

Abstract
A mutation in the centrosomal‐P4.1‐associated protein (CPAP) causes Seckel syndrome with microcephaly, which is suggested to arise from a decline in neural progenitor cells (NPCs) during development. However, mechanisms of NPCs maintenance remain unclear. Here, we report an unexpected role for the cilium in NPCs maintenance and identify CPAP as a negative regulator of ciliary length independent of its role in centrosome biogenesis. At the onset of cilium disassembly, CPAP provides a scaffold for the cilium disassembly complex (CDC), which includes Nde1, Aurora A, and OFD1, recruited to the ciliary base for timely cilium disassembly. In contrast, mutated CPAP fails to localize at the ciliary base associated with inefficient CDC recruitment, long cilia, retarded cilium disassembly, and delayed cell cycle re‐entry leading to premature differentiation of patient iPS‐derived NPCs. Aberrant CDC function also promotes premature differentiation of NPCs in Seckel iPS‐derived organoids. Thus, our results suggest a role for cilia in microcephaly and its involvement during neurogenesis and brain size control.
Keywords: brain organoids, CPAP, cilium, microcephaly, neural progenitor cell maintenance
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Development & Differentiation; Molecular Biology of Disease
Introduction
During interphase, the centriole within a centrosome nucleates the formation of the primary cilium, which is known to play key roles in signaling pathways (Berbari et al, 2009; Nigg & Raff, 2009; Jackson, 2011). The cilium is the primary microtubule‐based organelle that is dynamically regulated, with assembly occurring during cell cycle exit, and disassembly coinciding with cell cycle re‐entry (Jackson, 2011; Kim et al, 2011; Paridaen et al, 2013; Guemez‐Gamboa et al, 2014). Thus, a delay or failure in cilium disassembly could act as a brake, retaining cells in G0/G1 and transiently preventing cell cycle progression (Jackson, 2011; Kim et al, 2011; Li et al, 2011).
Mutations in centrosomal proteins cause developmental disorders such as primary microcephaly and Seckel syndrome (Thornton & Woods, 2009; Kalay et al, 2011; McIntyre et al, 2012; Lancaster et al, 2013; Alcantara & O'Driscoll, 2014; Bazzi & Anderson, 2014; Insolera et al, 2014; Martin et al, 2014). Microcephaly is a neurodevelopmental disorder leading to extreme reduction in brain size. In Seckel syndrome, the reduced brain size is coupled with pre‐ and postnatal reduction in body size. A mutation in the conserved centrosomal protein CPAP causes Seckel syndrome, characterized by dwarfism, low birthweight, microcephaly and intellectual disability (Al‐Dosari et al, 2010).
The cell biological basis for microcephaly‐linked disorders remains largely unclear. Most studies suggest a decline in the neural stem cell pool to eventually lead to a structurally normal but small‐sized brain (Thornton & Woods, 2009; Alcantara & O'Driscoll, 2014). In the developing neural epithelium, stem cell numbers are rapidly expanded through its own symmetrical division (Rakic, 1995). This step is crucial to generate a large enough pool of neural stem cells that undergo asymmetric division producing cells that form various cortical layers in later stages of development (Alcantara & O'Driscoll, 2014). By impairing stem cell division even mildly, the proportion of symmetric divisions is concomitantly reduced, depleting the progenitor pool and limiting the total number of neurons that can be generated (Thornton & Woods, 2009; Alcantara & O'Driscoll, 2014). On the other hand, altering the cell cycle length, in particular the G1 phase, contributes to switching of neural progenitors from proliferative to differentiating states (Calegari et al, 2005; Lange et al, 2009; Li et al, 2011).
Taken together, defective cellular division seems to underlie the depletion of a specific neural stem cell pool. However, how the centrosome or cilium regulates stem cell division to maintain a stem cell pool is largely unknown. Interestingly, till this day neither Seckel syndrome nor microcephaly disorders have been directly associated with clinical features of ciliopathies. Thus, the role of cilium in microcephaly disorders remains unclear. However, since centrosomes template ciliogenesis, it is conceivable that altered ciliogenesis could underlie the pathophysiology of microcephaly. Recent studies have uncovered that the proper timing of cilium disassembly mediated by Nde‐1 and TcTex‐1 is critically regulated during cell cycle (Kim et al, 2011; Li et al, 2011; Maskey et al, 2015). Interestingly, mutations in Nde‐1 and microtubule and kinetochore components causing microcephaly and ciliary defects highlight a possible role for the cilium in regulating NPCs to control the number of neurons generated during brain development (Alkuraya et al, 2011; Hu et al, 2014; Waters et al, 2015). However, the biological significance of these spatiotemporally interlinked processes namely cilium disassembly, cell cycle re‐entry and neural progenitor cells (NPCs) differentiation, and the molecular mechanisms linking these cellular events together remain unclear.
Here, we investigate the effects of cilium disassembly on differentiation of NPCs derived from Seckel patient‐induced pluripotent stem (iPS) cells. To model Seckel syndrome in a disease‐relevant tissue, we developed human Seckel iPS‐derived organoids. Our initial studies indicate Seckel cells to contain numerically normal centrosomes and mitotic spindle poles. However, upon further analysis, unusually long cilia, retarded cilia disassembly and delayed cell cycle re‐entry were observed. This promoted premature differentiation of NPCs, identifying a previously unknown role for CPAP‐mediated cilium disassembly in maintaining the neural stem cell pool. Our findings also uncover a yet‐unknown feature of NPCs differentiation that is coupled with a timely cilium disassembly.
Results
Seckel fibroblasts have long cilia and reduced proliferation
CPAP plays a role in centriole formation and provides a scaffold for protein complexes during centrosome assembly (Hung et al, 2004; Gopalakrishnan et al, 2011, 2012; Lin et al, 2013; Zheng et al, 2014). However, the consequences of how a CPAP mutation (Seckel variant CPAP) underlies Seckel syndrome are unknown. To address this, we examined the centrosomes of Seckel patient‐derived fibroblasts carrying a splice‐site mutation in CPAP introducing a homozygous G‐to‐C transition in the last nucleotide of intron 11 causing the deletion of exons 11, 12, and/or 13 (Appendix Fig S1A–C; Al‐Dosari et al, 2010).
In contrast to other microcephaly or Seckel cells that display numerically aberrant centrosomes, cells in the current study displayed correct numbers of centrosomes and mitotic spindle poles (Fig 1A; Rauch et al, 2008; Kalay et al, 2011; Hussain et al, 2012; Martin et al, 2014). However, interphase centrosomes displayed faint or no CPAP immunoreactivity. Importantly, compared to wild‐type (WT) fibroblasts, Seckel cells recruited similar levels of other centrosomal proteins (γ‐tubulin, pericentrin, and Cep152) (Fig 1A). These data suggest an altered function for the Seckel variant CPAP in an interphase‐specific process.
Figure 1. Seckel cells display overly long cilia and reduced proliferation.
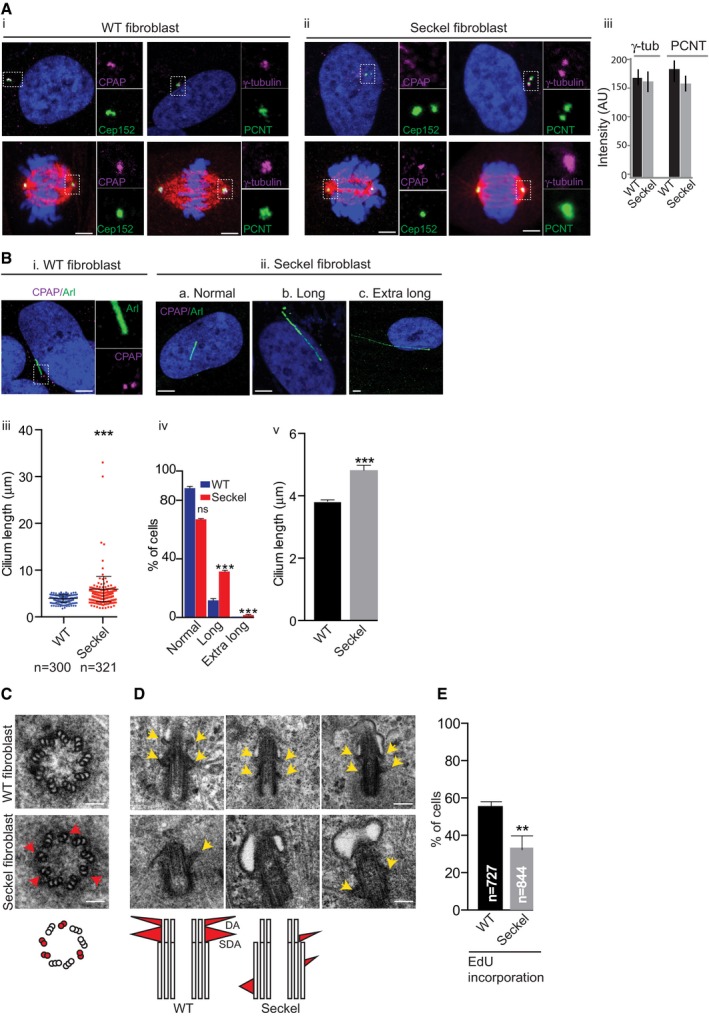
- Seckel cells contain normal centrosome numbers and mitotic spindle poles. Interphase centrosomes of Seckel cells have no CPAP immunoreactivity but recruit similar levels of centrosomal proteins (ii and iii). Error bars are SD. n = 3 independent experiments, > 60 cells in each condition. Scale bar, 1 μm.
- Seckel cells display overly long cilia. Arl13b labels cilium. Scale bar, 1 μm. (iii) Quantification of cilia length, scatter plot showing mean with ± SD; ***P < 0.001, t‐test; (iv) cells displaying normal (0–5 μm), long (5–10 μm), and extra long (> 10 μm) cilia. ***P < 0.001, ordinary two‐way ANOVA. (v) Average cilia length of WT and Seckel cells. n = 3 independent experiments, 228 cells for WT and 297 cells for Seckel. Error bars are SEM. ***P < 0.001, t‐test.
- Electron microscopy (EM) of Seckel and WT fibroblasts. In contrast to WT, Seckel basal body has disorganized microtubule of doublet and triplets (red arrows and red circles in schematics). Scale bar, 100 nm.
- Longitudinal serial‐sectioning EM of WT (top) Seckel fibroblasts (bottom). In contrast to WT basal bodies that have structurally normal distal and sub‐distal appendages (yellow arrows and red triangles of DA and SDA in schematics), Seckel basal body displays either lack or misplaced appendages. n > 15 basal bodies. Scale bar, 100 nm.
- Seckel cells show reduced EdU incorporation after 72 h. Error bars are SEM. **P < 0.01, t‐test; n = 4 independent experiments, 727 cells for WT and 844 cells for Seckel. Scale bar, 100 nm.
As centrioles template cilium formation in interphase, we examined the cilium in asynchronously proliferating cells cultured in the presence of serum and found a significant proportion of Seckel cells (134 out of 321 cells) to contain unusually long cilia measuring up to 30 μm in length with a median length of 4.46 μm compared to 3.74 μm for WT cells (Fig 1B and Appendix Fig S2A). Ultra‐structural analyses of Seckel cilia revealed disorganized abnormal ciliary appendages, and discontinuity of microtubular blades of basal bodies. These observations indicate aberrant ciliogenesis in CPAP‐mutated Seckel cells (Fig 1C and D).
In vertebrate cells, an elongated cilium is associated with suppression of cell division via a delay in G1‐S transition (Jackson, 2011; Kim et al, 2011; Li et al, 2011). As Seckel cells have long cilia, we speculated that they would also have a delayed G1‐S transition. Indeed, our 72‐h pulse labeling experiments using ethynyl‐deoxyuridine (EdU) revealed a reduced number of Seckel cells with EdU incorporation indicating that fewer cells are in S‐phase (Fig 1E). Furthermore, our fluorescence ubiquitination cell cycle indicator (FUCCI)‐based analyses revealed increased proportions of Seckel cells retained in G1 (Appendix Fig S2B).
To identify the transcript(s) responsible for the Seckel phenotype, individual transcripts were overexpressed in ciliated human retinal pigment epithelial cells (RPE1), causing long cilia accompanied with cell cycle delay, thus suggesting that all the transcripts might contribute to the observed phenotypes (Appendix Fig S3A and B). Importantly, to some extent, expression of WT CPAP using bacterial artificial chromosome (BAC) recombineering in Seckel fibroblasts rescued interphase CPAP localization and cilium lengths (Appendix Fig S3C).
These data suggest that the long cilium caused by a delay or failure in cilium disassembly acts as a brake, transiently preventing cell cycle progression, and might thus underlie the pathophysiology of Seckel syndrome.
Seckel cells display retarded cilium disassembly and cell cycle re‐entry
To study cilium disassembly and cell cycle re‐entry, we synchronized WT and Seckel fibroblasts at G0 by serum starvation for 96 h. Both Seckel and WT cells responded to this treatment acquiring equivalent ciliary length allowing us to estimate the rate of cilia disassembly upon serum stimulation (Fig 2A). Ciliated G0 WT fibroblasts displayed faint or no CPAP labeling at the base of the cilium (Fig 2B). However, CPAP levels were elevated 1 h after serum stimulation (Fig 2B and Appendix Fig S4B). After 24 h, CPAP was focused at centrosomes, concurrent with cilium disassembly and cell cycle re‐entry. Remarkably, the elevated levels of CPAP correlated with biphasic cilium disassembly, the first occurring 0–1 h after serum stimulation and the second after 4–24 h (Pugacheva et al, 2007; Fig 2B and Di–ii). In contrast, Seckel cells lacked dynamic CPAP localizations and exhibited retarded cilium disassembly and cell cycle re‐entry upon serum stimulation as indicated by the significant number of cells negative for cyclin A immuno‐reactivity, a G1‐S transition marker (Fig 2C and E, and Appendix Fig S4C). While most WT cells entered mitosis 24 h after serum stimulation, mitotic Seckel cells were only observed after 48 h. Interestingly, a fraction of these Seckel fibroblasts still harbored Arl13b‐positive ciliary remnants which are ciliary membrane structures observed near to a centriole (Paridaen et al, 2013; Fig 2F). These data suggest that CPAP plays a role in resorbing cilia before mitosis (Fig 2F).
Figure 2. CPAP plays a role in cilium disassembly.
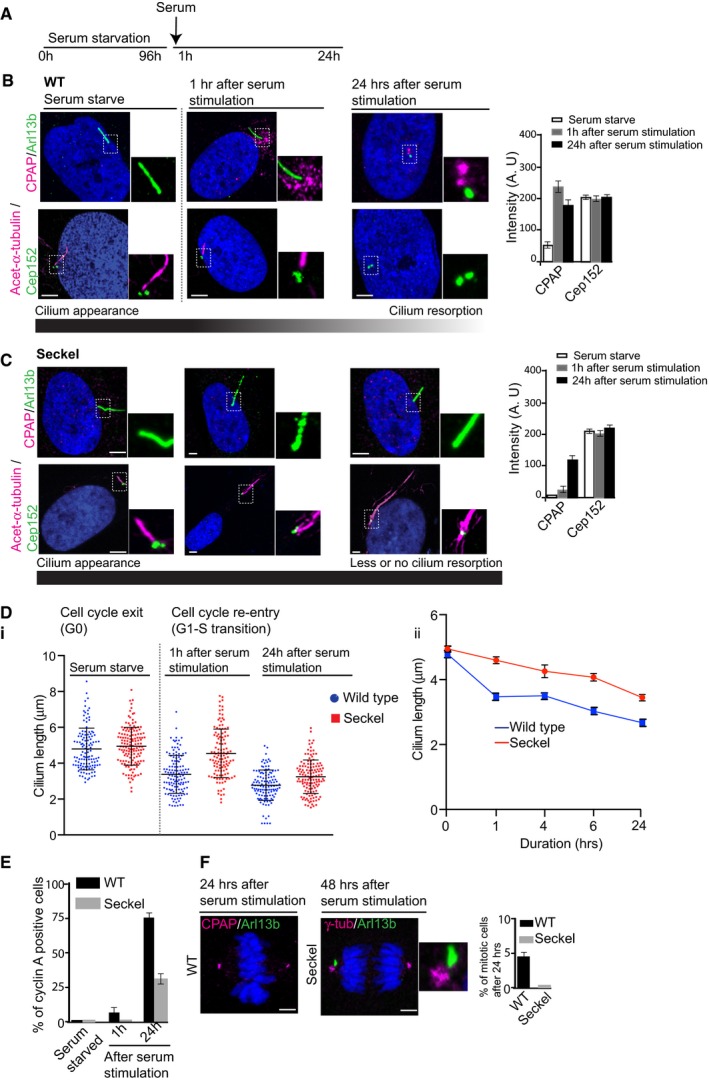
- Experimental plan. Both Seckel and WT cells were synchronized at G0 by serum starvation for 96 h. This treatment causes Seckel and WT cells to have equivalent cilia length allowing us to estimate the rate of cilia disassembly (see below and Appendix Fig S2A).
- Dynamic CPAP localization during cilium disassembly and cell cycle re‐entry. Cep152 levels remain unchanged. Arl13b (green) or acetylated α‐tubulin (magenta) marks the cilium. n > 100 cells. Bar diagram on the right shows semiquantitative intensity measurements. Error bars are ± SD. Scale bar, 1 μm.
- Seckel cells show neither CPAP dynamics nor cilium disassembly upon serum stimulation. Cep152 levels remain constant. n > 100 cells. Bar diagram on the right shows semiquantitative intensity measurements. Error bars are ± SD. Scale bar, 1 μm.
- Cilium disassembly dynamics in WT and Seckel cells. Relative cilium disassembly rate shows retarded cilium disassembly in Seckel cells (error bars are SEM) (ii). Quantification and distribution of cilia length in WT and Seckel fibroblasts. Scatter plot shows mean with ± SD (i).
- Seckel cells with retarded cilium disassembly show delayed G1‐S transition assessed by nuclear cyclin A immunoreactivity. n = 3 independent experiments, n > 200 cells for WT and Seckel. Error bars are SEM.
- Seckel cells show delayed mitotic entry. Note that an unresorbed Arl13b‐positive ciliary remnant (ciliary membrane, green) is present with one of the centrosomes (Paridaen et al, 2013). Bar diagram shows mitotic index of WT and Seckel cells 24 h after serum stimulation. n = 3 independent experiments, n > 300 cells for WT and Seckel. Error bars are SEM. Scale bar, 1 μm.
To establish the link between cilium disassembly and cell cycle progression, we overexpressed CPAP's CC5 domain, that is, the region of aa 1073–1159 deleted in Seckel variant CPAP, in RPE1 cells. We found a significant proportion of CC5‐GFP‐expressing cells to have either short or no cilia with a concurrent increase in EdU incorporation, suggesting that accelerating ciliary disassembly concomitantly accelerates cell cycle progression (Appendix Fig S5A).
CPAP provides a scaffold for the cilium disassembly complex
To gain insight into how CPAP coordinates cilium disassembly, we tested whether CPAP could interact with proteins, which are known to play roles in cilium disassembly such as Nde‐1, Aurora A, HDAC6 and OFD1 (Pugacheva et al, 2007; Kim et al, 2011; Tang et al, 2013; hereafter referred to as Cilium Disassembly Complex, CDC). Indeed, cytoplasmic CPAP co‐immunopurified with components of the CDC (CPAP‐CDC; Fig 3A). Interestingly, CDC component levels were elevated in WT fibroblasts specifically 1 h after serum stimulation (Fig 3B). The elevated levels of these proteins correlated with CPAP levels and expression of cyclin A (Figs 2B and 3B). All of these processes occur concomitantly with the onset of cilium disassembly (Figs 2B and 3B).
Figure 3. CPAP provides a scaffold for the cilium disassembly complex.
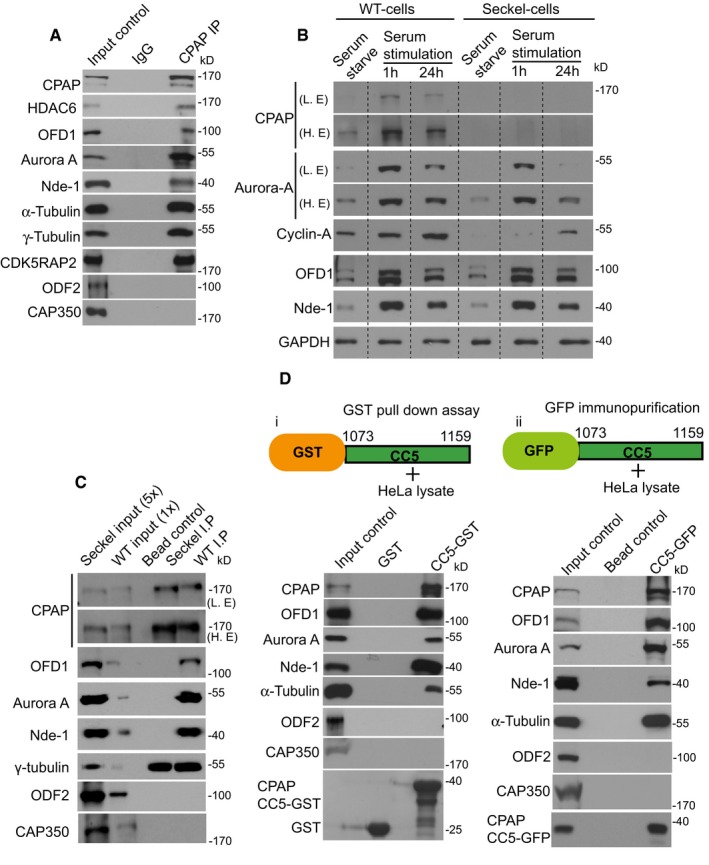
- CPAP co‐immunopurifies with CDC components from HeLa extracts. Centrosomal proteins Cap350 and ODF2 do not co‐purify with CPAP.
- Levels of CDC components (CPAP, Aurora A, OFD1, and Nde‐1) are differentially regulated in response to serum starvation and stimulation in WT and Seckel fibroblasts. Specifically, they are elevated 1 h after serum stimulation, thus enabling cilium disassembly. Elevated CDC levels in WT cells correlate with an elevated level of CPAP as shown in Fig 2 as well as with the expression of cyclin A, a G1‐S marker. GAPDH was used as a loading control indicating an equivalent amount of WT and Seckel fibroblast total proteins were loaded.
- WT CPAP (WT cells IP) but not Seckel variant CPAP (Seckel cells IP) co‐immunopurifies with CDC components from asynchronously grown WT and Seckel fibroblasts cell extracts. The IP samples were adjusted so that equivalent amounts of immunoprecipitated CPAP were used in samples. Seckel (5×) and WT (1×) fibroblast cell extracts were used as input controls to show the efficacy of the various antibodies. Note that CPAP abundance in Seckel cells is ˜5 times lower than that of WT cells. Centrosomal proteins Cap350 and ODF2 do not co‐purify with CPAP indicating the specificity of CPAP interaction with CDC components.
- GST pull‐down (i) and immunopurification assays (ii) of CPAP's CC5 domain demonstrate its sufficiency in mediating CPAP‐CDC protein interactions. The CC5 fragment is expressed as a GFP fusion protein in HeLa cells and immunopurified using anti‐GFP antibodies. HeLa extracts were used as input controls. CAP350 does not co‐purify with CPAP. Both Cap350 and ODF2 are not pulled down by CPAP's CC5 domain.
We then immunopurified endogenous cytoplasmic CPAP complexes from asynchronously proliferating WT and Seckel fibro‐blasts and found the Seckel variant CPAP to show reduced interaction with CDC components, although expression of CDC components was also increased by serum stimulation in Seckel cells (Fig 3B and C, and Appendix Fig S5D). The elevated levels of CDC components upon serum stimulation in Seckel cells indicate that they can respond to serum stimulation. Thus, the observed delay in cilium disassembly and cell cycle re‐entry is likely due to either Seckel variant CPAP's lack of scaffolding function or the absence of CPAP at the ciliary base causing inefficient targeting of CDC components (Appendix Fig S5B and C). The delayed cell cycle re‐entry is further reaffirmed by the low levels of nuclear cyclin A in Seckel cells even upon serum stimulation. By performing glutathione S‐transferase (GST) pull‐down and immunopurification assays with CC5, we found the CC5 domain to be sufficient to mediate these interactions (Fig 3D).
To confirm CC5‐dependent recruitment of CDC components during cilium disassembly, we analyzed CDC components in WT and Seckel fibroblasts synchronized by serum starvation and stimulation. In WT fibroblasts, CDC component levels were elevated after serum stimulation and recruited to the ciliary base, concurrent with cilium disassembly and cell cycle re‐entry. In contrast, although expression of CDC components was elevated in Seckel cells after serum stimulation, they failed to efficiently localize to the ciliary base, resulting in retarded cilium disassembly and cell cycle re‐entry (Fig 3B and Appendix Fig S6). These results suggest CPAP to play a specific role in spatiotemporal cilium disassembly by providing a scaffold for CDC components, independent of its role in centrosome biogenesis. Evidence for this is that Seckel cells display correct numbers of centrosomes and mitotic spindle poles (Fig 1A).
Retarded cilium disassembly causes premature NPCs differentiation
To further study the effects of cilium‐mediated cell cycle re‐entry delay on NPCs, we generated iPS cells, differentiated them into neuroectoderm followed by expansion into neural epithelium (Appendix Figs S7 and S8). Although control and Seckel neural epithelium developed neural rosettes (Perrier et al, 2004), Seckel rosettes were slightly decreased in size with disrupted structure (Appendix Fig S8B). Nonetheless, they could further be differentiated into NPCs. Interestingly, these Seckel rosettes already displayed the pan neuronal marker TUJ1 suggesting premature differentiation into neuronal cells (Appendix Fig S8B). Similar to our findings in fibroblasts, Seckel NPCs lacked CPAP at the base of the cilium and failed to efficiently recruit CDC components to the cilium (Fig 4A). As a result, an increased proportion of ciliated Seckel NPCs contained long cilia and exhibited reduced proliferation and delayed G1‐S transition (Fig 4A–D).
Figure 4. Retarded cilium disassembly causes premature neural differentiation.
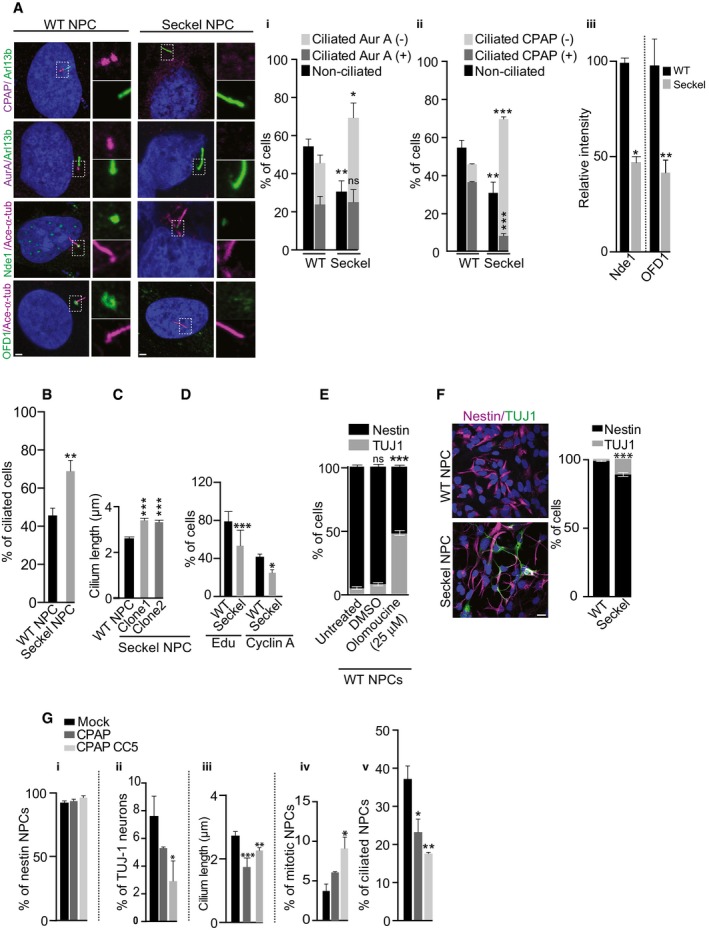
- Seckel NPCs fail to efficiently recruit CDC components. Fraction of non‐ciliated and ciliated WT and Seckel NPCs. Ciliated cells are further quantified and grouped based on whether they are positive or negative for Aurora A and CPAP at their basal bodies. Scale bar, 0.5 μm. (i–ii) Proportion of ciliated cells is higher in Seckel NPCs (**P < 0.01). Compared to WT, a higher proportion of Seckel NPCs lack Aurora A and CPAP in their basal bodies (*P < 0.05 for Aurora A and ***P < 0.001 for CPAP). Ordinary two‐way ANOVA followed by Sidak's multiple comparisons test. n = 3 independent experiments. 127 cells for WT and 97 cells for Seckel. Error bars are SEM. (iii) Seckel cells consistently contain Nde‐1 and Ofd1 with a significantly lesser intensity. Error bars are SEM; n = 3 independent experiments. 110 cells for WT and 100 cells for Seckel. *P < 0.05 for Nde‐1 and **P < 0.01 for Ofd1, t‐test.
- Proportion of ciliated cells is higher in Seckel NPCs. Error bars are SEM. **P < 0.01, t‐test. n = 3 independent experiments. 127 cells for WT and 97 cells for Seckel.
- Seckel NPCs exhibit longer cilia. Error bars are ± SEM. ***P < 0.001, t‐test. NPCs were derived from two independent iPS clones. n = 3 independent experiments. 146 cells for WT, 135 cells for Seckel (clone 1) and 202 for Seckel (clone 2).
- Seckel NPCs show reduced proliferation and delayed G1‐S transition. Error bars are ± SEM. ***P < 0.001 (EdU), *P < 0.05 (cyclin A), t‐test. For cyclin A, n = 3 independent experiments. For cyclin A, 270 cells for WT and 232 cells for Seckel cells. For EdU, 700 cells for WT and 1,630 cells for Seckel.
- Olomoucine triggers differentiation of WT NPCs into TUJ1‐positive neurons under non‐differentiating conditions. Error bars are SEM. ***P < 0.001, ordinary two‐way ANOVA followed by Tukey's multiple comparisons test. n = 3 independent experiments. 298 cells for untreated, 151 cells for DMSO, and 146 cells for olomoucine (25 μM).
- Seckel NPCs differentiated into TUJ1‐positive neurons under non‐differentiating conditions. Error bars show ± SEM. ***P < 0.001, t‐test. n = 3 independent experiments. 633 cells for WT and 275 cells for Seckel. Scale bar, 5 μm.
- Expression of CPAP‐GFP or CPAP‐CC5‐GFP in Seckel NPCs slightly increases nestin‐positive cells (i), decreases neuronal differentiation (for CC5 *P < 0.05) (ii), decreases ciliary length (for CPAP ***P < 0.001 and for CC5 **P < 0.01) (iii), elevates mitotic index (for CC5 *P < 0.05) (iv), and decreases the number of ciliated cells (for CPAP *P < 0.05 and for CC5 **P < 0.01) (v) compared to control (mock electroporation). Error bars show SEM. One‐way ANOVA followed by Tukey's multiple comparisons test. n = 3 independent experiments with > 350 (nestin staining, differentiation and mitotic index), and > 70 cells (cilium length) cells.
Altering G1‐S transition influences the switch of NPCs from proliferation to differentiation (Calegari et al, 2005; Lange et al, 2009). To corroborate this, we treated WT human NPCs with olomoucine, which lengthens the G1‐S transition of NPCs (Calegari & Huttner, 2003), and found this sufficient to trigger differentiation of NPCs into neurons (Fig 4E). We thus speculated that Seckel NPCs, exhibiting retarded cilium disassembly and extended G1‐S transition, undergo premature neural differentiation. Indeed, under non‐differentiating conditions, Seckel NPCs but not control NPCs differentiated into neurons at the expense of proliferating NPCs (Fig 4F).
To some extent, exogenous WT‐CPAP expression rescued this spontaneous differentiation. However, to our surprise, exogenous CC5 expression, not only restored, but also reversed the phenotypes of Seckel NPCs causing increased ciliary disassembly, enhanced cell proliferation and prevented Seckel NPCs differentiating into neurons (Fig 4G).
We then tested whether retarded cilium disassembly could be a common mechanism underlying a subset of microcephaly disorders. To do this, we first downregulated CDC components in WT NPCs using siRNA‐mediated depletion (for Nde‐1 and OFD1) or small molecule (VX680)‐mediated inhibition (for Aurora A). The treatments specifically caused retarded cilia disassembly, long cilia, premature differentiation of NPCs, accompanied by reduced cell cycle progression, suggesting that mutations in these genes might underlie NPC depletion resulting in microcephaly (Fig 5A–D). A mutation in Nde‐1 causes microcephaly for which the underlying mechanism remains unclear (Alkuraya et al, 2011). Together, these data suggest that cilia disassembly, cell cycle progression, and neuronal differentiation are causally related to each other.
Figure 5. Downregulation of CDC components or a CPAP natural microcephaly mutation causes neural differentiation.
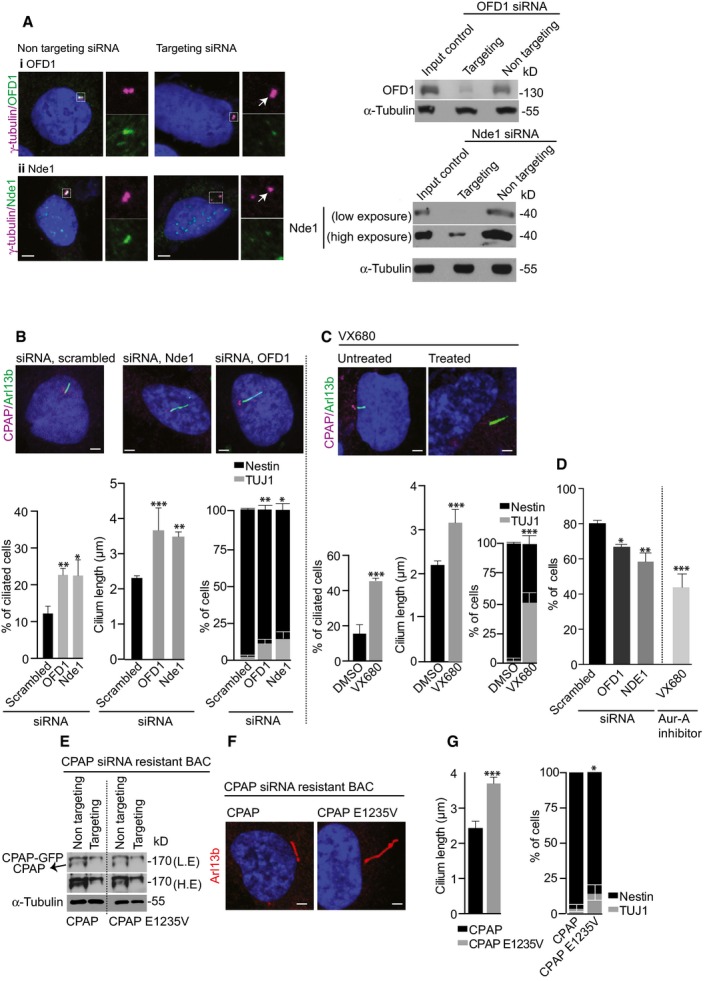
-
A, BsiRNA‐mediated downregulation of CDC components in WT NPCs causes longer cilia (Ofd1 ***P < 0.001, Nde1 **P < 0.01), retarded cilia disassembly as shown by increased numbers of ciliated cells (Ofd1 **P < 0.01, Nde1 *P < 0.05) and differentiation of NPCs into TUJ1‐positive cells (Ofd1 **P < 0.001, Nde1 *P < 0.01). In controls, a non‐targeting siRNA (scrambled) is used. Western blots on the right validate siRNAs‐mediated Nde1 and OFD1 depletion. Error bars are SEM. Ordinary two‐way ANOVA followed by Sidak's multiple comparisons test. n = 3 independent experiments. 275 cells for scrambled, 423 cells for Ofd1 siRNA, and 255 cells for Nde1 siRNA. Arrow marks the centrosome. Scale bar, 1 μm.
-
CVX680 (0.5 μM, a concentration that will specifically allow neuronal differentiation) was used to inhibit Aurora A activity. Bar diagrams below show quantifications. Compared to DMSO‐treated control NPCs, a higher proportion of VX680‐treated NPCs are ciliated (***P < 0.001), exhibiting longer cilia (***P < 0.001), and show Tuj1‐positive neurons (***P < 0.001). Error bars are SEM. For ciliated cells and cilium length: n = 3 independent experiments. 164 cells for DMSO‐treated and 77 cells for VX680‐treated. For NPC differentiation: n = 4 independent experiments. 286 cells for DMSO‐treated and 118 cells for VX680‐treated. Scale bar, 1 μm.
-
DCell cycle progression of siRNA‐ or VX680‐treated WT NPCs compared to control assessed by EdU incorporation for 24 h. Inhibition of Ofd1 (*P < 0.05), Nde1 (**P < 0.01) and Aurora A (***P < 0.001) caused significantly reduced incorporation of EdU. Error bars are SEM.
-
E–GWestern blots show that CPAP‐targeting siRNA treatment depletes endogenous CPAP. Both low and high exposures are given (E). Increased cilia length and NPC differentiation upon expression of RNAi‐resistant CPAP E1235V compared to WT CPAP (F, G). Error bars are SEM. ***P < 0.001 (cilia length), *P < 0.05 (differentiation), t‐test. For cilia length: n = 4 independent experiments. 25 cells for CPAP, 47 cells for CPAP E1235V. For differentiation: n = 4 independent experiments. 145 for CPAP and 81 for CPAP E1235V. Scale bar, 1 μm.
We then tested an additional patient mutation in CPAP, a homozygous mutation in its TCP domain (CPAP E1235V; Bond et al, 2005; Gul et al, 2006). To do this, we expressed CPAP E1235V using RNAi‐resistant BACs together with CPAP‐specific siRNA treatment in WT NPCs as previously described (Zheng et al, 2014). This resulted in premature differentiation of NPCs due to retarded cilia disassembly as indicated by the presence of long cilia (Fig 5E–G). Taken together, this suggests that defective cilium disassembly is not only limited to the Seckel mutation we describe here, but could potentially represent a common mechanism regulated by a group of microcephaly genes specifically implicated in ciliogenesis.
Human Seckel iPS‐derived organoids model microcephaly
To determine whether aberrant CDC function could lead to brain defects, we adopted the recently described three‐dimensional culture system to generate organoids in vitro (Lancaster et al, 2013; Mariani et al, 2015). Starting with the same number of WT and Seckel iPS cells, matrigel‐embedded droplets of neural epithelium were grown in spinner bioreactors (Appendix Fig S9A). We could obtain 2.5–3.5‐mm organoids exhibiting brain‐like architectures within 14–16 days (Appendix Fig S9A and B). Immunostaining revealed that organoids contained complex heterogeneous tissues displaying regions reminiscent of the ventricle zone (VZ) exhibiting proliferating apical progenitors (apical radial glial aRG) expressing nestin, Pax6, and phospho‐vimentin.
Importantly, we found that the morphology and sizes of ventricular zones of these iPS‐derived organoids, the region of our interest, were fairly consistent between organoids as was also demonstrated recently using iPS‐derived organoids (Mariani et al, 2015). While we observed proliferating RGs on the apical side toward the lumen, we observed neuronal cells exhibiting neuronal proteins such as doublecortin (DCX) and TUJ1 at the basal side away from the lumen (Fig 6A and B, Appendix Figs S9C and S10).
Figure 6. iPS modeling of microcephaly in 3D organoids.
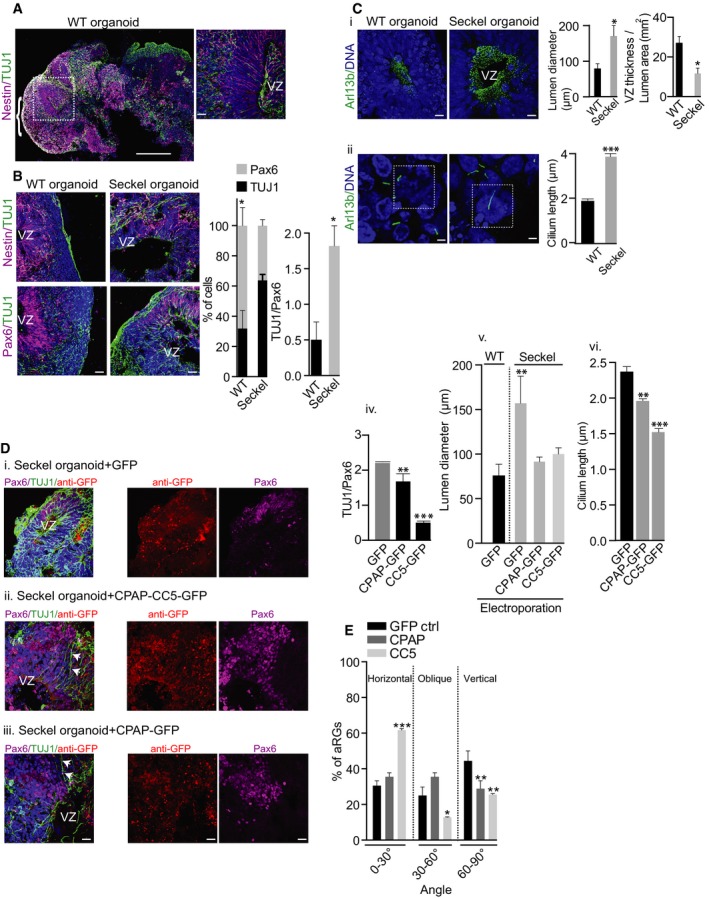
- iPS‐derived 14‐day‐old organoid displaying complex morphological structures of brain‐like tissues (parenthesis). The ventricular zone (VZ) with radial glial cells stained with nestin (magenta) and neurons stained with TUJ1 is shown at higher magnification. Scale bars, 500 μm (left) and 20 μm (right).
- Reduced Nestin‐/Pax‐6‐positive progenitors and increased TUJ1‐positive neurons in Seckel organoids. In ventricular zones of WT organoids, there are significantly more Pax6‐positive cells than TUJ1‐positive cells, which is not the case in Seckel organoids (*P < 0.05). TUJ1/Pax6 ratio in graph on the right shows significantly more TUJ1‐positive cells versus Pax6‐positive cells in Seckel organoid ventricular zones (*P < 0.05). Error bars are SEM. Ordinary two‐way ANOVA followed by Sidak's multiple comparisons test. n = 10 WT organoids (2,520 cells). Seckel n = 10 organoids (2,024 cells). Scale bar, 20 μm.
- (i) Seckel organoid tissues contain increased numbers of cilia with an obvious observation of enlarged lateral ventricular lumen (*P < 0.05). Graph at far right shows an increased lumen area as well as thinner ambient VZ in Seckel organoids (*P < 0.05). Error bars are SEM; t‐test. n = 6 for WT organoid and 6 for Seckel organoid. Scale bar, 20 μm. (ii) Seckel organoid cells display longer cilia. Error bars are SEM. ***P < 0.001, t‐test. n = 3 for WT organoids and 4 for Seckel organoids. Scale bar, 1 μm.
- Seckel organoids electroporated with constructs expressing GFP alone (i), GFP with CC5‐GFP (ii) and GFP with CPAP‐GFP (iii). Middle panel shows staining with anti‐GFP antibodies (red) to show the transfected area. Note the presence of increased Pax6‐positive (magenta) NPCs in CC5 and CPAP electroporated tissues compared to GFP control. Likewise, there is a larger neural epithelium with multiple RGs (arrowheads). Bar diagrams on the right show rescue effects of CPAP and CC5 electroporation compared with GFP alone resulting in (iv) lower TUJ1/Pax6 ratio (**P < 0.01 and ***P < 0.001, respectively), (v) smaller lumen diameter (**P < 0.01), and (vi) shorter cilia length (**P < 0.01 and ***P < 0.001, respectively). Error bars are SEM. One‐way ANOVA followed by Dunnett's multiple comparisons test. n = 3 organoids with 225 cells for GFP, 537 cells for CPAP, and 569 for CC5. At least 10 ventricles from 3 independent organoids were used to measure the lumen diameter. Error bars are SEM. Scale bar, 20 μm.
- Rescue effect of CC5 and CPAP electroporation on cleavage planes of phospho‐vimentin‐positive RGs in Seckel organoids. Compared to GFP control, electroporation of CC5 or CPAP increases the proportion of horizontally orientated Seckel RGs (for CC5 ***P < 0.001) and decreases the portion of obliquely (for CC5 *P < 0.05) or vertically oriented Seckel RGs (for CPAP and CC5 **P < 0.01). Error bars are SEM. Two‐way ANOVA followed by Tukey's multiple comparisons test. n = 4 organoids. 100 cells for GFP control, 112 cells for CPAP, and 99 cells for CC5.
We then developed a method to grow sliced organoids, generating matured cortical neurons with extended long‐range axons exhibiting complex branching and projections (Appendix Fig S11). Interestingly, these extended TUJ1‐positive structures showed distinct GAD67‐positive protrusions, dendritic spines of inhibitory GABAergic nature suggesting that our in vitro differentiating conditions are physiological (Guidotti et al, 2000).
Although Seckel organoids displayed distinct brain‐like regions, they were smaller in size, exhibiting reduced neural epithelial tissues and progenitor regions with disorganized VZ and larger lumen (Fig 6B and C and Appendix Fig S9A). These aspects to some extent were reminiscent of the small brain observed in microcephaly patients (Howanietz et al, 1989; Lancaster et al, 2013).
Analyzing cortical regions for RG cells and neurons, we found WT organoids to exhibit abundant neural epithelial tissues constituting nestin‐/PAX6‐positive progenitors. In contrast, Seckel organoids displayed neural epithelial tissues with diminished RGs and increased TUJ1‐positive neurons, suggesting premature neuronal differentiation. These findings are consistent with our NPCs experiments as well as with the observation of Tctex‐1‐mediated defective cilium disassembly triggering premature neuronal differentiation (Li et al, 2011; Fig 6B). Importantly, apoptotic cell deaths in Seckel and WT organoids did not differ which excludes the involvement of apoptotic cell death‐mediated NPCs loss (Marthiens et al, 2013; Bazzi & Anderson, 2014; Appendix Fig S12).
It is known that the horizontal orientation of the cleavage plane of mitotic apical progenitors is essential for early symmetric expansion of NPCs (Yingling et al, 2008; Li et al, 2011). After sufficient NPCs expansion, neurogenesis then begins, which requires switching of the cleavage plane from horizontal to vertical (Rakic, 1995; Lancaster et al, 2013). The defect in apical progenitor expansion at the ventricular zone via vertical orientation accompanied by premature neurogenesis has been shown to occur due to cell cycle delay (Calegari et al, 2005; Tapias et al, 2014).
When we examined the plane of division of mitotic apical progenitors (apical radial glial cells, aRGs) using p‐vimentin staining, Seckel organoids displayed increased numbers of mitotic aRGs with cleavage planes perpendicular to the ventricular surface suggesting a premature switching of the cleavage plane. These data agree with studies demonstrating that CPAP is critical for controlling the cleavage plane of mitotic neural progenitors (Insolera et al, 2014; Bazzi & Anderson, 2014; Garcez et al, 2015; Appendix Fig S10). Finally, when analyzing apical progenitor cilia extending into the lateral ventricle, Seckel organoids showed increased numbers and longer cilia compared to WT organoids, suggesting retarded cilium disassembly consistent with our results from fibroblasts and NPCs experiments (Fig 6C).
To test whether expressing WT CPAP or CC5 alone could rescue the phenotype, we performed co‐electroporation of CPAP‐GFP, CC5‐GFP, or GFP alone into 6‐day‐old Seckel organoids and analyzed them 5 days later. As control, GFP alone was electroporated. In analyzing regions of CPAP‐ or CC5‐electroporated tissues, we found CPAP to partially rescue certain aspects of Seckel phenotype. This partial rescue could be due to CPAP overexpression itself (Kohlmaier et al, 2009; Tang et al, 2009). However, the rescue effect was more prominent in CC5‐transfected Seckel organoids as similarly observed with our NPCs experiments (Fig 4G). In contrast to GFP‐electroporated regions, CPAP‐ and CC5‐electroporated regions displayed increased neural epithelium as well as PAX6‐positive progenitors. Accordingly, electroporation of CPAP and CC5 both proportionally increased the number of mitotic RGs with horizontal orientation and reduced the number of cells having vertical orientation. Similarly, CPAP and CC5 but not GFP‐electroporated regions displayed normal cilia length (Fig 6Div). From these results, we conclude that the observed phenotypes in Seckel organoids are specifically due to aberrant CDC function caused by the loss of CPAP's CC5 domain.
Discussion
A reduced NPC pool is an accepted cause underlying microcephaly‐linked disorders (Lancaster et al, 2013; Alcantara & O'Driscoll, 2014; Guemez‐Gamboa et al, 2014). However, the mechanisms involved in NPCs maintenance still remain unclear. The finding that several known microcephaly proteins are found in centrosomes suggested that a centrosomal mechanism can control neuronal number in a developing brain (Bond et al, 2005, 2005; Basto et al, 2006; Thornton & Woods, 2009, 2009; Gopalakrishnan et al, 2011; Hussain et al, 2012). Centrosomes are the major microtubule‐organizing centers of animal cells. Regulated centrosome assembly is essential for accurate cell division, organ/tissue homeostasis, and stem cell maintenance and for maintaining genome integrity. Thus, centrosomes are critical for animal development and their dysfunction results in many developmental disorders, including microcephaly.
Defects in centrosome biogenesis as it occurs in microcephaly‐related disorders result in premature switching of NPCs from symmetric to asymmetric division, which may cause premature depletion of progenitors (Thornton & Woods, 2009; Wollnik, 2010). For example, loss of MCPH1 results in a shorter G1 phase of the cell cycle through premature mitotic entry. As a result, the centrosomes do not have sufficient time to mature before the onset of division (Gruber et al, 2011). Likewise, loss of CDK5RAP2 directly affected centrosome maturation and thus immature centrosomes accumulate less PCM (Thornton & Woods, 2009). As a result, these centrosomes are compromised in generating astral microtubules. Astral microtubules contact the cell cortex and provide information guiding spindle orientation during division.
Interestingly, recent studies using mouse models have identified yet another mechanism involving apoptosis and resulting in neuronal loss and microcephaly in the absence of centrosomes. Specifically, Sas‐4/CPAP deletion in developing mouse brain caused loss of centrosomes, resulting in mis‐positioning of radial glial progenitors. These centrosome‐less cells underwent prolonged mitosis, p53 upregulation, and apoptosis (Insolera et al, 2014). On the other hand, centrosome amplification also causes microcephaly due to defects in extra centrosome clustering. Inefficient centrosome clustering then caused neural stem cells to undergo multipolar mitosis leading to aneuploidy. As a result, neural stem cells failed to proliferate efficiently, thus causing depletion of the neural progenitors pool (Marthiens et al, 2013).
While these studies have delineated underlying mechanisms of microcephaly that are mediated by centrosomes, our results illustrate a surprising role for the primary cilium in stem cell homeostasis, linking the importance of timely cilium disassembly mediated by CPAP‐CDC to cell cycle re‐entry for neurogenesis and brain size control.
Most studies to date have focused on analyzing centrosomes, as microcephaly‐related proteins are mainly centrosomal components, with clinical features of ciliopathies not reported to be associated with Seckel syndrome or microcephaly disorders. The results from our current study reveal that structurally and numerically aberrant centrosomes are not the only cause underlying microcephaly; instead, disrupted ciliary function could also lead to microcephaly‐linked disorders. Our results also rule out the possibility of apoptosis‐mediated NPCs loss in the currently studied microcephaly mutant. This suggests that disorders caused by defective cilium disassembly could potentially be grouped as ciliopathic diseases.
Although our experiments substantiate the previously unknown function of CPAP as a negative regulator of ciliary length independent of its role in centrosome biogenesis, what remains elusive is why Seckel CPAP lacking the CC5 domain results in long cilia. It is known that the CPAP‐tubulin interaction is required for centriole and cilium elongation (Tang et al, 2009; Wu & Tang, 2012). Evidence comes from a functional study where a non‐tubulin‐binding CPAP variant results in a short cilium, suggesting that CPAPs' N‐terminal tubulin binding capability could determine cilium length (Wu & Tang, 2012). From this observation, we speculate that the Seckel variant CPAP (where a part of its CC5 domain is deleted) could have an altered tubulin binding activity possibly accounting for an elongated cilium.
Alternatively, as shown by our ultra‐structural studies that Seckel cilia display disrupted ciliary appendages near the transition zone, we postulate that aberrant CDC recruitment could impair ciliary functions such as protein entry and exit from the cilium, thus regulating its length. Therefore, it would be important to study the ultra‐localization of CDC components at the basal body and how they interact with each other to form structurally normal cilia. It is noteworthy that proteins mutated in ciliopathies are localized to the transition zone (Garcia‐Gonzalo et al, 2011; Huang et al, 2011; Li et al, 2011).
Our results also suggest a model where retarded cilium disassembly‐mediated delay in cell cycle re‐entry is sufficient to trigger premature differentiation of NPCs. In this context, it appears that the cilium plays a crucial role in determining asymmetric cell division. How timely cilium disassembly directly or indirectly regulates the apical progenitor cleavage plane remains unclear, thus warranting further study. Since the primary cilium is an extracellular antenna of vertebrate cells playing key roles in signaling pathways, it is likely that it traffics specific signaling cues in a spatiotemporal manner for faithful asymmetric cell division. We hope that our current findings provide new insights into these open questions and substantiate the importance of the cilium in asymmetric cell division.
Our CPAP mutant phenotypes seem to overlap with those observed in other genes involved in Seckel syndrome and microcephaly (Bond et al, 2005; Gul et al, 2006; Kalay et al, 2011; Alcantara & O'Driscoll, 2014; Martin et al, 2014). Thus, it would be interesting to test whether the observed “cilium‐check point” in the current study serves as a common mechanism in organ size control. Indeed, our findings that depletion of CDC components leads to phenotypes similar to that observed in Seckel cells could represent a conserved mechanism among microcephaly causing genes that are implicated in ciliogenesis. This previously unknown role for cilium disassembly in fate determination of somatic stem cells may not only be restricted to NPCs but could also be true for other paradigms of organ size control and homeostasis during development.
Materials and Methods
Molecular biology and cell culture
WT skin fibroblast or RPE1 cells (Stemgent BJ Human Fibroblasts, Cat. No. 08‐0027, Stemgent, San Diego, USA), and skin fibroblasts derived from a Seckel patient were grown in fibroblast medium containing Dulbecco's modified Eagle's medium (DMEM), 10% (v/v) fetal bovine serum (FBS), 0.1 mM MEM non‐essential amino acids (NAA), 100 U/ml penicillin, 100 μg/ml streptomycin (all from Life Technologies GmbH, Darmstadt, Germany), and 0.05 mM β‐mercaptoethanol (Sigma‐Aldrich, Munich, Germany). HeLa cells were grown under standard conditions in DMEM supplemented with 10% FBS. For serum starvation experiments, cells were grown in fibroblast medium without serum for the described time period and stimulated with 10% FBS‐containing medium. For transfecting fibroblasts, the Neon Transfection System (Life Technologies) was used with the following parameters: 1 × 107 cells/ml in 10 μl buffer R containing 1 μg plasmid DNA or 250 ng BAC DNA per 1 × 106 cells, pulse voltage 1,650 V, pulse width 10 ms, and 3 pulses.
For lentiviral transductions, GFP‐tagged CPAP with different mutations were cloned into pLenti6.3 vector (3rd‐generation lentiviral plasmid) using Gateway cloning. For packaging virus, pMD2, pRSV, and pMDL plasmids were used. The packaged products were transfected in HEK293T along with pLenti plasmids using calcium chloride method. First batch of virus was collected after 24 h of transfection followed by second batch after 36 h. For transduction, pregrown RPE1 cells were transduced with viral particles with 1:10 dilution. Viral transductions were tested after 48 h and subsequently used for further analyses.
Western blot
Protein samples were resolved in 8 or 12% acrylamide gels and transferred to nitrocellulose membranes. After incubating with primary antibodies overnight at 4°C, the blots were treated with secondary antibodies at RT for 1 h. Super Signal West Pico or Femto Chemiluminescent substrates (Pierce) were used for detection. Antibody dilutions for Western blots: monoclonal mouse anti‐CPAP (1:50), rabbit‐CDK5RAP2 (1:3,000, T. K. Tang), mouse anti‐γ‐tubulin (1:3,000; Sigma‐Aldrich), mouse anti‐α‐tubulin (1:3,000; Sigma), mouse anti‐HDAC6 (1:1,000; Sigma‐Aldrich), rabbit anti‐Nde1 (1:1,000; Proteintech), mouse anti‐Aurora A (1:1,000; Cell Signaling), rabbit anti‐OFD1 (1:3,000; J. Reiter), mouse anti‐cyclin A (1:1,000; Sigma‐Aldrich), mouse anti‐GFP (1:3,000; Roche), and peroxidase‐conjugated secondary antibodies were used at 1:3,000 (Life Technologies).
Immunofluorescence and light microscopy
For immunofluorescence, mouse anti‐γ‐tubulin (1:500; Sigma‐Aldrich), rat anti‐α‐tubulin (1:50, Chemicon), mouse anti‐CPAP (1:25), rabbit anti‐Cep152 (1:500, E. Nigg), rabbit anti‐PCNT (ab4448, Abcam, Cambridge, USA; 1:1,000), rabbit anti‐Arl13b (1:100, Proteintech, Manchester, UK), mouse anti‐acetylated tubulin (1:500, Sigma‐Aldrich), rabbit anti‐Nde1 (1:50, Proteintech, Manchester, UK), mouse anti‐Nestin 4D11 (1:100, Novus Biologicals, Cambridge, USA), rabbit anti‐Tuj1 (1:100, Sigma‐Aldrich), mouse anti‐Oct4 (1:200, Santa Cruz), rabbit anti‐Nanog (1:200, Santa Cruz, Heidelberg, Germany), mouse anti‐Pax6 (1:50, DSHB, Iowa University, Iowa, USA), mouse anti‐cyclin A2 (ab16726, Abcam), mouse anti‐phosphorylated vimentin (1:100, 4A4, Biozol, Eching, Germany), rabbit anti‐OFD1 (J. Reiter; 1: 50), goat anti‐AFP (1:50 Santa Cruz), mouse anti‐MF‐20 (1:100, DHSB, Iowa) antibodies were used. Secondary antibodies Alexa Fluor dyes conjugated goat/donkey anti‐mouse or anti‐rabbit were used at 1:1,000 (Life Technologies). DAPI (1 μg/ml, Sigma) stained DNA.
For light microscopy, cells were fixed in 3.7% paraformaldehyde (PFA) or ice‐cold methanol, permeabilized with 0.5% Triton X‐100 in PBS for 10 min, and blocked with 0.5%% fish gelatin in PBS for 2 h at RT. Antibody labeling was performed for 1 h at RT or overnight at 4°C followed by three washes in PBS. Confocal images were collected using an Olympus Fluoview FV 1000 scanning confocal microscope and processed using Adobe Photoshop. Organoids were fixed in 3.7% PFA for 2 h, stored in 30% sucrose solution at 4°C overnight and then embedded in Tissue Tek (Weckert, Kitzingen, Germany), and stored at −80°C. Organoid cryosections were then cut in 20‐μm‐thin sections for immuno‐fluorescent stainings.
To count nestin‐positive neurons in organoid slices, we performed counting in Z‐serial 1‐μm slices. Special care was taken to prevent overlaps. When overlaps were seen, those cells were not considered. For clarity, we will describe it in the method section.
Statistics calculations
Statistical analyses, for example, Student's t‐test or one‐way ANOVA followed by Tukey's multiple comparisons were performed using GraphPad Prism version 6.0d.
Electron microscopy
Asynchronously grown fibroblasts were pelleted, fixed with 2% glutaraldehyde (Electron Microscopy Sciences), and processed for electron microscopy as previously described (Gopalakrishnan et al, 2010; Zheng et al, 2014). The embedded cell pellets were ultra‐thin‐sectioned (80 nm), counterstained, and visualized for ultra‐structural details of centrosomes using a Zeiss 10A electron microscope. Images were processed using Adobe Photoshop.
Pulse chasing experiments, chemical compounds, and RNAi treatments
For cell proliferation estimation, EdU was pulsed in cells for 24–72 h before fixation, and stained according to the manufacturer's instructions (Click‐iT EdU assay kit, Life Technologies). For inhibitor experiments, wherever necessary, NPCs were treated with the Cdk1 inhibitor, olomoucine (Sigma‐Aldrich; 25 μM), or VX680 (Selleckchem; 0.5 μM) for 4 days, fixed, and analyzed by microscopy.
For RNAi treatments, NPCs were electroporated using the Neon Transfection System with 100 nmol of siRNA targeting human NDE1 and OFD1 mRNA. NDE1: siGENOME Human NDE1 (54,820) siRNA‐SMARTpool (M‐020625‐00‐0005) OFD1: siGENOME Human OFD1 (8481) siRNA‐SMARTpool (M‐009300‐02‐0005). Negative controls were performed with non‐targeting siRNA (scrambled; 100 nmol, AllStars Negative control siRNA; Qiagen). Electroporation using 100 nmol siRNA was repeated on day 3. siRNA‐treated NPCs were fixed on day 7 for further analysis.
Protein expression, purification, and GST pull‐down assays
The region deleted in the Seckel syndrome patient (residues 1,073–1,159 of CPAP) was cloned into the pGEX vector (Amersham Biosciences) containing an N‐terminal GST tag and the eGFP‐C1 vector (Clontech). The protein was expressed in E. coli BL21(DE3) (New England Biolabs) and induced by 0.5 mM IPTG overnight at 18°C in LB medium. The recombinant protein was affinity‐purified from supernatant using GST‐sepharose beads (GE Healthcare). The conjugated protein was then treated with HeLa cell extracts and further analyzed by Western blots.
Immunopurification of CPAP complexes
Protein G beads were coated with anti‐CPAP antibodies overnight at 4°C, mixed with HeLa, WT, or Seckel fibroblast cell extracts, respectively, and incubated at 4°C for 4 h. Cell extracts were prepared in a buffer containing 80 mM BRB, 100 mM KCl, 1 mM MgCl2, 1 mM EGTA, protease inhibitor cocktails, and 1 mM PMSF. The extracts were centrifuged at 100,000 g, and the supernatant was collected. These high‐speed extracts were then used for immunopurification. The protein‐bound beads were washed with BRB buffer containing 0.1% Triton X‐100 and 100 mM NaCl and washed twice with high‐salt buffer containing 500 mM salt. After a final wash with buffer containing 100 mM NaCl, the samples were eluted using Laemmli buffer for Western blot analyses.
To immunopurify endogenous proteins that interact with CPAP 1073–1159, stable HeLa cell line expressing the fragment GFP‐1073‐1159 (CC5‐GFP) was generated. Cell extracts prepared from these stable cell lines were used for immunopurification using anti‐GFP‐coated magnetic beads as per the manufacturer's instructions. Complexes were then eluted for Western blot analyses.
Patient iPS cell reprogramming
Skin fibroblasts were transfected with pCXLE‐hOct3/4‐shp53, pCXLE‐hSox2‐hKlf4, pCXLE‐hLin28‐hL‐Myc (Addgene, Cambridge, USA) using the Neon Transfection System according to the manufacturer's protocol. After 5 days, the medium was replaced with DMEM/F12 medium supplemented with Glutamax, 20% knockout serum replacer, 1% NAA, 0.1 mmol/l β‐mercaptoethanol (Life Technologies), and 50 ng/ml bFGF (PeproTech, Hamburg, Germany). At day 10, cells were trypsinized and maintained on irradiated mouse embryonic fibroblasts (MEFs) in human iPS cell medium. Colonies were selected based on morphology (Okita et al, 2011; Yu et al, 2011), further passaged, and expanded. As quality control for iPS clones, we performed alkaline phosphatase (Vector Blue Alkaline Phosphatase Substrate Kit; Vector Laboratories, SK5300) and pluripotent markers staining.
Endodermal and mesodermal differentiation
Human iPS cells were grown in serum‐containing medium for 6 days, detached by accutase treatment, and then replated on collagen type I‐coated dishes. Endodermal differentiation was induced by low‐serum‐containing medium supplemented with HGF, Oncostatin M, and 100 nM dexamethasone for 10 days. Mesodermal differentiation was induced by serum‐free medium containing insulin and 10 μM SB431542 for 20 days.
Differentiation and characterization of NPCs
Similar amounts of feeder‐free human WT or patient iPS cells were differentiated into neural progenitor cells (NPCs) in STEMdiff Neural Induction Medium (Stem Cell Technologies Vancouver, Canada). After 5 days, formed neurospheres were collected and cultured on poly‐l‐ornithine (PLO)‐/laminin‐coated dishes with daily medium change. Neural rosettes were selected after 7 days using neural rosette selection medium and recultured. NPCs were split every 5–7 days and further maintained in STEMdiff Neural Progenitor Medium under non‐differentiating conditions.
Generation of organoids
Organoids were generated as described before (Lancaster et al, 2013) with slight modifications. Neurospheres were generated, collected after 5 days, and embedded in Matrigel (Corning, Tewksbury, USA) drops. The embedded droplets were differentiated in suspension culture in 1:1 DMEM/F12 and Neural Basal Medium (v:v), 1:200 N2, 1:100 B27 w/o vitamin A, 1:100 l‐glutamine, 0.05 mM MEM non‐essential amino acids (NAA), 100 U/ml penicillin, 100 μg/ml streptomycin, 1.6 g/l insulin (Sigma‐Aldrich), and 0.05 mM β‐mercaptoethanol (all from Life Technologies if not stated). After 4 days, matrigel‐embedded neurospheres were transferred to rotary suspension culture in spinner flasks (IBS, Integra biosciences) containing the same differentiation medium with addition of 0.5 μM dorsomorphin (Sigma‐Aldrich). Organoids were collected and analyzed after 14, 28, and 42 days of rotary culture.
To culture single slices of organoid, first organoid slices of about 100 μm were made using previously gelatin‐embedded whole cerebral organoids. The slices were grown in wells containing same media supplemented with 0.5 μM dorsomorphin, 10 ng/ml BDNF, and 10 ng/ml GDNF for 2 weeks until the extended neuronal structures are visually observed.
Organoid electroporation
Electroporation was carried out using two platinum electrodes and the Intracel TSS20 OVODYNE Electroporator with five square pulses of 80 V within 20 ms width. A total of 1 μg plasmid (CPAP or CPAP's CC5) was co‐electroporated with 2 μg/μl of GFP‐expressing vector mixed with Fast Green solution (Sigma) at a ratio of 2:1 into 6‐day‐old organoids at 3–4 different locations. After 24 h of reculture, transgenic tissue was tested for EGFP expression. Electroporated organoids were further grown in suspension cultures for 14 days for further analyses.
Author contributions
JG and EG conceived the project, designed experiments, and wrote the manuscript. EG performed experiments and analyzed the data. AW performed biochemical experiments. MLG and AR performed molecular biology. GC and MG ultra‐structural analyses. FC and FN supported with reagents and analyzed data. IP, AP, PK, and AAH for reagents and conceived experiments with BACs. NST and TS for reagents and experiments for iPS generation. JH and FSA analyzed the data. JG directed and supervised the project.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Review Process File
Acknowledgements
We are grateful to members of the laboratory for technical expertise and feedback, and Drs. Niessen, Noegel, and Schermer for critical comments and proof reading of the manuscript. We thank Dr. Nigg for Cep152 antibodies, Dr. Reiter for OFD1 antibodies, and Dr. T. K Tang for CDK5RAP2. We thank Dr. Rizwan Rehimi of ZMMK for helping us with electroporating cerebral organoids. We acknowledge the Anatomy Institute, University of Cologne, for EM experiments. We would especially like to thank the patient and patient's family for participating in this study. This work is supported by the Fritz Thyssen Foundation, Germany (to J.G. and E.G.) grant Az.10.14.2.152 and by the Deutsche Forschungsgemeinschaft (DFG) grant GO2301/2‐1. The work of I.P. at the laboratory of A. H. is supported by the European Community's Seventh Framework Programme (FP7/2007‐2013) under grant agreement of 241548 (MitoSys Project).”
The EMBO Journal (2016) 35: 803–819
References
- Alcantara D, O'Driscoll M (2014) Congenital microcephaly. Am J Med Genet C Semin Med Genet 166C: 124–139 [DOI] [PubMed] [Google Scholar]
- Al‐Dosari MS, Shaheen R, Colak D, Alkuraya FS (2010) Novel CENPJ mutation causes Seckel syndrome. J Med Genet 47: 411–414 [DOI] [PubMed] [Google Scholar]
- Alkuraya FS, Cai X, Emery C, Mochida GH, Al‐Dosari MS, Felie JM, Hill RS, Barry BJ, Partlow JN, Gascon GG, Kentab A, Jan M, Shaheen R, Feng Y, Walsh CA (2011) Human mutations in NDE1 cause extreme microcephaly with lissencephaly [corrected]. Am J Hum Genet 88: 536–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basto R, Lau J, Vinogradova T, Gardiol A, Woods CG, Khodjakov A, Raff JW (2006) Flies without centrioles. Cell 125: 1375–1386 [DOI] [PubMed] [Google Scholar]
- Bazzi H, Anderson KV (2014) Acentriolar mitosis activates a p53‐dependent apoptosis pathway in the mouse embryo. Proc Natl Acad Sci USA 111: E1491–E1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berbari NF, O'Connor AK, Haycraft CJ, Yoder BK (2009) The primary cilium as a complex signaling center. Curr Biol 19: R526–R535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond J, Roberts E, Springell K, Lizarraga SB, Scott S, Higgins J, Hampshire DJ, Morrison EE, Leal GF, Silva EO, Costa SM, Baralle D, Raponi M, Karbani G, Rashid Y, Jafri H, Bennett C, Corry P, Walsh CA, Woods CG (2005) A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat Genet 37: 353–355 [DOI] [PubMed] [Google Scholar]
- Calegari F, Huttner WB (2003) An inhibition of cyclin‐dependent kinases that lengthens, but does not arrest, neuroepithelial cell cycle induces premature neurogenesis. J Cell Sci 116: 4947–4955 [DOI] [PubMed] [Google Scholar]
- Calegari F, Haubensak W, Haffner C, Huttner WB (2005) Selective lengthening of the cell cycle in the neurogenic subpopulation of neural progenitor cells during mouse brain development. J Neurosci 25: 6533–6538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcez PP, Diaz‐Alonso J, Crespo‐Enriquez I, Castro D, Bell D, Guillemot F (2015) Cenpj/CPAP regulates progenitor divisions and neuronal migration in the cerebral cortex downstream of Ascl1. Nat Commun 6: 6474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Gonzalo FR, Corbit KC, Sirerol‐Piquer MS, Ramaswami G, Otto EA, Noriega TR, Seol AD, Robinson JF, Bennett CL, Josifova DJ, Garcia‐Verdugo JM, Katsanis N, Hildebrandt F, Reiter JF (2011) A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet 43: 776–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan J, Guichard P, Smith AH, Schwarz H, Agard DA, Marco S, Avidor‐Reiss T (2010) Self‐assembling SAS‐6 multimer is a core centriole building block. J Biol Chem 285: 8759–8770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan J, Mennella V, Blachon S, Zhai B, Smith AH, Megraw TL, Nicastro D, Gygi SP, Agard DA, Avidor‐Reiss T (2011) Sas‐4 provides a scaffold for cytoplasmic complexes and tethers them in a centrosome. Nat Commun 2: 359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan J, Frederick Chim YC, Ha A, Basiri ML, Lerit DA, Rusan NM, Avidor‐Reiss T (2012) Tubulin nucleotide status controls Sas‐4‐dependent pericentriolar material recruitment. Nat Cell Biol 14: 865–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber R, Zhou Z, Sukchev M, Joerss T, Frappart PO, Wang ZQ (2011) MCPH1 regulates the neuroprogenitor division mode by coupling the centrosomal cycle with mitotic entry through the Chk1‐Cdc25 pathway. Nat Cell Biol 13: 1325–1334 [DOI] [PubMed] [Google Scholar]
- Guemez‐Gamboa A, Coufal NG, Gleeson JG (2014) Primary cilia in the developing and mature brain. Neuron 82: 511–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidotti A, Auta J, Davis JM, Di‐Giorgi‐Gerevini V, Dwivedi Y, Grayson DR, Impagnatiello F, Pandey G, Pesold C, Sharma R, Uzunov D, Costa E (2000) Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch Gen Psychiatry 57: 1061–1069 [DOI] [PubMed] [Google Scholar]
- Gul A, Hassan MJ, Hussain S, Raza SI, Chishti MS, Ahmad W (2006) A novel deletion mutation in CENPJ gene in a Pakistani family with autosomal recessive primary microcephaly. J Hum Genet 51: 760–764 [DOI] [PubMed] [Google Scholar]
- Howanietz H, Frisch H, Jedlicka‐Kohler I, Steger H (1989) Seckel dwarfism based on a personal case. Klin Padiatr 201: 139–141 [DOI] [PubMed] [Google Scholar]
- Hu WF, Pomp O, Ben‐Omran T, Kodani A, Henke K, Mochida GH, Yu TW, Woodworth MB, Bonnard C, Raj GS, Tan TT, Hamamy H, Masri A, Shboul M, Al Saffar M, Partlow JN, Al‐Dosari M, Alazami A, Alowain M, Alkuraya FS et al (2014) Katanin p80 regulates human cortical development by limiting centriole and cilia number. Neuron 84: 1240–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Szymanska K, Jensen VL, Janecke AR, Innes AM, Davis EE, Frosk P, Li C, Willer JR, Chodirker BN, Greenberg CR, McLeod DR, Bernier FP, Chudley AE, Muller T, Shboul M, Logan CV, Loucks CM, Beaulieu CL, Bowie RV et al (2011) TMEM237 is mutated in individuals with a Joubert syndrome related disorder and expands the role of the TMEM family at the ciliary transition zone. Am J Hum Genet 89: 713–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung LY, Chen HL, Chang CW, Li BR, Tang TK (2004) Identification of a novel microtubule‐destabilizing motif in CPAP that binds to tubulin heterodimers and inhibits microtubule assembly. Mol Biol Cell 15: 2697–2706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain MS, Baig SM, Neumann S, Nurnberg G, Farooq M, Ahmad I, Alef T, Hennies HC, Technau M, Altmuller J, Frommolt P, Thiele H, Noegel AA, Nurnberg P (2012) A truncating mutation of CEP135 causes primary microcephaly and disturbed centrosomal function. Am J Hum Genet 90: 871–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insolera R, Bazzi H, Shao W, Anderson KV, Shi SH (2014) Cortical neurogenesis in the absence of centrioles. Nat Neurosci 17: 1528–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson PK (2011) Do cilia put brakes on the cell cycle? Nat Cell Biol 13: 340–342 [DOI] [PubMed] [Google Scholar]
- Kalay E, Yigit G, Aslan Y, Brown KE, Pohl E, Bicknell LS, Kayserili H, Li Y, Tuysuz B, Nurnberg G, Kiess W, Koegl M, Baessmann I, Buruk K, Toraman B, Kayipmaz S, Kul S, Ikbal M, Turner DJ, Taylor MS et al (2011) CEP152 is a genome maintenance protein disrupted in Seckel syndrome. Nat Genet 43: 23–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Zaghloul NA, Bubenshchikova E, Oh EC, Rankin S, Katsanis N, Obara T, Tsiokas L (2011) Nde1‐mediated inhibition of ciliogenesis affects cell cycle re‐entry. Nat Cell Biol 13: 351–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlmaier G, Loncarek J, Meng X, McEwen BF, Mogensen MM, Spektor A, Dynlacht BD, Khodjakov A, Gonczy P (2009) Overly long centrioles and defective cell division upon excess of the SAS‐4‐related protein CPAP. Curr Biol 19: 1012–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA (2013) Cerebral organoids model human brain development and microcephaly. Nature 501: 373–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange C, Huttner WB, Calegari F (2009) Cdk4/cyclinD1 overexpression in neural stem cells shortens G1, delays neurogenesis, and promotes the generation and expansion of basal progenitors. Cell Stem Cell 5: 320–331 [DOI] [PubMed] [Google Scholar]
- Li A, Saito M, Chuang JZ, Tseng YY, Dedesma C, Tomizawa K, Kaitsuka T, Sung CH (2011) Ciliary transition zone activation of phosphorylated Tctex‐1 controls ciliary resorption, S‐phase entry and fate of neural progenitors. Nat Cell Biol 13: 402–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Chang CW, Hsu WB, Tang CJ, Lin YN, Chou EJ, Wu CT, Tang TK (2013) Human microcephaly protein CEP135 binds to hSAS‐6 and CPAP, and is required for centriole assembly. EMBO J 32: 1141–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani J, Coppola G, Zhang P, Abyzov A, Provini L, Tomasini L, Amenduni M, Szekely A, Palejev D, Wilson M, Gerstein M, Grigorenko EL, Chawarska K, Pelphrey KA, Howe JR, Vaccarino FM (2015) FOXG1‐dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders. Cell 162: 375–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marthiens V, Rujano MA, Pennetier C, Tessier S, Paul‐Gilloteaux P, Basto R (2013) Centrosome amplification causes microcephaly. Nat Cell Biol 15: 731–740 [DOI] [PubMed] [Google Scholar]
- Martin CA, Ahmad I, Klingseisen A, Hussain MS, Bicknell LS, Leitch A, Nurnberg G, Toliat MR, Murray JE, Hunt D, Khan F, Ali Z, Tinschert S, Ding J, Keith C, Harley ME, Heyn P, Muller R, Hoffmann I, Daire VC et al (2014) Mutations in PLK4, encoding a master regulator of centriole biogenesis, cause microcephaly, growth failure and retinopathy. Nat Genet 46: 1283–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskey D, Marlin MC, Kim S, Kim S, Ong EC, Li G, Tsiokas L (2015) Cell cycle‐dependent ubiquitylation and destruction of NDE1 by CDK5‐FBW7 regulates ciliary length. EMBO J 34: 2424–2440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre RE, Lakshminarasimhan Chavali P, Ismail O, Carragher DM, Sanchez‐Andrade G, Forment JV, Fu B, Del Castillo Velasco‐Herrera M, Edwards A, van der Weyden L, Yang F, Sanger Mouse Genetics P, Ramirez‐Solis R, Estabel J, Gallagher FA, Logan DW, Arends MJ, Tsang SH, Mahajan VB, Scudamore CL et al (2012) Disruption of mouse Cenpj, a regulator of centriole biogenesis, phenocopies Seckel syndrome. PLoS Genet 8: e1003022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg EA, Raff JW (2009) Centrioles, centrosomes, and cilia in health and disease. Cell 139: 663–678 [DOI] [PubMed] [Google Scholar]
- Okita K, Matsumura Y, Sato Y, Okada A, Morizane A, Okamoto S, Hong H, Nakagawa M, Tanabe K, Tezuka K, Shibata T, Kunisada T, Takahashi M, Takahashi J, Saji H, Yamanaka S (2011) A more efficient method to generate integration‐free human iPS cells. Nat Methods 8: 409–412 [DOI] [PubMed] [Google Scholar]
- Paridaen JT, Wilsch‐Brauninger M, Huttner WB (2013) Asymmetric inheritance of centrosome‐associated primary cilium membrane directs ciliogenesis after cell division. Cell 155: 333–344 [DOI] [PubMed] [Google Scholar]
- Perrier AL, Tabar V, Barberi T, Rubio ME, Bruses J, Topf N, Harrison NL, Studer L (2004) Derivation of midbrain dopamine neurons from human embryonic stem cells. Proc Natl Acad Sci USA 101: 12543–12548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA (2007) HEF1‐dependent Aurora A activation induces disassembly of the primary cilium. Cell 129: 1351–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P (1995) A small step for the cell, a giant leap for mankind: a hypothesis of neocortical expansion during evolution. Trends Neurosci 18: 383–388 [DOI] [PubMed] [Google Scholar]
- Rauch A, Thiel CT, Schindler D, Wick U, Crow YJ, Ekici AB, van Essen AJ, Goecke TO, Al‐Gazali L, Chrzanowska KH, Zweier C, Brunner HG, Becker K, Curry CJ, Dallapiccola B, Devriendt K, Dorfler A, Kinning E, Megarbane A, Meinecke P et al (2008) Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science 319: 816–819 [DOI] [PubMed] [Google Scholar]
- Tang CJ, Fu RH, Wu KS, Hsu WB, Tang TK (2009) CPAP is a cell‐cycle regulated protein that controls centriole length. Nat Cell Biol 11: 825–831 [DOI] [PubMed] [Google Scholar]
- Tang Z, Lin MG, Stowe TR, Chen S, Zhu M, Stearns T, Franco B, Zhong Q (2013) Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature 502: 254–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapias A, Zhou ZW, Shi Y, Chong Z, Wang P, Groth M, Platzer M, Huttner W, Herceg Z, Yang YG, Wang ZQ (2014) Trrap‐dependent histone acetylation specifically regulates cell‐cycle gene transcription to control neural progenitor fate decisions. Cell Stem Cell 14: 632–643 [DOI] [PubMed] [Google Scholar]
- Thornton GK, Woods CG (2009) Primary microcephaly: do all roads lead to Rome? Trends Genet 25: 501–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters AM, Asfahani R, Carroll P, Bicknell L, Lescai F, Bright A, Chanudet E, Brooks A, Christou‐Savina S, Osman G, Walsh P, Bacchelli C, Chapgier A, Vernay B, Bader DM, Deshpande C, O'Sullivan M, Ocaka L, Stanescu H, Stewart HS et al (2015) The kinetochore protein, CENPF, is mutated in human ciliopathy and microcephaly phenotypes. J Med Genet 52: 147–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollnik BA (2010) Common mechanism for microcephaly. Nat Genet 42: 923–924 [DOI] [PubMed] [Google Scholar]
- Wu KS, Tang TK (2012) CPAP is required for cilia formation in neuronal cells. Biol Open 1: 559–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yingling J, Youn YH, Darling D, Toyo‐Oka K, Pramparo T, Hirotsune S, Wynshaw‐Boris A (2008) Neuroepithelial stem cell proliferation requires LIS1 for precise spindle orientation and symmetric division. Cell 132: 474–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Chau KF, Vodyanik MA, Jiang J, Jiang Y (2011) Efficient feeder‐free episomal reprogramming with small molecules. PLoS ONE 6: e17557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Gooi LM, Wason A, Gabriel E, Mehrjardi NZ, Yang Q, Zhang X, Debec A, Basiri ML, Avidor‐Reiss T, Pozniakovsky A, Poser I, Saric T, Hyman AA, Li H, Gopalakrishnan J (2014) Conserved TCP domain of Sas‐4/CPAP is essential for pericentriolar material tethering during centrosome biogenesis. Proc Natl Acad Sci USA 111: E354–E363 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Review Process File