the degradation of Beclin‐1, we examined the protein stability of Flag‐Beclin‐1 in the presence of the translational inhibitor cycloheximide (CHX), which blocks protein synthesis (Nazio et al, 2013). We observed that the degradation of Flag‐Beclin‐1 could be rescued by the proteasome inhibitor (MG132) and USP19 overexpression, but not by the autophagy inhibitor (3MA) or autophagolysosome inhibitor (CQ) (Fig 3H). Collectively, these results suggest that USP19 protects Beclin‐1 from proteasomal degradation through its protease activity.
USP19 removes the K11‐linked ubiquitin chains of Beclin‐1
To determine whether USP19 functions as a bona fide Beclin‐1 deubiquitinase, we analyzed the ubiquitination of Beclin‐1 by immunoprecipitation with anti‐Beclin‐1 antibody, followed by immunoblot analysis with antibody to total ubiquitin. We reasoned that USP19 regulates Beclin‐1 through deubiquitination. Indeed, silencing of USP19 significantly increased the poly‐ubiquitination of Beclin‐1 (Fig 4A). Since different types of poly‐ubiquitination processes, including lysine 48 (K48)‐, K11‐, K63‐, and K27‐linked ubiquitination, have been implicated to deliberately regulate protein fate (Zinngrebe et al, 2014; Michel et al, 2015), we set out to examine which types of Beclin‐1 ubiquitination USP19 could affect. Overexpression of USP19 markedly inhibited WT and K11‐linked ubiquitination of Beclin‐1, but had no appreciable effect on the ubiquitination of Beclin‐1 with other linkages (K6, K27, K29, K33, K48, or K63) (Fig 4B). Consistent with this observation, the K11‐linked ubiquitination of Beclin‐1 was increased by USP19 knockdown (Fig 4C). We also demonstrated that USP19 specifically regulated the ubiquitination of Beclin‐1, but not other members of Beclin‐1 complex, such as ATG14L (Appendix Fig S2). To investigate whether deubiquitination of Beclin‐1 by USP19 is required for its protease activity, we transfected the cells with WT USP19, or its mutants and found that only WT USP19 cleaved the K11‐linked poly‐ubiquitin chains on Beclin‐1 (Fig 4D). We further performed in vitro deubiquitination assay to show that USP19 CS mutant failed to affect the K11‐linked ubiquitination of Beclin‐1 as the WT USP19 did (Fig 4E). Taken together, these results suggest that the catalytic activity of USP19 is essential for Beclin‐1 deubiquitination through a K11 linkage.
Figure 4. USP19 removes K11‐linked ubiquitination of Beclin‐1.
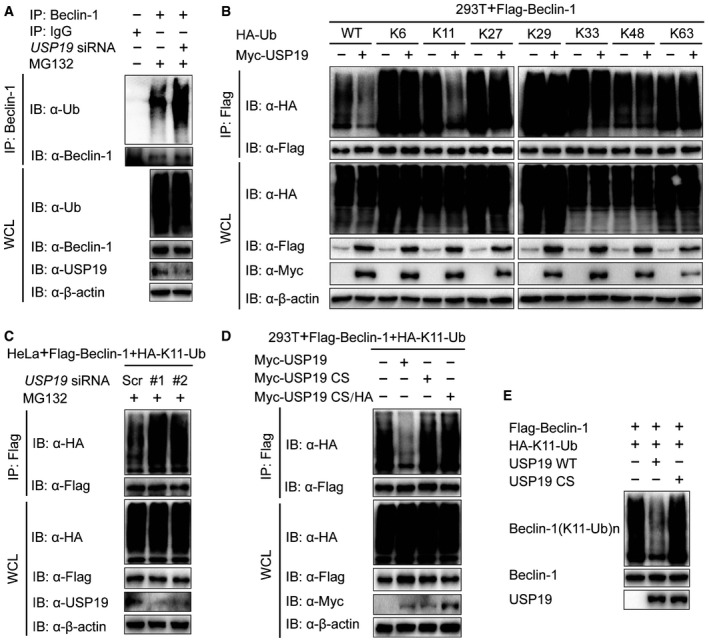
- HeLa cells were transfected with USP19 siRNA or scrambled siRNA and protein extracts were harvested after MG132 (10 μM) treatment for 3 h. Protein extracts were immunoprecipitated using anti‐Beclin‐1 antibody or with IgG as a negative control and analyzed by immunoblot using anti‐ubiquitin and anti‐Beclin‐1 antibodies.
- Lysates of 293T cells transfected with plasmid expressing Flag‐Beclin‐1 and HA‐tagged ubiquitin (HA‐Ub (wild type), K6‐linked‐Ub, K11‐linked‐Ub, K27‐linked‐Ub, K29‐linked‐Ub, K33‐linked‐Ub, K48‐linked‐Ub, or K63‐linked‐Ub), together with the empty vector or expression vector of Myc‐USP19 and treated with MG132 (10 μM) for 3 h, were immunoprecipitated with anti‐Flag and immunoblotted with anti‐HA.
- HeLa cells were transfected with USP19‐specific siRNA and plasmid encoding Flag‐Beclin‐1, plus K11‐linked‐Ub. Protein was harvested after MG132 (10 μM) treatment for 3 h and immunoprecipitated with anti‐Flag and immunoblotted with anti‐HA.
- 293T cells were transfected with plasmid for Flag‐Beclin‐1 and HA‐K11‐Ub, together with the empty vector or expression vector for Myc‐USP19 (WT, CS, or CS/HA) and treated with MG132 (10 μM). Cell lysates were subjected to immunoprecipitation with anti‐Flag and immunoblot analysis with anti‐HA.
- Purified ubiquitinated Beclin‐1 was incubated with immunopurified Flag‐USP19 in vitro in deubiquitinating buffer. The immunoblot was probed with anti‐HA.
Lys437 in Beclin‐1 is essential for its ubiquitination and degradation
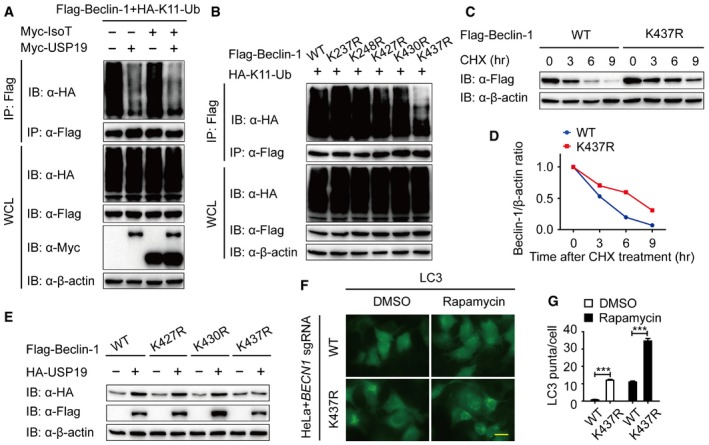
It has been reported that unanchored ubiquitin chains act as critical mediators in the cellular signal transduction (Xia et al, 2009). We examined whether Beclin‐1 is covalently attached to anchored ubiquitin chains or noncovalently with unanchored ubiquitin chains. We observed IsoT, an isopeptidase which can specifically degrade unanchored ubiquitin chains (Rajsbaum et al, 2014), did not influence K11‐linked ubiquitination of Beclin‐1, suggesting that Beclin‐1‐associated K11‐linked ubiquitin chains are primarily covalently attached (Fig 5A). In addition, we confirmed that USP19 mainly cleaved the anchored K11‐linked poly‐ubiquitin chains of Beclin‐1 in the presence of IsoT (Fig 5A). As K11‐linked poly‐ubiquitin chain is directly attached to Beclin‐1, we moved on to identify which lysine site of Beclin‐1 is associated with this modification. We made a series of mutant constructs by substituting lysine residues with arginines (R) with a focus on key, lysine residues that are conserved in the orthologs of Beclin‐1 (Fig EV3A), and performed in vivo ubiquitination assay to find that only the K437R Beclin‐1 mutant displayed reduced K11‐linked ubiquitination (Fig 5B). We further showed that starvation‐induced autophagy promoted the deubiquitination of WT Beclin‐1, but not K437R Beclin‐1 mutant (Fig EV3B), which suggesting that K437 functions as a critical K11‐linked ubiquitination site of Beclin‐1.
Figure 5. The K437 site is critical for Beclin‐1 degradation.
-
ALysates of 293T cells transfected with plasmid for Flag‐Beclin‐1 and HA‐K11‐linked‐Ub, together with empty vector or expression vector for Myc‐USP5 or Myc‐USP19, were immunoprecipitated with anti‐Flag and immunoblotted with anti‐HA.
-
BImmunoprecipitation and immunoblot analysis of 293T cells transfected with vectors expressing Flag‐Beclin‐1 and HA‐K11‐linked ubiquitin.
-
C, D293T cell were transfected with Flag‐Beclin‐1 (WT or K437R) and treated with CHX (100 μg/ml) for the indicated time points. The expression levels of WT and K437R Flag‐Beclin‐1 were analyzed by immunoblot. Quantification of the expression levels of WT and K437R Flag‐Beclin‐1 is shown in (D).
-
EImmunoblot analysis of protein extracts of 293T cells transfected with empty vector or vector for HA‐USP19, together with plasmid expressing WT or K427R, K430R, or K437R mutant of Flag‐Beclin‐1.
-
F, GHeLa cells transfected with BECN1 sgRNA and restored with WT and K437R Flag‐Beclin‐1 were stained by anti‐LC3 antibody. Scale bar, 200 μm. Quantification of LC3 puncta is shown in (G). Bars represent mean ± SEM of triplicate samples. ***P < 0.001 (two‐tailed Student's t‐test).
Figure EV3. The K437 site is important for Beclin‐1 degradation.
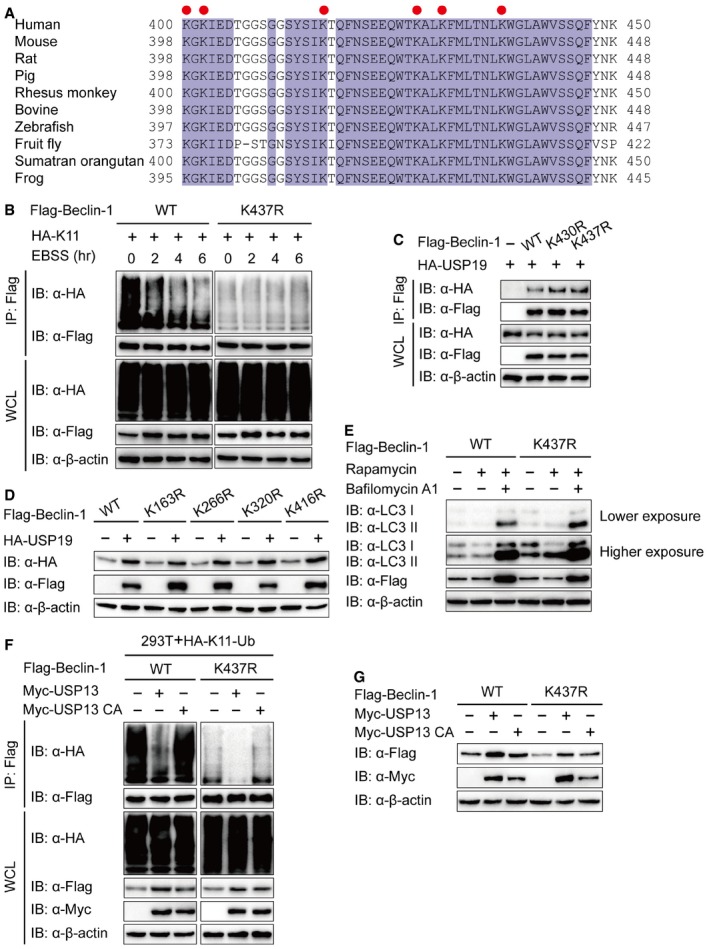
- Alignment of Beclin‐1 sequences of different species. Conserved lysine residues are indicated with a red circle.
- Lysates of 293T cells transfected with plasmids for Flag‐Beclin‐1 (WT or K437R) and HA‐K11‐Ub were incubated with EBSS medium for the indicated time points, then immunoprecipitation with anti‐Flag beads and immunoblot analysis with anti‐HA.
- 293T cells were transfected with plasmids encoding HA‐USP19 and Flag‐Beclin‐1 (WT, K430R, or K437R), followed by immunoprecipitation with anti‐Flag beads and immunoblot analysis with anti‐HA.
- Immunoblot analysis of extracts of 293T cells transfected with empty vector or vector for HA‐USP19, together with plasmids for Flag‐Beclin‐1 (WT, K163R, K266R, K320R, or K416R).
- BECN1 KO 293T cells were transfected with WT or K437R Flag‐Beclin‐1, and lysates were analyzed by immunoblot.
- 293T cells were transfected with plasmid for Flag‐Beclin‐1 and HA‐K11‐Ub, together with the constructs of USP13 or USP13 enzyme inactivity mutant (C345A (CA)), and the lysates were immunoprecipitated with anti‐Flag and immunoblotted with anti‐HA.
- 293T cells were transfected with empty vector or vector for Myc‐USP13 (WT or CA), together with plasmid for Flag‐Beclin‐1 (WT or K437R), and the lysates were analyzed with each antibody.
We next investigated whether K11‐linked poly‐ubiquitin chains on K437 serve as a degradation signal for Beclin‐1. We used a CHX‐chase assay to determine the degradation rate of WT Beclin‐1 and K437R Beclin‐1 mutant and observed the degradation rate of K437R Beclin‐1 mutant was slower compared to that of WT Beclin‐1 (Fig 5C and D). Additionally, we found that, even though USP19 can interact with K437R Beclin‐1 (Fig EV3C), USP19 failed to stabilize the K437R Beclin‐1 mutant as did with WT or other Beclin‐1 mutants (Figs 5E and EV3D). These results indicate that USP19 specifically removes the K11‐linked ubiquitin chains on K437 in Beclin‐1 to prevent it from degradation.
Given our observation that K437R Beclin‐1 has a longer half‐life time, we reasoned that it might induce a stronger autophagic flux when compared to WT Beclin‐1. To test this hypothesis, BECN1 KO cells were transfected with WT or K437R Beclin‐1. Consistent with our hypothesis, compared to WT, K437R Beclin‐1 mutant dramatically increased the formation of LC3 puncta and accumulation of LC3 II (Figs 5F and G, and EV3E). These results suggested that K437 in Beclin‐1 is an essential residue for USP19‐mediated K11‐linked deubiquitination and stabilization of Beclin‐1, thereby regulating autophagic flux. As we observed in Fig 5B, the K11‐linked ubiquitination of K437R Beclin‐1 was not absolutely disappeared. It implies that some other lysines may also associate with K11‐linked ubiquitination of Beclin‐1. We then determined the K11‐linked ubiquitination of Beclin‐1 in the presence of USP13 and observed the K11‐linked ubiquitin chains on Beclin‐1 can also be cleaved by USP13 (Fig EV3F). Moreover, USP13 removed the K11‐linked ubiquitin chains of K437R Beclin‐1 mutant. We then studied the stability of K437R Beclin‐1 mutant with the existence of USP13 and found that unlike USP19, USP13 could still stabilize K437R Beclin‐1 mutant (Fig EV3G). Taken together, our results indicate that in addition to K437, there are some other lysine(s) responsible for K11‐linked ubiquitination of Beclin‐1, which are regulated by USP13, but not USP19.
USP19 inhibits RNA virus‐induced type I IFN activation
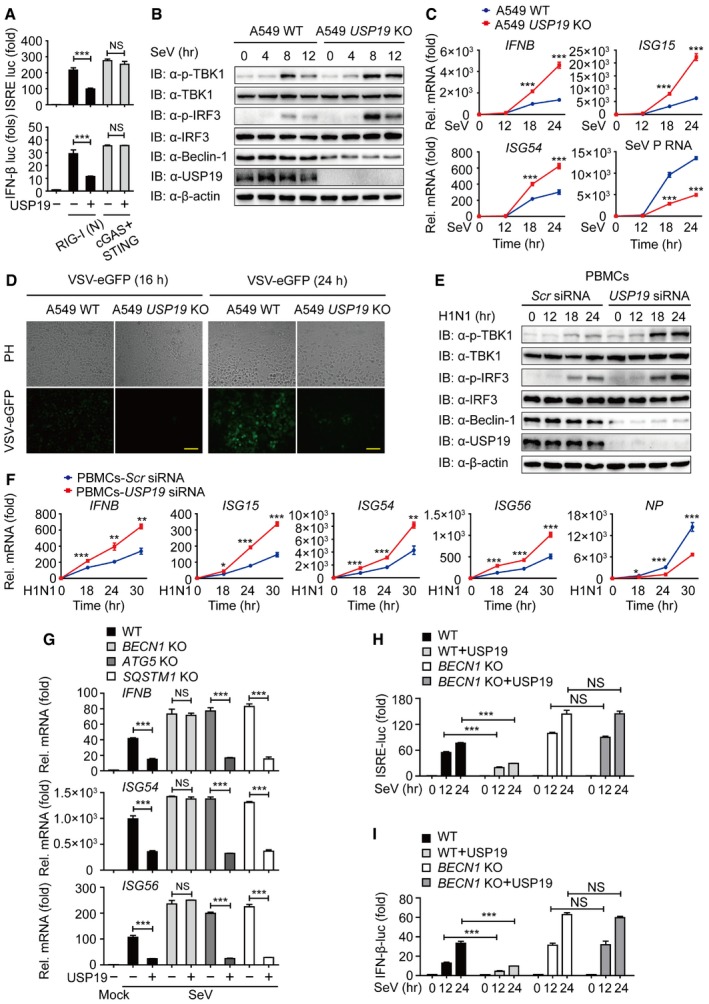
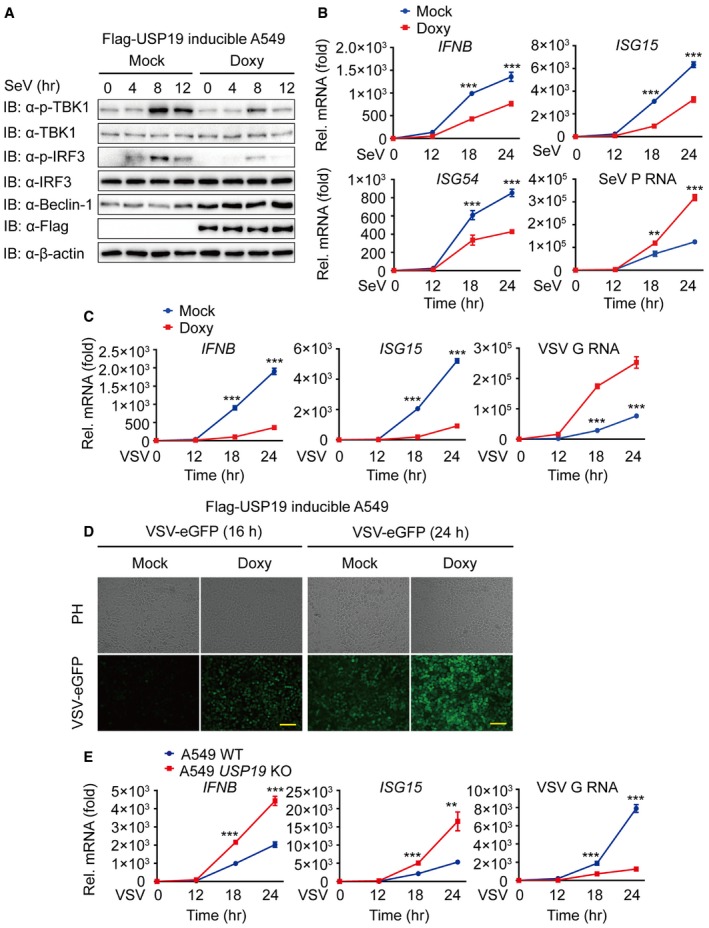
Several recent studies show that many key regulators of autophagy, including Atg5‐Atg12, Atg9, and ULK1, also play important roles in antiviral immunity (Saitoh et al, 2009; Deretic et al, 2013; Konno et al, 2013). To determine whether USP19 is involved in the regulation of type I IFN signaling, we performed functional assays and found that USP19 specifically inhibits RIG‐I‐mediated, but not cGAS‐mediated type I IFN activation (Fig 6A), suggesting that USP19 also serves as a negative regulator of RNA virus‐induced antiviral immunity. To confirm the inhibitory effect of USP19 on type I IFN activation, we assessed the phosphorylation status of TBK1 and IRF3 by overexpressing USP19 in Flag‐USP19‐inducible A549 cells and found that USP19 potently blocked the phosphorylation of endogenous TBK1 and IRF3 after Sendai virus (SeV) infection (Fig EV4A). By contrast, USP19 deficiency in A549 cells markedly increased TBK1 and IRF3 phosphorylation levels (Fig 6B). To further demonstrate the effect of USP19 on the expression of IFN‐responsive genes, we treated Flag‐USP19‐inducible A549 cells with Doxy. Real‐time PCR analysis revealed that overexpression of USP19 resulted in blunted induction of IFNΒ, as well as the interferon‐stimulating genes ISG15, ISG54, and ISG56 by both SeV and vesicular stomatitis virus (VSV, a negative‐stranded RNA virus) infections (Fig EV4B and C). In agreement with these results, we found that infection with SeV or VSV strongly increased the transcription of IFNB, ISG15, ISG54, and ISG56 genes in USP19 KO cells (Figs 6C and EV4E).
Figure 6. USP19 suppresses type I IFN signaling in a Beclin‐1‐dependent manner.
-
ALuciferase activity in 293T cells transfected with an ISRE (left) or IFN‐β (right) promoter‐driven luciferase reporter and together with plasmid encoding RIG‐I (N) or cGAS and STING.
-
BProtein lysates of WT and USP19 KO A549 cells infected with SeV at the indicated time points were immunoblotted with the indicated antibodies.
-
CRT–PCR analysis of gene transcription in WT and USP19 KO A549 cells after infection with SeV at the indicated time points.
-
DPhase‐contrast (PH) and fluorescence microscopy images of WT and USP19 KO A549 cells infected with VSV‐eGFP at an MOI of 0.01 for the indicated time length. Scale bar, 50 μm.
-
EHuman PBMCs were transfected with control or USP19‐specific siRNA, followed by treatment with influenza A/Puerto Rico/8/34 (H1N1) (PR8) (MOI = 5) at different time points, and the lysates were analyzed with each antibody.
-
FHuman PBMCs were transfected with control or USP19‐specific siRNA and infected with H1N1 virus (MOI = 5) at different time points. Relative expression levels of IFNB, ISG15, ISG54, ISG56 mRNA, and H1N1 nucleoprotein (NP) RNA were measured by real‐time PCR.
-
GRelative expression levels of IFNB mRNA and interferon‐stimulated genes, including ISG54 and ISG56 in WT, BECN1, ATG5, and SQSTM1 KO cells with or without USP19.
-
H, ILuciferase activity in WT and BECN1 KO 293T cells transfected with an ISRE (H) or IFN‐β (I) promoter‐driven luciferase reporter after infection with SeV for the indicated time points.
Figure EV4. USP19 negatively regulates type I IFN signaling.
-
AFlag‐USP19‐inducible A549 cells were incubated with Doxy (200 ng/ml) overnight and infected with SeV for the indicated time points. Lysates of cells were immunoblotted with the indicated antibodies.
-
B, CFlag‐USP19‐inducible A549 cells were treated with Doxy (200 ng/ml) overnight. The transcription levels of IFNB and interferon‐stimulated genes (including ISG15 and ISG54) after SeV (B) or VSV (C) infection were analyzed by quantitative RT–PCR.
-
DPhase‐contrast (PH) and fluorescence microscopy analyses of Flag‐USP19‐inducible A549 cells with or without Doxy treatment infected with VSV‐eGFP at an MOI of 0.01 at the indicated time. Scale bar, 50 μm.
-
ERT–PCR analysis of gene transcription in WT and USP19 KO A549 cells after infection with VSV at the indicated time points.
Data information: Data are representative of three independent experiments, n = 3. Error bars indicate the SEM. **P < 0.01, ***P < 0.001 (two‐tailed Student's t‐test).
To demonstrate the consequence of reduced type I IFN expression mediated by USP19 over antiviral immunity, we treated USP19‐inducible cells with Doxy, then infected the cells with VSV‐eGFP (multiplicities of infection (MOI) = 0.01), and monitored viral infection based on GFP expression. Overexpression of USP19 (with Doxy) rendered the cells more susceptible to viral infection and resulted in considerably more GFP+ (virus‐infected) cells than mock cells (Fig EV4D). Real‐time PCR analysis also revealed that overexpression of USP19 resulted in more replication of VSV‐eGFP (Fig EV4C). Consistently, USP19 KO cells were more resistant to VSV‐eGFP infection and showed considerably fewer GFP+ cells than WT cells (Fig 6D). The inhibition of antiviral immunity by USP19 was further confirmed by SeV infection (Figs 6C and EV4B).
Influenza A is a segmented, single‐stranded, negative‐sense RNA virus that has adapted to infect multiple species (Shapira et al, 2009). Once infected by influenza, host cells recognize viral RNA through pathogen sensors, such as RIG‐I and induce type I IFNs production to initiate an antiviral responses (He et al, 2011). To further study the functional contribution of USP19 by influenza infection, we measured the phosphorylation levels of TBK1 and IRF3 after human influenza virus A (H1N1) infection. We found that the phosphorylation levels of TBK1 and IRF3 were significantly higher in human PBMCs transfected with USP19‐specific siRNA than that with control siRNA (Fig 6E). Real‐time PCR analysis revealed that depletion of USP19 resulted in higher transcription of IFNΒ, ISG15, ISG54, and ISG56 by H1N1 infection (Fig 6F). We further measured the replication of H1N1 virus by detecting the influenza virus nucleoprotein (NP) RNA levels, and found that USP19 depletion resulted in less influenza virus replication in human PBMCs (Fig 6F). Taken together, these results suggest that USP19 is a negative regulator of RIG‐I‐mediated type I IFN signaling as well as antiviral immunity.
USP19 suppresses type I IFN signaling in a Beclin‐1‐dependent manner
To determine whether the inhibition of type I IFN signaling by USP19 was dependent on autophagy process, we examined the mRNA level of IFNB and several ISG genes in BECN1, ATG5, and SQSTM1 KO cells. We found that USP19 lost the ability to suppress the transcription of these genes only in BECN1 KO cells, but not in ATG5 or SQSTM1 KO cells (Fig 6G). These results indicated that the inhibition of type I IFN signaling by USP19 is associated with Beclin‐1, rather than with autophagy. We then performed the luciferase reporter assay to test whether USP19 inhibits type I IFN activation in a Beclin‐1‐dependent manner and observed the inhibition of ISRE and IFN‐β promoter‐driven luciferase activation by USP19 was almost abolished in BECN1 KO cells (Fig 6H and I). Collectively, our results suggested that USP19 suppresses type I IFN signaling, a process that relies on the non‐autophagic role of Beclin‐1.
Beclin‐1 and USP19 inhibit the interaction between RIG‐I and MAVS
To identify the non‐canonical role of Beclin‐1 in antiviral immunity, we performed luciferase assay and revealed that BECN1 deficiency promoted the activation of the ISRE and IFN‐β luciferase reporter induced by SeV, while restored expression of Beclin‐1 suppressed the activation of both luciferase reporters in BECN1 KO cells (Fig 7A). We then examined the effect of Beclin‐1 on the phosphorylation of TBK1 and IRF3. As shown in Fig 7B, the phosphorylation levels of TBK1 and IRF3 in BECN1 KO cells were much higher than those in WT cells from 8 to 12 h after SeV infection.
Figure 7. Beclin‐1 inhibits the interaction between RIG‐I and MAVS .
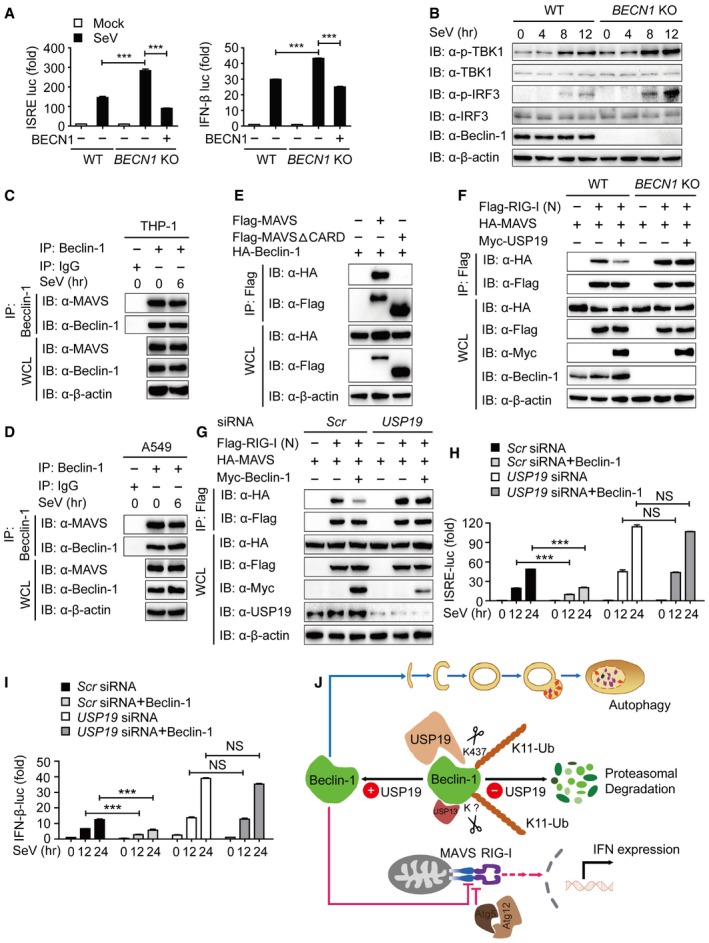
- Luciferase activity in WT and BECN1 KO 293T cells transfected with an ISRE (left) or IFN‐β (right) promoter‐driven luciferase reporter after infection with SeV for 12 h.
- WT and BECN1 KO 293T cells were infected with SeV for the indicated time points, then protein extracts were analyzed by immunoblot using the indicated antibodies.
- Protein extracts of THP‐1 cells infected with SeV for various time points (upper lanes) were subjected to immunoprecipitation with anti‐Beclin‐1 and immunoblot analysis with antibodies shown on the left.
- Coimmunoprecipitation and immunoblot analysis of protein lysates of A549 cells infected with SeV.
- 293T cells were transfected with vectors encoding HA‐Beclin‐1 and Flag‐MAVS or Flag‐MAVSΔCARD, followed by IP with anti‐Flag beads and immunoblot analysis with anti‐HA.
- Coimmunoprecipitation and immunoblot analysis of WT and BECN1 KO 293T cells transfected with HA‐MAVS plasmid along with vector encoding Flag‐RIG‐I (N) in the absence or presence of USP19.
- Protein extracts of scramble or USP19‐specific siRNA‐treated 293T cells transfected with HA‐MAVS plasmid along with vector encoding for Flag‐RIG‐I (N) in the absence or presence of Beclin‐1 were subjected to immunoprecipitation and immunoblot analysis.
- Luciferase activity in 293T cells transfected with scramble or USP19‐specific siRNA and ISRE luciferase reporter after SeV infection for the indicated time points with or without Beclin‐1.
- Luciferase activity in 293T cells transfected with scramble or USP19‐specific siRNA and IFN‐β luciferase reporter after SeV infection for the indicated time points with or without Beclin‐1.
- A proposed working model to illustrate how the USP19‐Beclin‐1 axis positively regulates autophagy and negatively regulates type I IFN signaling.
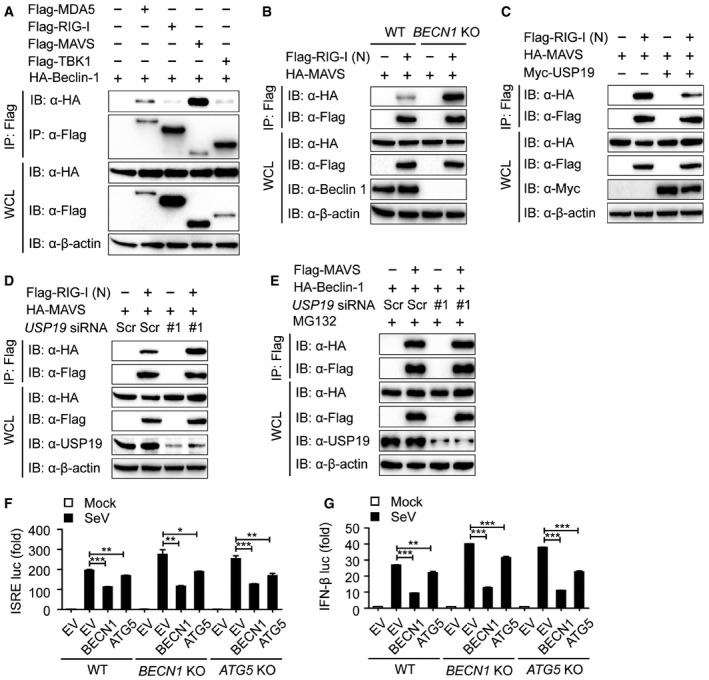
To gain insights into the mechanisms underlying Beclin‐1‐mediated inhibition in type I IFN signaling, we examined the interaction between Beclin‐1 and the upstream proteins of type I IFN signaling. Co‐IP and immunoblot analyses revealed that Beclin‐1 strongly interacted with MAVS (mitochondrial antiviral signaling, also known as IPS‐1, CARDIF, and VISA) (Fig EV5A), which can be further recapitulated in A549 and THP‐1 cells (Fig 7C and D). MAVS is an essential adaptor protein of RIG‐I like receptor (RLR)‐mediated type I IFN signaling, which contains the similar CARD domain as the RLRs. Once RIG‐I and MDA5 recognizes viral RNAs, they expose their CARD domains and activate MAVS by the homotypic interaction of their CARD domains (Cui et al, 2014; Schneider et al, 2014; Wu & Hur, 2015). Upon depletion of the CARD domain of MAVS, we observed that the interaction between Beclin‐1 and MAVS was abolished (Fig 7E ). Our results implied that Beclin‐1 interacts with the CARD domain of MAVS and blocks the interaction between MAVS and RIG‐I. We further checked the association of the RIG‐I (N) (CARD domain of RIG‐I) with MAVS in BECN1 KO cells. Interestingly, we observed that knockout of BECN1 enhanced the interaction between the RIG‐I (N) and MAVS (Fig EV5B). We next investigated whether USP19 inhibits type I IFN signaling in a similar way as Beclin‐1 does. As shown in Fig EV5C and D, USP19 markedly blocked the association of RIG‐I (N) with MAVS. Due to the upstream regulation of Beclin‐1 by USP19, we further determined whether the inhibition of RIG‐I‐MAVS axis by USP19 relied on Beclin‐1 and observed that USP19 could no longer suppress RIG‐I‐MAVS interaction in BECN1 KO cells (Fig 7F). In addition, USP19 depletion resulted in the dysfunction Beclin‐1 in the regulation of RIG‐I‐MAVS axis (Fig 7G). Our results also showed that inhibition of Beclin‐1 in the activation of the ISRE and IFN‐β luciferase reporter was almost abolished by USP19 depletion (Fig 7H and I). As USP19 interacted with Beclin‐1 and protected it from proteasomal degradation, we then checked whether USP19 affects the interaction between Beclin‐1 and MAVS. Figure EV5E showed that association of Beclin‐1 and MAVS remained unchanged in USP19 knockdown cells under the treatment of MG132. Taken together, our results suggest that USP19‐mediated stabilization of Beclin‐1 suppresses RIG‐I‐MAVS axis and further negatively regulates type I IFN signaling.
Figure EV5. USP19‐Beclin‐1 suppresses RIG‐I‐MAVS association.
-
A293T cells were transfected with vectors encoding HA‐Beclin‐1 and Flag‐MDA5, Flag‐RIG‐I, Flag‐MAVS, or Flag‐TBK1, followed by immunoprecipitation with anti‐Flag beads and immunoblot analysis with anti‐HA.
-
BCoimmunoprecipitation and immunoblot analysis of WT and BECN1 KO 293T cells transfected with HA‐MAVS plasmid along with vector encoding for Flag‐RIG‐I (N).
-
CCoimmunoprecipitation and immunoblot analysis of 293T cells transfected with HA‐MAVS plasmid along with vector encoding for Flag‐RIG‐I (N) in the absence or presence of USP19.
-
DExtracts of scramble or USP19‐specific siRNA‐treated 293T cells transfected with HA‐MAVS plasmid along with vector encoding for Flag‐RIG‐I were subjected to immunoprecipitation and immunoblot analysis.
-
E293T cells transfected with scramble or USP19‐specific siRNA and then transfected with HA‐Beclin‐1 plasmid along with plasmid encoding for Flag‐MAVS with MG132 treatment for 4 h were subjected to immunoprecipitation and immunoblot analysis.
-
F, GLuciferase activity in 293T cells transfected with plasmid encoding for Beclin‐1 or ATG5 and ISRE (F) or IFN‐β (G) luciferase reporter after SeV infection for 24 h. Data are representative of three independent experiments, n = 3. Error bars indicate the SEM. *P < 0.05, **P < 0.01, ***P < 0.001 (two‐tailed Student's t‐test).
Since it has been known that Atg5‐Atg12 conjugate negatively regulates the type I IFN signaling by association with the RIG‐I and MAVS through CARD (Jounai et al, 2007), we studied whether Beclin‐1 inhibits RIG‐I‐mediated signaling in an Atg5‐dependent manner. We found that the Beclin‐1 could still inhibit type I IFN activation in ATG5 KO cells (Fig EV5F and G). These results indicate that Beclin‐1 suppresses RIG‐I‐mediated signaling pathway in an Atg5‐independent manner.
Based on our findings, we propose the following working model to explain how USP19‐Beclin‐1 regulates autophagy and type I IFN signaling. K11‐linked ubiquitination controls the degradation of Beclin‐1 through proteasome pathway, while USP13 and USP19 reverse this modification and rescue Beclin‐1 degradation. USP19 interacts with Beclin‐1 and removes its K11‐linked ubiquitin chains at K437. In RLR‐mediated type I IFN signaling, Atg5‐Atg12 conjugate associates with RIG‐I, blocking its interaction with MAVS; meanwhile, Beclin‐1 interacts with CARD domain of MAVS. The stabilized Beclin‐1 promotes autophagic flux, while suppresses RIG‐I‐MAVS interaction, thereby inhibiting the activation of type I interferon signaling (Fig 7J).
Discussion
Beclin‐1 is mammalian ortholog of yeast ATG6, and biallelic loss of BECN1 results in early embryonic lethality in mice. Mice with monoallelic deletion of BECN1 spontaneously develop various malignancies, including lymphomas as well as lung and liver carcinomas, and are more susceptible to parity‐associated and Wnt1‐driven mammary carcinogenesis than their wild‐type counterparts (Galluzzi et al, 2015). Due to the pivotal role of Beclin‐1 in autophagy, the protein amount of Beclin‐1, as well as its activity, is tightly regulated by multiple post‐translational modifications (PTM). Ubiquitination is an essential PTM process which is involved in the regulation of many cellular activities (Zinngrebe et al, 2014). Although it has been reported that USP10 and USP13 can regulate the stability of Beclin‐1/VPS34 complex (Liu et al, 2011), the molecular mechanism underlying the ubiquitination of Beclin‐1 in the regulation of autophagy is still unclear. Recently, RNF216 has been shown to inhibit autophagy through promoting proteasome‐dependent degradation of Beclin‐1 via K48‐linked ubiquitination (Xu et al, 2014). In this report, we demonstrated that USP19 is required to stabilize Beclin‐1 during autophagy. Our results indicated that USP19 maintains Beclin‐1 stability through deubiquitination of Beclin‐1 in a USP13‐independent manner. We also demonstrated that USP19 cleaves the K11‐linked poly‐ubiquitin chains on Beclin‐1 at Lys437. For this atypical ubiquitin linkage type, its cellular role is largely unclear. Previous study revealed that K11‐linked poly‐ubiquitin chains are important in cell cycle regulation, where they seem to constitute an alternative proteasomal degradation signal (Michel et al, 2015). Our results suggest that K11‐linked ubiquitination is a signal for Beclin‐1 degradation and USP19 reverses this modification. Furthermore, USP19 overexpression is sufficient to promote autophagic flux. Thus, USP19 functions as a guardian for Beclin‐1 stabilization and autophagosome turnover.
Acetylation and ubiquitination are both associated with lysine residue. Recently, K437 site of Beclin‐1 has been identified as an acetylation site. Beclin‐1 acetylation inhibits autophagosome maturation and endocytic trafficking by promoting the recruitment of Rubicon. In tumor xenografts, the expression of 2KR mutant Beclin‐1 (substitution of K430 and K437 to arginines) leads to enhanced autophagosome maturation and tumor growth suppression (Xia et al, 2013; Sun et al, 2015). In our results, lysine 437 also serves as a critical site for K11‐linked ubiquitination and degradation of Beclin‐1, while USP19 reverses this process. In addition, we found that the K11‐linked ubiquitination of WT Beclin‐1 is decreased during starvation‐induced conditions, whereas the ubiquitination of K437R mutant remains unaffected (Fig EV3B). These results indicate that multiple PTMs on lysine 437 of Beclin‐1 might tightly regulate its amount as well as activity.
Autophagy is a fundamental eukaryotic pathway that has multiple effects on immunity. Accumulating evidence indicated the cross talk between autophagy and type I interferon signaling. A recent study has revealed that Beclin‐1 interacts with cGAS and suppresses cGAMP synthesis by treatment with microbial DNA (Liang et al, 2014). In our study, we found that Beclin‐1 functions in RNA virus infection by association with MAVS CARD domain, which is indispensable for aggregation or oligomerization of MAVS and plays the central role in of RIG‐I‐/MDA5‐ and MAVS‐mediated signaling pathway (Wu & Hur, 2015). Through binding to the CARD domain of MAVS, Beclin‐1 acts as a negative regulator in type I interferon signaling by suppressing RIG‐I‐MAVS interaction. Our results are consistent with previous reports that autophagy can have a pro‐infective role (Saitoh et al, 2009; Martins et al, 2015). Beclin‐1 suppresses not only the cGAS‐mediated IFN signaling but also MAVS‐mediated antiviral responses. Considering that USP19 can still inhibit the activation of type I IFN signaling in ATG5 and SQSTM1 KO cells, the function of USP19 and Beclin‐1 in RLR‐mediated antiviral responses is not associated with autophagy (Fig 6F). Interestingly, unlike Atg5‐Atg12 conjugate, Beclin‐1 mainly targets MAVS (Fig EV5A). In ATG5 KO cells, the activation of luciferase reporters can be rescued only by overexpression of Beclin‐1 (Fig EV5F and G). It seems that Beclin‐1 is more flexible in the regulation of RLR‐mediated type I IFN signaling than Atg5‐Atg12 conjugate. Beclin‐1 and Atg5‐Atg12 conjugate function independently on each other, leading to the integration of autophagy and type I IFN signaling and broadening the multilayer regulation of the cross talk.
Taken together, USP19 serves as a deubiquitinating enzyme directly cleaving K11‐linked ubiquitin chains of Beclin‐1 at K437 and protecting it from degradation through proteasome pathway. USP19 coordinately controls type I IFN production and autophagic flux by targeting Beclin‐1. Through Beclin‐1 stabilization, USP19 promotes autophagic flux and represses RIG‐I‐MAVS association, thereby inhibiting the activation of TBK1/IRF3 and antiviral immunity. Our finding provides novel evidence to support the potential cross talk between autophagy and antiviral responses and expands our understanding of such interplays that are required in the therapeutic application.
Materials and Methods
Cell culture and reagents
HEK293T (human embryonic kidney 293T), HeLa, and A549 cells were cultured in DMEM (Gibco) with 10% (vol/vol) fetal bovine serum (Gibco). Human PBMCs and THP‐1 cells were maintained in RPMI‐1640 medium (Gibco) with 10% fetal bovine serum. To induce starvation, cells were washed with phosphate‐buffered saline (PBS) and incubated in EBSS (Gibco). Rapamycin, bafilomycin A1, and chloroquine were purchased from Sigma.
Plasmids and siRNA transfection
Constructs coding for USP19 and its fragment were cloned in the pcDNA3.1 vector for transient expression and into the FG‐EH‐DEST (provided by Xiaofeng Qin laboratory) for retroviral expression. Chemically synthesized 21‐nucleotide siRNA duplexes were obtained from Invitrogen and transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. RNA oligonucleotides used in this study are as follows:
NC: 5′‐GUUAUCGCAACGUGUCACGUA‐3′;
USP19 siRNA #1: 5′‐GACAGGGUCUCGAUAUGUUGC‐3′;
USP19 siRNA #2: 5′‐GAUCAAUGACUUGGUGGAGUU‐3′.
Immunoprecipitation and immunoblot analysis
For immunoprecipitation, whole‐cell extracts were prepared after transfection or stimulation with appropriate ligands, followed by incubation overnight with the appropriate antibodies plus Protein A/G beads (Pierce). For immunoprecipitation with anti‐Flag or anti‐HA, anti‐Flag or anti‐HA agarose gels (Sigma) were used. Beads were then washed five times with low‐salt lysis buffer, and immunoprecipitates were eluted with 2× SDS Loading Buffer and resolved by SDS–PAGE. Proteins were transferred to PVDF membranes (Bio‐Rad) and further incubated with the appropriate antibodies. Immobilon Western Chemiluminescent HRP Substrate (Millipore) was used for protein detection.
In vitro deubiquitination assay
Ubiquitinated Beclin‐1 was isolated from 293T cells transfected with expression vectors for HA‐K11 Ub and Flag‐Beclin‐1. Ubiquitinated Beclin‐1 was purified from the cell extracts with anti‐Flag affinity column in Flag‐lysis buffer (50 mM Tris–HCl (pH 7.8), 137 mM NaCl, 10 mM NaF, 1 mM EDTA, 1% Triton X‐100, 0.2% Sarcosyl, 1 mM DTT, 10% glycerol, and fresh proteinase inhibitors). After extensive washing with the Flag‐lysis buffer, the proteins were eluted with Flag peptides (Sigma). The recombinant Flag‐USP19 and its mutant were expressed in 293T cells and purified using Flag affinity column and eluted with Flag peptide. For in vitro deubiquitination assay, ubiquitinated Beclin‐1 protein was incubated with recombinant USP19 or its mutant in the deubiquitination buffer (50 mM Tris–HCl (pH 8.0), 50 mM NaCl, 1 mM EDTA, 10 mM DTT, 5% glycerol) for 2 h at 37°C.
Fluorescence microscopy
Cells were cultured on a glass‐bottomed dish and directly observed. For examination by immunofluorescence microscopy, cells grown on gelatin‐coated coverslips were fixed with 4% paraformaldehyde, permeabilized using 50 μg/ml digitonin, and then stained with specific antibodies. Colocalization images were examined under a Zeiss LSM 700 confocal microscope equipped with a × 100 (NA 1.4) oil‐immersion objective, and the LC3 puncta images were detected by Leica DMI3000 B with a × 100 oil‐immersion objective.
Virus infection
Human influenza virus A/Puerto Rico/8/34 (H1N1) (PR8) was kindly provided by Dr. Hui Zhang (Zhongshan Medical School, Sun Yat‐sen University). Sendai virus and VSV‐eGFP were kindly provided by Dr. Xiaofeng Qin (Sun Yat‐sen University). Cells were infected at various MOI, as previously described by Shapira et al (2009) and Cui et al (2014).
Statistical analyses
Data are represented as mean ± SD unless otherwise indicated, and Student's t‐test was used for all statistical analyses with the GraphPad Prism 5 software. Differences between two groups were considered significant when P‐value was < 0.05.
Author contributions
SJ, ST, and YC performed the experiments and analyzed the results. CZ and WX provided technical help. CZ, XX, and WX provided technical help. R‐FW and JC initiated and designed the project, directed the research, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
This work was supported by National Key Basic Research Program of China (2015CB859800 and 2014CB910800), National Natural Science Foundation of China (31370869, 31522018), Guangdong Natural Science Funds for Distinguished Young Scholar (S2013050014772), Guangdong Innovative Research Team Program (NO. 2011Y035 and 201001Y01046872443), and the Fundamental Research Funds for the Central Universities (15lgjc02), and NCI, NIH (CA090327, CA101795, and CA121191 to RFW).
The EMBO Journal (2016) 35: 866–880
References
- Choi AMK, Ryter SW, Levine B (2013) Autophagy in human health and disease. N Engl J Med 368: 651–662 [DOI] [PubMed] [Google Scholar]
- Cui J, Song YX, Li YY, Zhu QY, Tan P, Qin YF, Wang HY, Wang RF (2014) USP3 inhibits type I interferon signaling by deubiquitinating RIG‐I‐like receptors. Cell Res 24: 400–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic V, Saitoh T, Akira S (2013) Autophagy in infection, inflammation and immunity. Nat Rev Immunol 13: 722–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Pietrocola F, Bravo‐San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J, Gewirtz DA, Karantza V (2015) Autophagy in malignant transformation and cancer progression. EMBO J 34: 856–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassink GC, Zhao B, Sompallae R, Altun M, Gastaldello S, Zinin NV, Masucci MG, Lindsten K (2009) The ER‐resident ubiquitin‐specific protease 19 participates in the UPR and rescues ERAD substrates. EMBO Rep 10: 755–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W, Han H, Wang W, Gao B (2011) Anti‐influenza virus effect of aqueous extracts from dandelion. Virol J 8: 538–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husnjak K, Dikic I (2012) Ubiquitin‐binding proteins: decoders of ubiquitin‐mediated cellular functions. Annu Rev Biochem 81: 291–322 [DOI] [PubMed] [Google Scholar]
- Jounai N, Takeshita F, Kobiyama K, Sawano A, Miyawaki A, Xin K‐Q, Ishii KJ, Kawai T, Akira S, Suzuki K (2007) The Atg5‐Atg12 conjugate associates with innate antiviral immune responses. Proc Natl Acad Sci 104: 14050–14055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, Guan K‐L (2013) Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 152: 290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno H, Konno K, Barber GN (2013) Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell 155: 688–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuballa P, Nolte WM, Castoreno AB, Xavier RJ (2012) Autophagy and the immune system. Annu Rev Immunol 30: 611–646 [DOI] [PubMed] [Google Scholar]
- Lamb CA, Yoshimori T, Tooze SA (2013) The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol 14: 759–774 [DOI] [PubMed] [Google Scholar]
- Levine B, Liu R, Dong X, Zhong Q (2015) Beclin orthologs: integrative hubs of cell signaling, membrane trafficking, and physiology. Trends Cell Biol 25: 533–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Q, Seo GJ, Choi YJ, Kwak M‐J, Ge J, Rodgers MA, Shi M, Leslie BJ, Hopfner K‐P, Ha T (2014) Crosstalk between the cGAS DNA sensor and Beclin‐1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe 15: 228–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JL, Xia HG, Kim M, Xu LH, Li Y, Zhang LH, Cai Y, Norberg HV, Zhang T, Furuya T, Jin MZ, Zhu ZM, Wang HC, Yu J, Li YX, Hao Y, Choi A, Ke HM, Ma DW, Yuan JY (2011) Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 147: 223–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell MA, Jain A, Arko‐Mensah J, Chauhan S, Kimura T, Dinkins C, Silvestri G, Munch J, Kirchhoff F, Simonsen A, Wei YJ, Levine B, Johansen T, Deretic V (2014) TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev Cell 30: 394–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins JD, Liberal J, Silva A, Ferreira I, Neves BM, Cruz MT (2015) Autophagy and inflammasome interplay. DNA Cell Biol 34: 274–281 [DOI] [PubMed] [Google Scholar]
- Mei YD, Hahn AA, Hu SM, Yang XL (2011) The USP19 deubiquitinase regulates the stability of c‐IAP1 and c‐IAP2. J Biol Chem 286: 35380–35387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel MA, Elliott PR, Swatek KN, Simicek M, Pruneda JN, Wagstaff JL, Freund SM, Komander D (2015) Assembly and specific recognition of K29‐and K33‐linked poly‐ubiquitin. Mol Cell 58: 95–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, Gretzmeier C, Dengjel J, Piacentini M, Fimia GM, Cecconi F (2013) mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self‐association and function through AMBRA1 and TRAF6. Nat Cell Biol 15: 406–416 [DOI] [PubMed] [Google Scholar]
- Rajsbaum R, Versteeg GA, Schmid S, Maestre AM, Belicha‐Villanueva A, Martinez‐Romero C, Patel JR, Morrison J, Pisanelli G, Miorin L, Laurent‐Rolle M, Moulton HM, Stein DA, Fernandez‐Sesma A, tenOever BR, Garcia‐Sastre A (2014) Unanchored K48‐linked poly‐ubiquitin synthesized by the E3‐ubiquitin ligase TRIM6 stimulates the interferon‐IKKepsilon kinase‐mediated antiviral response. Immunity 40: 880–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogov V, Dötsch V, Johansen T, Kirkin V (2014) Interactions between autophagy receptors and ubiquitin‐like proteins form the molecular basis for selective autophagy. Mol Cell 53: 167–178 [DOI] [PubMed] [Google Scholar]
- Russell RC, Tian Y, Yuan HX, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL (2013) ULK1 induces autophagy by phosphorylating Beclin‐1 and activating VPS34 lipid kinase. Nat Cell Biol 15: 741–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahtoe DD, Sixma TK (2015) Layers of DUB regulation. Trends Biochem Sci 40: 456–467 [DOI] [PubMed] [Google Scholar]
- Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, Matsunaga K, Kageyama S, Omori H, Noda T, Yamamoto N, Kawai T, Ishii K, Takeuchi O, Yoshimori T, Akira S (2009) Atg9a controls dsDNA‐driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci USA 106: 20842–20846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider WM, Chevillotte MD, Rice CM (2014) Interferon‐stimulated genes: a complex web of host defenses. Annu Rev Immunol 32: 513–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira SD, Gat‐Viks I, Shum BO, Dricot A, de Grace MM, Wu L, Gupta PB, Hao T, Silver SJ, Root DE (2009) A physical and regulatory map of host‐influenza interactions reveals pathways in H1N1 infection. Cell 139: 1255–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi CS, Kehrl JH (2010) TRAF6 and A20 regulate lysine 63‐linked ubiquitination of Beclin‐1 to control TLR4‐induced autophagy. Sci Signal 3: ra42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simicek M, Lievens S, Laga M, Guzenko D, Aushev VN, Kalev P, Baietti MF, Strelkov SV, Gevaert K, Tavernier J, Sablina AA (2013) The deubiquitylase USP33 discriminates between RALB functions in autophagy and innate immune response. Nat Cell Biol 15: 1220–1230 [DOI] [PubMed] [Google Scholar]
- Sun T, Li X, Zhang P, Chen W, Zhang H, Li D, Deng R, Qian X, Jiao L, Ji J (2015) Acetylation of Beclin 1 inhibits autophagosome maturation and promotes tumour growth. Nat Commun 6: 7215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaram P, Pang ZY, Miao M, Yu L, Wing SS (2009) USP19‐deubiquitinating enzyme regulates levels of major myofibrillar proteins in L6 muscle cells. Am J Physiol Endocrinol Metab 297: E1283–E1290 [DOI] [PubMed] [Google Scholar]
- Wu B, Hur S (2015) How RIG‐I like receptors activate MAVS. Curr Opin Virol 12: 91–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia ZP, Sun L, Chen X, Pineda G, Jiang X, Adhikari A, Zeng W, Chen ZJ (2009) Direct activation of protein kinases by unanchored poly‐ubiquitin chains. Nature 461: 114–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia P, Wang S, Du Y, Zhao Z, Shi L, Sun L, Huang G, Ye B, Li C, Dai Z (2013) WASH inhibits autophagy through suppression of Beclin 1 ubiquitination. EMBO J 32: 2685–2696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia PY, Wang S, Huang GL, Du Y, Zhu PP, Li M, Fan ZS (2014) RNF2 is recruited by WASH to ubiquitinate AMBRA1 leading to downregulation of autophagy. Cell Res 24: 943–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu CF, Feng K, Zhao XN, Huang SQ, Cheng YJ, Qian L, Wang YN, Sun HX, Jin M, Chuang TH, Zhang YY (2014) Regulation of autophagy by E3 ubiquitin ligase RNF216 through BECN1 ubiquitination. Autophagy 10: 2239–2250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinngrebe J, Montinaro A, Peltzer N, Walczak H (2014) Ubiquitin in the immune system. EMBO Rep 15: 28–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File