

Abstract
Aims
This study aimed to develop a population pharmacokinetic model for quantitative evaluation of the influence of genetic variants in metabolic enzymes and transporters on lamotrigine pharmacokinetics while taking into account the influence of various clinical, biochemical and demographic factors.
Methods
We included 100 patients with epilepsy on stable dosing with lamotrigine as mono or adjunctive therapy. Lamotrigine and lamotrigine N‐2‐glucuronide concentrations were determined in up to two plasma samples per patient. Patients were genotyped for UGT1A4, UGT2B7, ABCB1 and SLC22A1. Population pharmacokinetic analysis was performed by non‐linear mixed effects modelling. Prior knowledge from previous pharmacokinetic studies was incorporated to stabilize the modelling process. A parent–metabolite model was developed to get a more detailed view on the covariate effects on lamotrigine metabolism.
Results
With a base model absorption rate (interindividual variability) was estimated at 1.96 h−1 (72.8%), oral clearance at 2.32 l h−1 (41.4%) and distribution volume at 77.6 l (30.2%). Lamotrigine clearance was associated with genetic factors, patient's weight, renal function, smoking and co‐treatment with enzyme inducing or inhibiting drugs. In patients with UGT2B7–161TT genotype clearance was lower compared with GT and GG genotypes. Clearance was particularly high in patients with UGT2B7 372 GG genotype (compared with AA genotype it was 117%; 95% CI 44.8, 247% higher).
Conclusions
Variability in lamotrigine pharmacokinetics is large and quantification of its sources may lead to more precise individual treatment. Genotyping for UGT2B7 may be useful in various clinical settings.
Keywords: epilepsy, lamotrigine, metabolism, pharmacogenetics, population pharmacokinetics
What is Already Known about this Subject
Interindividual variability in lamotrigine pharmacokinetics is large and therapeutic drug monitoring may be warranted.
Lamotrigine disposition is mediated by UDP‐glucuronosyltransferases, mainly by UGT1A4 and UGT2B7. ABCB1 and SLC22A1 transporters appear to be also involved.
Polymorphisms in UGT1A4, UGT2B7, ABCB1 and SLC22A1 genes affect protein activity and expression.
What this Study Adds
Influences of polymorphisms of UGT2B7, patient's weight, renal function, smoking and co‐treatment with enzyme inducing or inhibiting drugs on lamotrigine pharmacokinetics were quantified.
No significant association of lamotrigine pharmacokinetics with UGT1A4, SLC22A1 and ABCB1 polymorphisms was observed.
Clearance of lamotrigine‐N‐2‐glucuronide depends on renal function and body weight.
Introduction
Lamotrigine (LTG) is a second generation anti‐epileptic drug (AED) that has been widely used for focal and generalized seizures in adults and children as monotherapy or in combination with other AEDs 1, 2. Moreover, because of its safety profile it has been accepted as a first line drug for treatment of women of childbearing age and during pregnancy 3.
LTG exhibits first order linear pharmacokinetics. Following oral administration it is rapidly and completely absorbed into the systemic circulation with a maximum concentration in plasma after 1–3 h 4. The predominant route of LTG elimination is hepatic metabolism. Renal excretion of unchanged LTG accounts for less than 10% 5. Metabolic inactivation is catalyzed by UDP‐glucuronosyltransferases (UGT), mainly by UGT1A4 and UGT2B7 6. The main metabolite, LTG‐N‐2‐glucuronide (LTG‐glu) is excreted in urine 4. Previous studies suggest that LTG metabolism and distribution is mediated also with some transporter proteins such as ATP‐binding cassette protein B1 (ABCB1) 7 and solute carrier family 22, member 1 (SLC22A1) alias organic cation transporter 1 (OCT1) 8.
There is a large inter‐individual variability in the pharmacokinetics of LTG which is influenced by patient's age, weight, co‐medications, gender, liver and renal function, and specific physiological states such as pregnancy 9. These characteristics suggest that LTG use may be facilitated by therapeutic drug monitoring (TDM). The reference range of serum LTG concentration in patients treated with therapeutic doses is 2.5–15 mg l−1 10. Concomitant treatment with enzyme inducing AEDs increases clearance and shortens half‐life of LTG. On the other hand, in patients taking valproic acid, the half‐life of LTG is substantially prolonged [10]. LTG has been extensively evaluated in pregnancy and in women taking oral contraceptives because of the reported decrease in serum LTG concentrations 11, 12, 13, 14, 15. LTG glucuronidation appears to be induced with estradiol [16]. Moreover, in the UGT1A4 17, 18, UGT2B7 19, 20 and ABCB1 genes [21], polymorphisms were described which affect protein expression and activity. These polymorphisms may also affect the clearance of LTG and its concentration in blood plasma and the central nervous system and consequently the effectiveness and safety of treatment. A significant association of UGT2B7–161C > T (rs7668258) with LTG concentration : dose ratio was found in a multivariate analysis in a Hispanic population 20. In another study the influence of UGT1A4 polymorphisms on LTG plasma concentration was investigated and a 52% decrease in the concentration of LTG was found in patients carrying the UGT1A4 142G > T (rs2011425) polymorphism compared with patients with wild‐type alleles 18. Similar findings on the effect of this polymorphism were also obtained in Chinese patients with epilepsy 22, 23. On the other hand, a previous study showed no association of UGT1A4 142G > T polymorphism with LTG pharmacokinetics, whereas UGT2B7–161C > T significantly influenced LTG oral clearance [19]. In addition, Lovric et al. studied the association of LTG serum concentration with polymorphisms of ABCB1 (1236C > T, 2677G > T/A and 3435C > T) and found that patients with ABCB1 1236CC genotype have higher LTG concentration compared with groups with CT and TT genotype 21.
The majority of published pharmacogenetic studies with LTG evaluated variation in concentration to dose ratio and its association with single gene variability, and did not take into account variation in other factors such as dosing regimen, blood sampling times and patient specific covariates including demographics, co‐medication and biochemical parameters. There is only one study in a Thai population that investigated the influence of specific genetic variants of UGT1A4 and UGT2B7, along with some non‐genetic factors, on LTG pharmacokinetics using the population pharmacokinetic approach 19. The available published studies provide limited information on the association of LTG pharmacokinetics with genetic factors.
Furthermore, the majority of previous population pharmacokinetic studies with LTG were based on very sparse concentration measurements (trough concentration only). There is only one study 24 where all pharmacokinetic parameters and their inter‐individual variability were estimated. In the remaining published studies parameters related to LTG absorption rate and distribution were fixed to literature values or only their typical population values without inter‐individual variability were estimated. There is a concern with such analyses as the precision of the prior estimates is not taken into account.
The aim of the present study was to develop a population pharmacokinetic model for quantitative evaluation of the influence of genetic variability in LTG metabolizing enzymes (UGT1A4 and UGT2B7) and transporters (ABCB1 and OCT1) on LTG pharmacokinetics while taking into account the influence of various clinical, biochemical and demographic factors. In the analysis we incorporated external data from preceding pharmacokinetic studies to stabilize the modelling process in a non‐linear mixed effects modelling analysis of poorly informative data. To obtain informative priors for the analysis we conducted a meta‐analysis of published population pharmacokinetic studies. Moreover, in the analysis we incorporated measurements of LTG‐glu to obtain more precise estimates of the covariate effects on LTG metabolism. To our knowledge this is the first population pharmacokinetic study with LTG incorporating the metabolite data.
Methods
Patients and blood sampling
This prospective study was conducted in adult patients with epilepsy on stable LTG treatment for at least 2 months at the Department of Neurology, University Medical Centre Ljubljana, Slovenia during their regular ambulatory visits. Patients of both genders older than 18 years with confirmed epilepsy who were on oral LTG treatment either as a mono‐ or combination therapy were eligible for the study. Chronic renal and hepatic diseases and pregnancy were exclusion criteria for the study. All enrolled patients were previously informed about the purpose of the study and provided written informed consent. The study was approved by the National Medical Ethics Committee of the Republic of Slovenia (25p/04/12) and was carried out according to the Declaration of Helsinki.
Demographic data (age, weight, height, gender), smoking status, adverse event data, duration of current AED therapy, concomitant drug therapy, co‐morbidities and LTG dosing regimen were collected at the time of enrolment into the study. Biochemical parameters, including serum creatinine (Crs), aspartate aminotransferase (AST) and alanine transaminase (ALT) were obtained from the patient's charts. Patient compliance with LTG treatment was assessed by the attending neurologist and all subjects suspected of non‐compliance were excluded from the study. Patients were asked to record the time of the last dose before the ambulatory visit. Two blood samples were drawn, approximating trough (immediately before dosing) and peak (after 2–4 h) steady‐state concentrations. Exact blood sampling times were recorded by the laboratory personnel. The collected blood samples were immediately centrifuged at 3000 g for 10 min, transported to a central storage facility and stored at −80°C until analysis.
Drug assay
For plasma concentration measurements of LTG and LTG‐glu, an h.p.l.c. method with u.v. detection from Saracino et al. was adapted 25. The assay was calibrated over the concentration range of 0.25–20 mg l−1 (r 2 > 0.9997) for LTG and 0.25–15 mg l−1 (r 2 > 0.9998) for LTG‐glu. The accuracy of the method expressed as bias was better than 13.1% and the precision (RSD) was below 3.8%. The lower limit of quantification was 0.25 mg l−1 for both analytes.
Genotyping
Isolation of genomic DNA from peripheral blood leukocytes was performed using the Qiagen FlexiGene kit (Qiagen, Hilden, Germany). The genotyping of UGT1A4 70C > A (rs 6 755 571), UGT2B7–161C > T (rs7668258) and 372 A > G (rs28365063) was performed by real‐time PCR genotyping using the Taqman Allelic Discrimination Assays (Applied Biosystems, Foster City, CA, USA) assay on the ABI 7500 instrument (Applied Biosystems, Foster City, California, USA). Genotyping of SLC22A1 1222G > A (rs628031), ABCB1 3435C > T (rs1045642) and 1236C > T (rs1128503) polymorphisms was carried out using a fluorescence‐based competitive allele‐specific (KASPar) assay (KBiosciences, Herts, UK). ABCB1 2677G > T/A (rs2032582) polymorphism was determined using PCR amplification followed by the analysis of PCR fragments on agarose gel as previously described 26. The genotype distributions were evaluated for Hardy–Weinberg equilibrium (HWE) by χ2 test applying Yates' correction.
Meta‐analysis of previous population pharmacokinetic studies with LTG
Since pharmacokinetic data in this study did not provide information on all structural and variability parameters, we used prior information from population pharmacokinetic studies with LTG to stabilize the model. For calculation of priors we performed a literature search through MEDLINE database for studies published until December 2014. The search strategy was: lamotrigine[tw] AND pharmacokinetics[tw] AND (nonmem[tw] OR population[tw]). Searching was limited to studies in adults published in English language. From all identified studies we extracted information on typical values (geometric means) of pharmacokinetic parameters in the population of patients, their interindividual variability (IIV) and relative standard errors (RSE) of all estimates. Before the analysis the data on IIV of the parameters were transformed into log‐domain, i.e. ω2 were calculated from the reported coefficient of variation (CV%) values. Meta‐analysis of the parameters was performed using a random effects model approach, taking into consideration heterogeneity of the studies 27. Normal inverse Wishart distribution for the fixed effects and interindividual variability was used. Degrees of freedom (DF) for the inverse Wishart distribution on ω2 were calculated using equation (1) 28.
| (1) |
Model development
Population pharmacokinetic analysis was performed by a non‐linear mixed effects modelling approach using nonmem software (ver. 7.3, Icon Development Solutions, Ellicott City, MD, USA). Model building steps and graphical presentations were managed using Perl‐speaks‐nonmem (PsN)® (ver. 3.7.6; http://psn.sourceforge.net/) and Xpose® (ver. 4.4.1; http://xpose.sourceforge.net). Pharmacokinetic analysis was performed in two steps. Initially, only the measurements of LTG concentration (parent drug) were taken into analysis. The structural model used was a one compartment model with first order absorption and elimination (ADVAN2/TRANS2 PREDPP subroutine). Since absolute bioavailability cannot be estimated based on oral data only, the estimated parameters were absorption rate constant (K a), apparent volume of distribution (V) and apparent clearance after oral dosing (CL). A log‐normal distribution of individual parameter values was assumed and an exponential model was used to estimate IIV. Additive, proportional and combination (additive + proportional) error models were evaluated to describe the residual intra‐individual variability of the LTG concentration. Prior information from meta‐analyses was incorporated into the model using nonmem prior functionality 29. The first order conditional method with interaction (FOCE‐I) was used for parameter estimation.
Stepwise covariate modelling procedure was used to build the final model. Categorical covariates including patient's gender, smoking status, co‐therapy with inducers (carbamazepine, oxcarbazepine, phenytoin, phenobarbital, oral contraceptives) or inhibitors (valproic acid (VPA), sertraline) of LTG metabolism, and genetic polymorphisms in UGTs, SLC22A1 and ABCB1 were tested. Association with genetic polymorphisms were tested using a co‐dominant model. This model compares heterozygous and homozygous variant genotypes to the homozygous wild‐type. Additionally, dominant and recessive models were evaluated. Moreover, the effect of ABCB1 haplotype was tested. Continuous covariates considered for inclusion were patient's weight, ideal body weight (IBW) calculated using the Devine equation, age, Crs, Cockroft–Gault estimate of creatinine clearance (CLcr), liver enzymes (AST, ALT), and LTG daily dose. Continuous covariates were centred to the population mean covariate values and were incorporated using linear and power models. Missing covariate data were substituted with the mean covariate value (continuous) or most frequent value (categorical covariates). In the forward addition step criteria for covariate inclusion into the model were set at P < 0.05. A difference in objective function value (OFV) is approximately χ2 distributed, with degrees of freedom equal to the difference in the number of parameters between the two hierarchical models. The final model was determined by backward elimination of covariates from the full model using a more stringent criterion (P < 0.01) to compensate for multiple comparisons. An additional criterion for the retention of a covariate was reduction in the unexplained IIV. The model adequacy was checked by standard diagnostic plots of predicted vs. observed concentration and weighted residuals vs. observed concentration or time. The final pharmacokinetic model was further evaluated for precision by a bootstrap procedure. The bootstrap re‐sampling method with replacement using 1000 replications was applied to calculate parameter standard errors and non‐parametric 95% confidence intervals (95% CI). Internal predictive ability was evaluated by visual predictive check (VPC) applying prediction and variability correction. Multiple imputation analysis described by Johansson et al. [30] was used to evaluate the influence of the missing covariate data on smoking status.
To get further insight into disposition of LTG, the data on LTG‐glu concentration measurements were included into analysis in the second step. An additional compartment was introduced for LTG‐glu plasma concentration. We assumed that LTG was completely converted to LTG‐glu. This assumption seems reasonable as mass balance studies with LTG indicate that renal elimination of unchanged LTG is insignificant and accounts for less than 10% and that conversion to LTG‐N‐5‐glucuronide is a minor route of LTG metabolism 5. The model was coded by differential equations employing nonmem's ADVAN 6 subroutine. Pharmacokinetic parameters estimated were apparent LTG‐glu volume of distribution (V LTG‐glu) and LTG‐glu elimination clearance (CLLTG‐glu), in addition to the parameters related to disposition of LTG. Covariate effects on the pharmacokinetic parameters CL, V, CLLTG‐glu and V LTG‐glu were tested as described above.
Sensitivity analysis
Sensitivity of the model parameters to specification of the priors was evaluated by re‐estimation of the final model by variation of the prior mean and variance by −50% and +50%. Additionally, we evaluated the sensitivity of the parameters to informativeness of the prior using more and less informative prior. For this purpose we altered the precision of the prior by changing standard errors to −50% and +50%.
Results
Patients
We included 100 patients with epilepsy on monotherapy with LTG (n = 54) or in combination with other AEDs (n = 46), including carbamazepine, oxcarbazepine, valproic acid, phenytoin, phenobarbital, levetiracetam, topiramate, lacosamide, zonisamide, pregabalin, clonazepam and clobazam. All patients were on stable AED therapy for at least 2 months; therefore we assumed that LTG pharmacokinetics were in steady‐state and that potential induction or inhibition of LTG metabolism with other AEDs was complete. A total of 195 LTG and 195 LTG‐ glu concentration measurements were available for analysis. The measured concentrations ranged from 0.7–23.18 mg l−1 and 0.32–8.52 mg l−1 for LTG and LTG‐glu, respectively. Characteristics of the patients included in the study are summarized in Table 1.
Table 1.
Summary of patient characteristics
| Patient characteristic | Value |
|---|---|
| Demographic data | |
| Number of patients (n) | 100 |
| Gender (female/male) | 70 (70)/30 (30) |
| Age (years) | 39.8 (20.7–80.4) |
| Weight (kg) | 70 (50–124) |
| Body mass index ( kg m −2 ) | 24.5 (18.7–41.3) |
| Height (m) | 1.67 (1.52–1.90) |
| Biochemical assessment | |
| ALT (μkat l −1 ) | 0.33 (0.14–1.57) |
| AST (μkat l −1 ) | 0.31 (0.16–0.91) |
| Cr s (μmol l −1 ) | 74 (39–125) |
| CL cr (ml min−1) | 107.6 (40.84–246) |
| Co‐medication | |
| Inducers | |
| Carbamazepine (n) | 4 (4) |
| Oxcarbazepine (n) | 6 (6) |
| Phenytoin (n) | 2 (2) |
| Phenobarbital (n) | 2 (2) |
| Oral contraceptives (n) | 2 (2) |
| Inhibitors | |
| Valproic acid (n) | 13 (13) |
| Sertraline (n) | 2 (2) |
| Other | |
| LTG dose (mg day –1 ) | 200 (50–600) |
| Somatic diseases (Yes/No) | 18 (19)/78 (79) |
| Smokers/Non‐smokers | 17 (22)/59 (78) |
Continuous data are presented as median (range) and categorical data as the number of patients (%). ALT, alanine transaminase; AST, aspartate transaminase; CLcr, Cockroft–Gault estimate of creatinine clearance; Crs, serum creatinine.
Genotyping
Genotypes and allele frequencies are presented in Table 2. ABCB1 2677GA genotype was observed in one patient and was analyzed in the ABCB1 2677GT group. All genotype frequencies were in Hardy–Weinberg equilibrium (P > 0.05). Linkage disequilibrium was observed between all three ABCB1 loci (D’(2677G > T/A, 3435C > T) = 0.79, D’(2677G > T/A, 1236C > T) = 0.89, D’(3435C > T, 1236C > T) = 0.72, all P < 0.001). ABCB1 1236C > T‐2677G > T‐3435C > T haplotypes were constructed. ABCB1 T–T‐T haplotype was observed in 53% of patients, followed by C‐G‐C (23%) and C‐G‐T (14%). Frequencies of other ABCB1 haplotypes were minor, accounting for <3%.
Table 2.
Genotype and allele frequency
| Polymorphism | Genotype frequency (%) | Allele frequency* | p(HWE)† | |||
|---|---|---|---|---|---|---|
| Homozygous wild type | Heterozygous | Homozygous variant | p | q | 0.861 | |
| SLC22A1 | ||||||
| rs628031 1222G > A | 39 (39.8) | 45 (45.9) | 14 (14.3) | 0.628 | 0.372 | 0.867 |
| UGT2B7 | ||||||
| rs7668258 ‐161C > T | 21 (22.3) | 46 (48.9) | 27 (28.7) | 0.468 | 0.532 | 0.921 |
| rs28365063 372 A > G | 72 (72.7) | 25 (25.3) | 2 (2.0) | 0.854 | 0.146 | 0.753 |
| UGT1A4 | ||||||
| rs6755571 70C > A | 91 (93.8) | 6 (6.2) | 0 (0.0) | 0.969 | 0.031 | 0.851 |
| ABCB1 | ||||||
| rs2032582 2677G > T/A | 40 (42.6) | 42 (44.7) | 12 (12.8) | 0.649 | 0.351 | 0.173 |
| rs1045642 3435C > T | 28 (28.0) | 56 (56.0) | 16 (16.0) | 0.560 | 0.440 | 0.484 |
| rs1128503 1236C > T | 40 (40.0) | 49 (49.0) | 11 (11.0) | 0.645 | 0.355 | 0.861 |
Hardy–Weinberg notation for allele frequencies: p, frequency of wild‐type allele; q, frequency of variant allele;
χ2‐test for deviation from Hardy–Weinberg equilibrium.
Calculation of priors
Eleven eligible studies were identified by the systematic literature search (Table 3) and were used for calculation of priors. Collectively, all relevant studies estimated the typical value of CL (θCL) and its IIV (IIVCL). Typical values of V were estimated only in six studies while in other studies it was fixed to a literature value. Typical values of K a were estimated in only three studies and most of the studies used a fixed value, previously estimated by Grasela et al. 24. Moreover, this is the only study which estimated IIVKa. IIVV was estimated in three population analyses. In a study by Milovanovic et al. a one compartment model with first‐order elimination without absorption (intravenous bolus) was used 31, while in a study by Gidal et al. a steady‐state pharmacokinetic model (continuous i.v. infusion) was used for the analysis 32. The Q‐statistic (P < 0.0001) demonstrated heterogeneity among studies in all parameters, except IIVV. Graphical summary of the meta‐analysis, prior estimates used for population pharmacokinetic analysis and nonmem code are given in the online supporting information.
Table 3.
Pharmacokinetic parameters from reference studies
| Reference | Patients (n) | Typical values | Interindividual variability (CV%) | ||||
|---|---|---|---|---|---|---|---|
| CL (l h−1) | V (l) | K a (h−1) | IIVCL | IIVV | IIVKa | ||
| Brzakovic et al. 33 | 53 | 4.23 (8.2) | 84*, * | 3.5* | 42.2 (13) | / | / |
| Wegner et al. 12 | 23 | 3.17 (12) | 132† | 1.3† | 35 (61) | / | / |
| Mallaysamy et al. 38 | 95 | 2.27 (4.1) | 56.3 (14) | 0.379 (17) | 28.8 (8.4) | 30.6 (21) | / |
| Singkham et al. 19 | 75 | 2.49 (8.4) | 156 (47) | 1.3† | 22.4 (50) | / | / |
| Wegner et al. 39 | 809 | 2.56 (4.2) | 105† | 1.3† | 43 (11) | / | / |
| Punyawudho et al. 35 | 148 | 2.39 (20) | 115 (15) | 3.5* | 34.2 (17) | / | / |
| Milovanovic et al. 31, ‡ | 38 | 1.98 (17) | 78.9 (17) | / | 30 (16) | / | / |
| Rivas et al. 36 | 600 | 1.96 (2.1) | 105† | 1.3† | 27.5 (9.4) | / | / |
| Gidal et al. 32, § | 62 | 2.25 (7.0) | / | / | 24.1 (26) | / | / |
| Grasela et al. 24 | 527 | 4.23 (12) | 132 (12) | 1.30 (17) | 32.2 (11) | 44.3 (44) | 77.5 (137) |
| Hussein et al. 43 | 163 | 2.28 (2.9) | 77.4 (6.6) | 3.18 (14) | 32.1 (16) | 33.6 (43) | / |
| Meta‐analysis | 2.55 (5.1) | 89.8 (12.4) | 1.54 (40.4) | 31.8 (9.5) | 31.8 (17.7) | 77.5 (137) | |
Data reported as parameter estimates (% relative standard error);
Parameter not estimated, fixed to a literature value (Chan et al. [46]);
Parameter not estimated, fixed to a literature value (Grasela et al. [24]);
Absorption rate not estimated, compartmental model with bolus intravenous injection was used;
Steady‐state pharmacokinetic model (continuous intravenous infusion) was used for analysis.
Population pharmacokinetic analysis of lamotrigine
A total of 100 pharmacokinetic profiles were available for analysis. With the base model, the population geometric means of K a, CL and V and their IIV (CV%) were estimated at 1.96 h−1 (72.8%), 2.32 l h−1 (41.4%) and 77.6 l (30.2%), respectively. Residual variability was most adequately described using a proportional error model and was estimated at 19.8%. Graphical exploration of the relationship between individual Bayesian estimates of the base model and various patient specific covariates indicated a trend of higher LTG CL in patients who smoke and patients who are concomitantly treated with enzyme inducing AEDs including carbamazepine, phenobarbital and phenytoin, but not with oxcarbazepine. Since induction magnitude of carbamazepine, phenobarbital and phenytoin appeared similar and the number of patients on combination treatment was relatively small, we combined these patients into one group (Figure 1). Additionally, there was a trend of higher CL in patients taking oral contraceptives, but the number of these patients (n = 2) was too small to be tested within nonmem. On the other hand, there was a trend towards lower LTG CL in patients taking VPA and sertraline. Again, the magnitude of these two effects was comparable. Among genetic factors evaluated, the effect of UGT2B7 polymorphisms was evident. Interestingly, LTG CL appeared lower in patients with UGT2B7–161TT genotype compared with patients who were not carriers of this allele. However, with UGT2B7 372 A > G patients homozygous for the variant allele (372GG genotype) had higher CL than patients with the 372AA genotype, while patients who were heterozygous (372AG) were in‐between. There was no evident genotype–smoking or genotype–co‐treatment with inhibitors of metabolism interaction (online supporting information).
Figure 1.
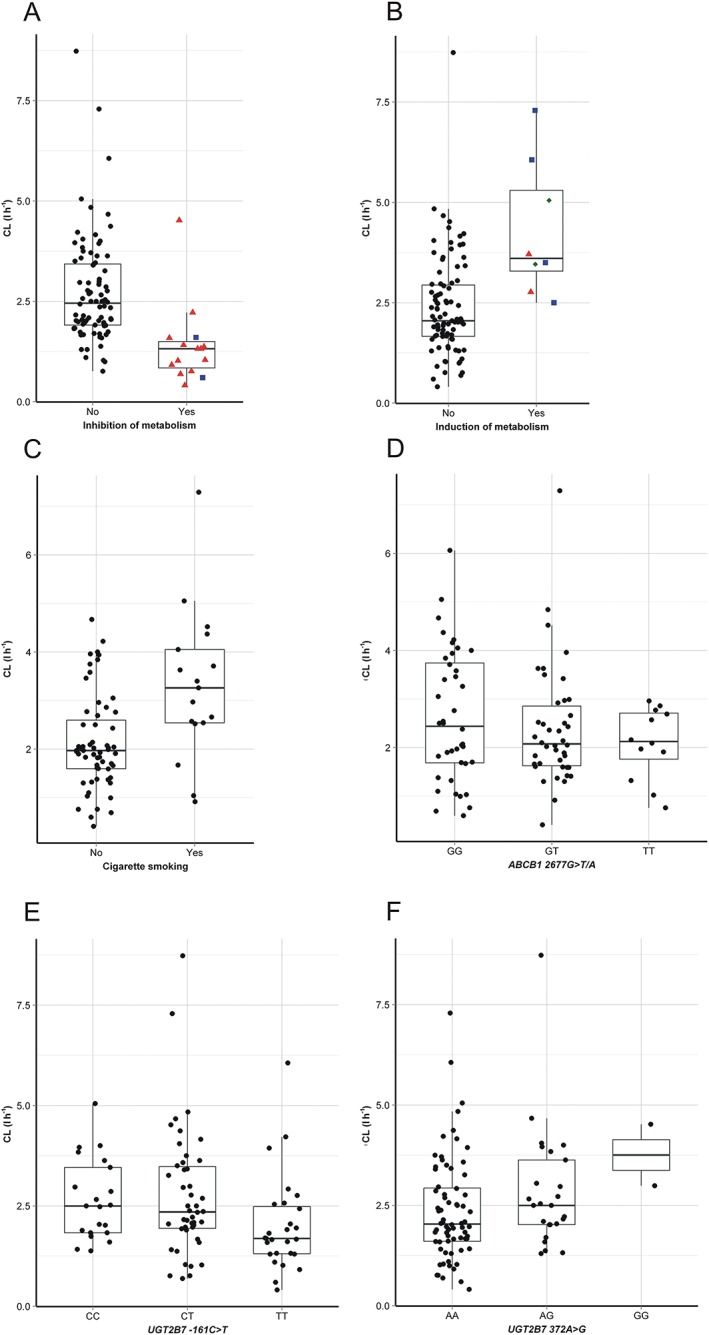
Post hoc estimates of individual patient's lamotrigine clearance (CL) with the base population model according to co‐treatment with inhibitors of metabolism (A:  None,
None,  Sertraline,
Sertraline,  Valproic acid), inducers (B:
Valproic acid), inducers (B:  None,
None,  Carbamazepine,
Carbamazepine,  Phenobarbital,
Phenobarbital,  Phenytoin), cigarette smoking (C), ABCB1 2677G > C/A genotype (D), UGT2B7–161C > T genotype (E) and UGT2B7 372 A > G genotype (F)
Phenytoin), cigarette smoking (C), ABCB1 2677G > C/A genotype (D), UGT2B7–161C > T genotype (E) and UGT2B7 372 A > G genotype (F)
With stepwise covariate modelling, during forward inclusion CL was found to be significantly (P < 0.05) affected by weight, cigarette smoking, LTG DD, CLcr, co‐therapy with inducers and inhibitors, genetic polymorphisms of UGT2B7 (−161C > T and 372 A > G) and ABCB1 (2677G > T/A). Furthermore, V was found to be influenced by patient weight. During the backward elimination step the influences of LTG DD and ABCB1 genotype were found insignificant (P > 0.01) and were excluded from the model. Of the continuous covariates included in the final model the effect of patient weight on LTG CL was described with the power model, while the effects of weight on V and CLcr on CL were more adequately described with the linear model. Parameter estimates of the final model along with the bootstrap results are presented in Table 4.
Table 4.
Parameters of the final pharmacokinetic model of lamotrigine and results of the bootstrap analysis
| Parameter | Estimate | Bootstrap | ||
|---|---|---|---|---|
| Mean | RSE (%) | 95% CI | ||
| K a ( h −1 ) | 1.96 | 1.97 | 6.69 | (1.72, 2.24) |
| CL (l h −1 ) | 2.40 | 2.40 | 2.13 | (2.30, 2.50) |
| Covariate effects on CL | ||||
| Co‐treatment with inhibitors * | −0.579 | −0.582 | 8.52 | (−0.674, 0.483) |
| Body weight | 0.938 | 0.937 | 20.9 | (0.558, 1.32) |
| Cigarette smoking | 0.340 | 0.331 | 33.5 | (0.147, 0.590) |
| Co‐treatment with inducers † | 0.546 | 0.567 | 49.6 | (0.114, 1.21) |
| UGT2B7 372 A > G genotype | ||||
| AG vs. AA | 0.194 | 0.199 | 47.4 | (0.0331, 0.392) |
| GG vs. AA | 1.17 | 1.26 | 43.2 | (0.448, 2.47) |
| UGT2B7–161C > T genotype | ||||
| CT vs. CC | −0.0358 | −0.0338 | 204 | (−0.161, 0.098) |
| TT vs. CC | −0.204 | −0.197 | 38.7 | (−0.336, −0.0364) |
| CL cr | 0.00328 | 0.00333 | 44.7 | (0.000550, 0.00640) |
| V (l) | 76.2 | 75.8 | 5.60 | (66.9, 84.2) |
| Covariate effects on V | ||||
| Body weight | 0.0181 | 0.0178 | 19.0 | (0.0102, 0.0239) |
| Interindividual variability | ||||
| IIV Ka (%) | 71.1 | 74.4 | 32.4 | (0.329, 0.691) |
| IIV CL (%) | 33.1 | 32.6 | 5.00 | (0.0921, 0.112) |
| IIV V (%) | 30.1 | 30.3 | 5.40 | (0.0831, 0.0959) |
| Residual variability (%) | 18.0 | 17.2 | 19.3 | (10.4, 23.5) |
CL(l h−1) = 2.40 × (1 − 0.579 Inh) × (Wt/70)0.938 × (1 + 0.340 Tob) × (1 + 0.546 Ind) × (1 − 0.0358 UGT2B7−161CT) × (1 − 0.204 UGT2B7−161TT) × (1 + 0.194 UGT2B7372AG) × (1 + 1.17 UGT2B7372GG) × (1 + 0.00328 (CLcr − 110))
V(l) = 76.2 × (1 + 0.0181(Wt − 70))
Valproic acid or sertraline (Inh);
Carbamazepine, phenobarbital or phenytoin (Ind); Patient weight (Wt); Tobacco smoking (Tob); Creatinine clearance (CLcr).
Categorical covariates including patient genotype are assigned a value of 1 (present) or zero (absent).
Results of the bootstrap indicate that the parameters are estimated with reasonable precision. In comparison with the base model, unexplained IIV of CL decreased from 41.4 to 33.1%. Diagnostic plots of population predicted vs. observed concentrations and individual predicted vs. observed concentrations (Figure 2) demonstrate adequate performance of the model. Compared with the base model, the final model showed an improved fit. Internal predictive performance of the final model was adequate (Figure 3).
Figure 2.
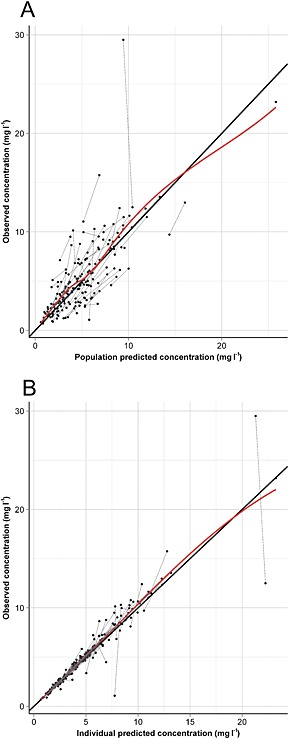
Diagnostic plots of the final lamotrigine population pharmacokinetic model. Population predicted vs. observed concentrations (A) and individual predicted vs. observed concentrations (B) with line of identity (solid) and LOESS fit (dashed)
Figure 3.
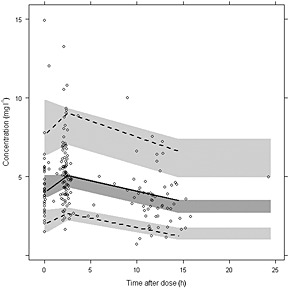
Visual predictive check of the final model with prediction and variability correction. Median (solid line), 5th and 95th percentiles (dashed lines) of the binned observed data (circles) with corresponding 95% confidence intervals of the simulations (shaded areas)
Except for smoking status (24%) the proportion of missing covariate data was low (maximum 6% per individual covariate). To ensure that handling of the missing smoking status did not affect the results of the final model we applied multiple imputation analysis (online supporting information). The mean estimate of the effect of smoking on LTG CL obtained with multiple imputation was 0.418 with the relative standard error (RSE) of 22.7% and was in good agreement with the estimate obtained when non‐smoking status was imputed to missing smoking data (Table 4).
Sensitivity analysis
Sensitivity analysis of the final model revealed that the effect of the prior uncertainty on parameter estimates and the modelling conclusions is minor. By changing standard errors of the priors to +50% and −50%, parameter estimates remained within ±15% range. Moreover, using less or more informative priors on all structural and variability parameters of the model did not importantly influence the covariate relationships, as all of the identified relationships remained significant. However, the influence of prior means on structural parameters K a and V was more profound. Changing the prior means on these parameters to +50% and −50% resulted in estimates between +30% and −18% compared with the reference parameter estimates. The effect of prior mean on CL was more complex. Altering the prior mean on CL to +50% and −50% shifted this parameter to +37% and −24%, respectively. The impact on the magnitude of covariate effects was even more profound. However, the influence of the covariates on CL stayed significant. More detailed analysis of these results revealed that when patients were stratified according to the covariate values, LTG CL in each patient stratum was relatively unaffected. The covariate effects resulting from alteration of prior mean on CL therefore compensated for the change in the typical estimate of CL. As expected, changing the prior on IIV of K a and V resulted in almost identical change in the estimates of these parameters, indicating that there is little information on the IIV of these parameters in our data.
Population pharmacokinetic analysis of the lamotrigine‐N‐2‐glucuronide
To confirm our findings we incorporated the data on LTG‐glu plasma concentration in the population pharmacokinetic analysis. Typical value of CLLTG‐glu was estimated at 3.16 l h−1 and was found affected by the patient's renal function (linear model, a decrease of 0.759% per 1 ml min−1 decrease in CLcr) and patient weight (power model, exponent 1.01). Unexplained IIV on CLLTG‐glu was 41.7%. Distribution volume of the metabolite (V LTG‐glu) was estimated at 110 l. Due to sparse concentration measurements and no prior data on pharmacokinetics of the metabolite we were not able to estimate the IIV of V LTG‐glu. The residual (intra‐individual) variability of LTG‐glu concentration was 13.8%. The estimates of the pharmacokinetic parameters and covariate relationships related to the parent LTG obtained with this model were very similar to those obtained with the parent drug model.
Discussion
The majority of published population pharmacokinetic studies with LTG is based on sparse concentration measurements. In most of these studies parameters related to LTG absorption rate and distribution were fixed to literature values or only their typical population values without IIV were estimated. There is a concern with such analyses as the precision of the previous estimates is not taken into account. In the present analysis plasma concentration measurements of LTG and LTG‐glu were insufficient to estimate all parameters. Therefore we used prior information from previous population pharmacokinetic studies with LTG to stabilize the model using nonmem prior functionality. With this approach the objective function based on the current data is augmented by a penalty term which is a representation of previous knowledge with regards to model parameters 29.
Data for elicitation of priors for the one compartment pharmacokinetic model parameters of LTG were derived from 11 studies. In comparison with the priors, our estimates of the typical values of K a, V and CL obtained with the base model were 1.96 vs. 1.54 h−1, 77.6 vs. 89.8 l and 2.32 vs. 2.55 l h−1, respectively. This suggests that our data were informative on the typical values of pharmacokinetic parameters. On the other hand, our estimates of the IIV of K a and V were very similar to the priors used for the analysis, 72.8% vs. 77.5% for K a and 30.2% vs. 31.7% for V, while our estimate of the IIV on CL was considerably larger, 41.4% vs. 31.8%. This suggests either that the priors on IIV of K a and V accurately described our data or that our data contained very little information on IIV of these two parameters. Collectively, comparison with priors indicates that our data are informative on CL, which is important from the modelling perspective as we were primarily interested in covariate relationships with CL. For all the estimated parameters RSE was less than 10%, except for IIVKa (32.4%).
Previous published studies showed that various factors such as weight 31, 32, 33, 34, 35, 36, 37, 38, age 39, 40, 41, pregnancy 13, smoking 42, blood urea nitrogen : serum creatinine ratio 35, race 24, 43, LTG daily dose [31], oral contraceptives 11, 12, 14, 44 and use of concurrent AEDs 13, 24, 31, 32, 33, 34, 35, 36, 37, 38, 41, 45, 46, 47 influence LTG plasma concentrations. Additionally, genetic polymorphisms in genes encoding drug‐metabolizing enzymes UGT1A4 18, 19, 22, 23, UGT2B7 19, 20 and ABCB1 transporter [21] were also suggested to contribute to the IIV of LTG CL. However, the relative contribution of these factors to the variability in LTG pharmacokinetics is controversial. In this study we systematically explored the influence of various covariates on LTG pharmacokinetics by a non‐linear mixed effects modelling approach. Furthermore, in the analysis we incorporated metabolite data to obtain more precise estimates of the covariate effects on LTG metabolism. To our knowledge this is the first population pharmacokinetic study with LTG which systematically studied various demographic, genetic and clinical factors and incorporated in the analysis the data on disposition of the metabolite.
Results from our study provide evidence for the influence of UGT2B7 polymorphisms, weight, creatinine clearance, smoking and co‐therapy with enzyme inducers and inhibitors on LTG pharmacokinetics. Additionally, we found that LTG‐glu clearance was affected by the patient's renal function and patient's weight.
We confirmed previously a demonstrated effect of smoking on LTG metabolism 42. Compared with non‐smokers, LTG clearance was 34% (95% CI 14.7, 59.0%) higher in patients who smoked. Since LTG is not a CYP 450 substrate and UGT1A4 activity may not be affected by nicotine [42, 48] it is suggested that the effect of tobacco smoking is mediated by UGT2B7. Considering the magnitude of the effect of smoking on LTG CL and the fact that smoking is still a widely spread habit we recommend smoking habits to be taken into account in the evaluation of treatment with this drug.
We confirmed that LTG CL and V depended on patient's body weight. Similar results have been reported by other authors in paediatric and adult populations of epileptic patients 31, 33, 35, 36, 37. In accordance with the allometric scaling approach the effect of weight on V was more adequately described with the linear model, while a power model was more appropriate for the relationship of weight with CL. The exponent for the effect of weight on CL was 0.932 and the 95% CI (0.558, 1.32) included the theoretical value of 0.75. Accordingly, a 100 kg patient would require a 40% larger dose to achieve similar steady‐state LTG concentrations found in a 70 kg patient, while in a lean 45 kg patient LTG dose should be reduced by 34%. However, the dose per body weight is very similar, as the estimated exponent for the effect of weight is close to 1. Additionally, we have found that patient's body weight also has an effect on CL of LTG‐glu.
Concomitant administration of inhibitors or inducers of LTG metabolism proved to have an effect on LTG CL, as would be expected for a drug mainly eliminated by metabolism. Concomitant administration of VPA and sertraline, which are commonly prescribed, results in reduction of LTG CL by 58%. The mechanism behind the interaction with VPA is competitive inhibition of glucuronidation by VPA 49. Previous reports on lamotrigine interaction with sertraline are conflicting 49, 50. Our data suggest that co‐treatment with sertraline decreases LTG CL. We hypothesize that sertraline inhibits LTG glucuronidation through competitive inhibition with resultant increased plasma concentration. On the other hand, LTG CL in patients concomitantly treated with inducers of hepatic metabolism (carbamazepine, phenytoin and phenobarbital) was 55% higher. This is consistent with the previous reports 24, 33, 35, 36, 38. The current guidelines for LTG dosing account for the clearance alteration in patients when using enzyme inducing or inhibiting drugs 52.
Moreover, CL of LTG and LTG‐glu was influenced by the patient's renal function. Since renal excretion of unchanged drug is a minor route of LTG elimination the influence of CLcr on CLLTG is relatively small (a decrease by 0.328% per 1 ml deviation from a standard CLcr of 110 ml min−1). Based on this estimate a decrease in CLLTG up to 31% is predicted in patients with stage 4 chronic kidney disease (CLcr 15–30 ml min−1). The influence of renal function on CLLTG‐glu is more pronounced (a decrease by 0.759% per 1 ml deviation from a standard CLcr of 110 ml min−1). Collectively these results indicate that impaired renal function has little effect on the plasma concentrations of LTG achieved for a given dosing regimen which is in accordance with a previous report 53. Nevertheless, due to limited clinical experience, LTG should be used with caution in patients with significant impairment of renal function 52.
LTG seems to be primarily metabolized by UGT1A4 which explains its interaction with hormonal contraceptives. Nevertheless, previous in vitro studies indicate that UGT2B7 is also involved in LTG glucuronidation and that inhibition of this metabolic route is the mechanism behind the observed interaction with VPA 6. In our study, patients carrying the UGT2B7–161 TT genotype had 20.4% lower CLLTG compared with patients with CC genotype which is in accordance with the previously published data 19, 20. In patients who were heterozygous for UGT2B7–161C > T the difference in CLLTG was only minor (−3.7%) and insignificant in comparison with the patients with the UGT2B7–161 CC genotype. Contrary to the previous studies investigating the effects of UGT2B7 genotypes 19, 20 we observed a significant effect of UGT2B7 372 A > G on CLLTG. In patients carrying the UGT2B7 372 GG genotype CLLTG was 117% higher compared with patients with the UGT2B7 372 AA genotype, while in patients who were heterozygous CLLTG was 19.4% higher compared with patients who were homozygous for the wild type allele. The frequency of patients having the UGT2B7–161 TT and UGT2B7 372 AG genotype in our population was relatively high (48.9% and 25.3%, respectively), suggesting that these polymorphisms could be considered to facilitate LTG dosage adjustments. UGT2B7 372 GG genotype is rare, accounting for 2% in our population. Nevertheless, having in mind the magnitude of its effect, LTG treatment could be ineffective in patients with this genotype due to underdosing. The results from our study are consistent with those of previous studies which reported no association of UGT1A4 70C > A polymorphism with LTG pharmacokinetics 18, 19, 23.
To our knowledge this is the first study that simultaneously evaluated the influence of ABCB1 and SLC22A1 polymorphisms on LTG pharmacokinetics. In contrast to the study by Lovric et al. 21 who investigated the association of ABCB1 genotypes with LTG concentration and LTG concentration to dose ratio, we observed no significant influence of ABCB1 1236C > T on LTG pharmacokinetics. In our data, compared with patients with the ABCB1 2677 TT genotype we observed a trend of 22% and 9% lower CLLTG in patients with ABCB1 2677 GG and ABCB1 2677 GT genotypes, respectively. However, during the backward elimination step this effect was insignificant. Futhermore, our analysis did not reveal any association of ABCB1 haplotypes with LTG pharmacokinetics despite the fact that haplotype frequencies in our study were very similar to those reported by Lovric et al. 21.
Previous in vitro studies indicated that LTG is a substrate for OCT1 at brain endothelial cells 8. In a systematic study on genetic determinants of LTG dosing, Grant [54] studied the association between SLC22A1 1222G > A and maintenance dose of LTG in 96 epilepsy patients. This is a coding non‐synonymous polymorphism which results in a Met408Val amino acid substitution in the OCT1 protein. In the multivariate analysis (not corrected for multiple comparisons) Grant observed that patients who were homozygous for a major allele (GG) require a higher maintenance dose of LTG than the heterozygous group (GA) and the homozygous‐minor allele group (AA). The geometric mean ratios of maintenance doses were 1.28 for GG vs. GA genotype and 0.895 for AA vs. GA genotype. We observed similar non‐significant trends in our group of patients (P = 0.600). However, the magnitude of the influence on CLLTG in our study was smaller. Compared with patients with GA genotype CL in patients with GG genotype was 8.58% higher, while in patients with AA genotype it was 3.06% lower. These data reveal that SLC22A1 1222G > A has a very minor influence on LTG pharmacokinetics. However, since this transporter is also expressed in the blood brain barrier it may be involved in the disposition of LTG in the central nervous system.
The major limitation of our study was the relatively low number of patients to determine association of the genetic polymorphisms other than UGT2B7. Another limitation of the study is sparse sampling strategy which did not support precise estimation of K a and its IIV. Nevertheless, with inclusion of the prior knowledge we were able to overcome this limitation. Moreover, our analysis indicates that our results are relatively robust to the choice of priors. Additionally, the effects of each enzyme‐inducing (carbamazepine, phenytoin, phenobarbital, oral contraceptives) and enzyme‐inhibiting drug (VPA, sertraline) were not quantified separately for each of these drugs, as the number of patients using phenytoin, phenobarbital, oral contraceptives and sertraline was small. Nevertheless, we were able to quantify the contribution of genetic, demographic and clinical factors to the IIV of LTG pharmacokinetics. Our results are further supported by the pharmacokinetic analysis of the LTG metabolite. Although the metabolite is not active, it provides additional evidence on kinetics of LTG metabolism.
In conclusion, to our knowledge this is the first population pharmacokinetic study which systematically evaluated the effects of genetic, demographic and clinical factors on LTG pharmacokinetics. This study indicates that there is a considerable variability in LTG pharmacokinetics and therefore TDM may be warranted. Genotyping could be useful in various clinical settings for therapy individualization. The population pharmacokinetic model could be used for Bayesian estimation of the patient's individual pharmacokinetic parameters based on sparse plasma sampling and for selection of the optimal dosing regimen in routine patient care. Nevertheless, the clear benefit of TDM and genotyping, like decrease of risk for serious adverse effects, is yet to be demonstrated in a clinical study.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare no support from any organization for the submitted work, no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work.
We are grateful to medical technician Aleš Kranjc from the Department of Neurology, University Medical Centre Ljubljana for his valuable contribution with blood sampling and the handling of blood samples. DM received doctoral funding from Ad Futura Scientific and Educational Foundation of Slovenia.
Contributors
DM, BL, TV, VD and IG conceived and designed the study. BL was the principal investigator. DM and BL contributed to patient recruitment and collection of clinical data. DM and TV performed the chromatographic analyses. MK and VD genotyped the patients. IG and DM analyzed the data. DM and IG drafted the first manuscript. All authors reviewed and approved the final manuscript.
Supporting information
S1 Pharmacokinetic priors used for the analysis
S2 Graphical summary of the meta‐analysis. Parameter estimates (symbols) with 95% CI (error bars); Relative study weight is presented by horizontal bars
S3 nonmem control stream
S4 Influence of smoking on post‐hoc estimates of individual patient's lamotrigine clearance (CL) with the base population model across various genotype groups
S5 Influence of co‐treatment with inhibitors of metabolism on post‐hoc estimates of individual patient's lamotrigine clearance (CL) with the base population model across various genotype groups
S6 Multiple imputation of missing smoking status
S7 Conditional density of smoking given lamotrigine oral clearance (left) and ROC curve for logistic regression (right)
Supporting info item
Milosheska, D. , Lorber, B. , Vovk, T. , Kastelic, M. , Dolžan, V. , and Grabnar, I. (2016) Pharmacokinetics of lamotrigine and its metabolite N‐2‐glucuronide: Influence of polymorphism of UDP‐glucuronosyltransferases and drug transporters. Br J Clin Pharmacol, 82: 399–411. doi: 10.1111/bcp.12984.
References
- 1. Pellock JM. Lamotrigine. J Child Neurol 1997; 12: S1. [DOI] [PubMed] [Google Scholar]
- 2. Cohen AF, Land GS, Breimer DD, Yuen WC, Winton C, Peck AW. Lamotrigine, a new anticonvulsant: pharmacokinetics in normal humans. Clin Pharmacol Ther 1987; 42: 535–41. [DOI] [PubMed] [Google Scholar]
- 3. EURAP Study Group . Seizure control and treatment in pregnancy: observations from the EURAP epilepsy pregnancy registry. Neurology 2006; 66: 354–60. [DOI] [PubMed] [Google Scholar]
- 4. Garnett WR. Lamotrigine: pharmacokinetics. J Child Neurol 1997; 12: S10–5. [DOI] [PubMed] [Google Scholar]
- 5. Doig MV, Clare RA. Use of thermospray liquid chromatography‐mass spectrometry to aid in the identification of urinary metabolites of a novel antiepileptic drug, lamotrigine. J Chromatogr 1991; 554: 181–9. [DOI] [PubMed] [Google Scholar]
- 6. Rowland A, Elliot DJ, Williams JA, Mackenzie PI, Dickinson RG, Miners JO. In vitro characterization of lamotrigine N2‐glucuronidation and the lamotrigine‐valproic acid interaction. Drug Metab Dispos 2006; 34: 1055–62. [DOI] [PubMed] [Google Scholar]
- 7. Luna‐Tortos C, Fedrowitz M, Loscher W. Several major antiepileptic drugs are substrates for human P‐glycoprotein. Neuropharmacology 2008; 55: 1364–75. [DOI] [PubMed] [Google Scholar]
- 8. Dickens D, Owen A, Alfirevic A, Giannoudis A, Davies A, Weksler B, et al. Lamotrigine is a substrate for OCT1 in brain endothelial cells. Biochem Pharmacol 2012; 83: 805–14. [DOI] [PubMed] [Google Scholar]
- 9. Johannessen SI, Tomson T. Pharmacokinetic variability of newer antiepileptic drugs: when is monitoring needed? Clin Pharmacokinet 2006; 45: 1061–75. [DOI] [PubMed] [Google Scholar]
- 10. Patsalos PN, Berry DJ, Bourgeois BF, Cloyd JC, Glauser TA, Johannessen SI, et al. Antiepileptic drugs – best practice guidelines for therapeutic drug monitoring: a position paper by the subcommission on therapeutic drug monitoring, ILAE Commission on Therapeutic Strategies. Epilepsia 2008; 49: 1239–76. [DOI] [PubMed] [Google Scholar]
- 11. Sabers A, Ohman I, Christensen J, Tomson T. Oral contraceptives reduce lamotrigine plasma levels. Neurology 2003; 61: 570–1. [DOI] [PubMed] [Google Scholar]
- 12. Wegner I, Wilhelm AJ, Lambrechts DA, Sander JW, Lindhout D. Effect of oral contraceptives on lamotrigine levels depends on comedication. Acta Neurol Scand 2014; 129: 393–8. [DOI] [PubMed] [Google Scholar]
- 13. Polepally AR, Pennell PB, Brundage RC, Stowe ZN, Newport DJ, Viguera AC, et al. Model‐based lamotrigine clearance changes during pregnancy: clinical implication. Ann Clin Transl Neurol 2014; 1: 99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reimers A, Helde G, Brathen G, Brodtkorb E. Lamotrigine and its N2‐glucuronide during pregnancy: the significance of renal clearance and estradiol. Epilepsy Res 2011; 94: 198–205. [DOI] [PubMed] [Google Scholar]
- 15. Wegner I, Edelbroek PM, Bulk S, Lindhout D. Lamotrigine kinetics within the menstrual cycle, after menopause, and with oral contraceptives. Neurology 2009; 73: 1388–93. [DOI] [PubMed] [Google Scholar]
- 16. Chen H, Yang K, Choi S, Fischer JH, Jeong H. Up‐regulation of UDP‐glucuronosyltransferase (UGT) 1A4 by 17beta‐estradiol: a potential mechanism of increased lamotrigine elimination in pregnancy. Drug Metab Dispos 2009; 37: 1841–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou J, Argikar UA, Remmel RP. Functional analysis of UGT1A4(P24T) and UGT1A4(L48 V) variant enzymes. Pharmacogenomics 2011; 12: 1671–9. [DOI] [PubMed] [Google Scholar]
- 18. Gulcebi MI, Ozkaynakci A, Goren MZ, Aker RG, Ozkara C, Onat FY. The relationship between UGT1A4 polymorphism and serum concentration of lamotrigine in patients with epilepsy. Epilepsy Res 2011; 95: 1–8. [DOI] [PubMed] [Google Scholar]
- 19. Singkham N, Towanabut S, Lertkachatarn S, Punyawudho B. Influence of the UGT2B7‐161C > T polymorphism on the population pharmacokinetics of lamotrigine in Thai patients. Eur J Clin Pharmacol 2013; 69: 1285–91. [DOI] [PubMed] [Google Scholar]
- 20. Blanca Sanchez M, Herranz JL, Leno C, Arteaga R, Oterino A, Valdizan EM, et al. UGT2B7_‐161C > T polymorphism is associated with lamotrigine concentration‐to‐dose ratio in a multivariate study. Ther Drug Monit 2010; 32: 177–84. [DOI] [PubMed] [Google Scholar]
- 21. Lovric M, Bozina N, Hajnsek S, Kuzman MR, Sporis D, Lalic Z, et al. Association between lamotrigine concentrations and ABCB1 polymorphisms in patients with epilepsy. Ther Drug Monit 2012; 34: 518–25. [DOI] [PubMed] [Google Scholar]
- 22. Wang Q, Liang M, Dong Y, Yun W, Qiu F, Zhao L, et al. Effects of UGT1A4 genetic polymorphisms on serum lamotrigine concentrations in Chinese children with epilepsy. Drug Metab Pharmacokinet 2015. [DOI] [PubMed] [Google Scholar]
- 23. Chang Y, Yang LY, Zhang MC, Liu SY. Correlation of the UGT1A4 gene polymorphism with serum concentration and therapeutic efficacy of lamotrigine in Han Chinese of Northern China. Eur J Clin Pharmacol 2014; 70: 941–6. [DOI] [PubMed] [Google Scholar]
- 24. Grasela TH, Fiedler‐Kelly J, Cox E, Womble GP, Risner ME, Chen C. Population pharmacokinetics of lamotrigine adjunctive therapy in adults with epilepsy. J Clin Pharmacol 1999; 39: 373–84. [DOI] [PubMed] [Google Scholar]
- 25. Saracino MA, Bugamelli F, Conti M, Amore M, Raggi MA. Rapid HPLC analysis of the antiepileptic lamotrigine and its metabolites in human plasma. J Sep Sci 2007; 30: 2249–55. [DOI] [PubMed] [Google Scholar]
- 26. Kastelic M, Koprivsek J, Plesnicar BK, Serretti A, Mandelli L, Locatelli I, et al. MDR1 gene polymorphisms and response to acute risperidone treatment. Prog Neuropsychopharmacol Biol Psychiatry 2010; 34: 387–92. [DOI] [PubMed] [Google Scholar]
- 27. DerSimonian R, Laird N. Meta‐analysis in clinical trials. Control Clin Trials 1986; 7: 177–88. [DOI] [PubMed] [Google Scholar]
- 28. Bauer R. NONMEM Users Guides. Introduction to NONMEM 7.2.0. Ellicott City, Maryland, USA: Icon Development Solutions, 2011. [Google Scholar]
- 29. Gisleskog PO, Karlsson MO, Beal SL. Use of prior information to stabilize a population data analysis. J Pharmacokinet Pharmacodyn 2002; 29: 473–505. [DOI] [PubMed] [Google Scholar]
- 30. Johansson ÅM, Karlsson MO. Comparison of methods for handling missing covariate data. AAPS J 2013; 15: 1232–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Milovanovic JR, Jankovic SM. Population pharmacokinetics of lamotrigine in patients with epilepsy. Int J Clin Pharmacol Ther 2009; 47: 752–60. [DOI] [PubMed] [Google Scholar]
- 32. Gidal BE, Anderson GD, Rutecki PR, Shaw R, Lanning A. Lack of an effect of valproate concentration on lamotrigine pharmacokinetics in developmentally disabled patients with epilepsy. Epilepsy Res 2000; 42: 23–31. [DOI] [PubMed] [Google Scholar]
- 33. Brzakovic B, Vucicevic K, Kovacevic SV, Miljkovic B, Prostran M, Martinovic Z, et al. Pharmacokinetics of lamotrigine in paediatric and young adult epileptic patients – nonlinear mixed effects modelling approach. Eur J Clin Pharmacol 2014; 70: 179–85. [DOI] [PubMed] [Google Scholar]
- 34. Chen C. Validation of a population pharmacokinetic model for adjunctive lamotrigine therapy in children. Br J Clin Pharmacol 2000; 50: 135–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Punyawudho B, Ramsay RE, Macias FM, Rowan AJ, Collins JF, Brundage RC, et al. Population pharmacokinetics of lamotrigine in elderly patients. J Clin Pharmacol 2008; 48: 455–63. [DOI] [PubMed] [Google Scholar]
- 36. Rivas N, Buelga DS, Elger CE, Santos‐Borbujo J, Otero MJ, Dominguez‐Gil A, et al. Population pharmacokinetics of lamotrigine with data from therapeutic drug monitoring in German and Spanish patients with epilepsy. Ther Drug Monit 2008; 30: 483–9. [DOI] [PubMed] [Google Scholar]
- 37. He DK, Wang L, Qin J, Zhang S, Lu W, Li L, et al. Population pharmacokinetics of lamotrigine in Chinese children with epilepsy. Acta Pharmacol Sin 2012; 33: 1417–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mallaysamy S, Johnson MG, Rao PG, Rajakannan T, Bathala L, Arumugam K, et al. Population pharmacokinetics of lamotrigine in Indian epileptic patients. Eur J Clin Pharmacol 2013; 69: 43–52. [DOI] [PubMed] [Google Scholar]
- 39. Wegner I, Wilhelm AJ, Sander JW, Lindhout D. The impact of age on lamotrigine and oxcarbazepine kinetics: a historical cohort study. Epilepsy Behav 2013; 29: 217–21. [DOI] [PubMed] [Google Scholar]
- 40. Arif H, Svoronos A, Resor SR Jr, Buchsbaum R, Hirsch LJ. The effect of age and comedication on lamotrigine clearance, tolerability, and efficacy. Epilepsia 2011; 52: 1905–13. [DOI] [PubMed] [Google Scholar]
- 41. Reimers A, Skogvoll E, Sund JK, Spigset O. Lamotrigine in children and adolescents: the impact of age on its serum concentrations and on the extent of drug interactions. Eur J Clin Pharmacol 2007; 63: 687–92. [DOI] [PubMed] [Google Scholar]
- 42. Reinsberger C, Dorn T, Kramer G. Smoking reduces serum levels of lamotrigine. Seizure 2008; 17: 651–3. [DOI] [PubMed] [Google Scholar]
- 43. Hussein Z, Posner J. Population pharmacokinetics of lamotrigine monotherapy in patients with epilepsy: retrospective analysis of routine monitoring data. Br J Clin Pharmacol 1997; 43: 457–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Christensen J, Petrenaite V, Atterman J, Sidenius P, Ohman I, Tomson T, et al. Oral contraceptives induce lamotrigine metabolism: evidence from a double‐blind, placebo‐controlled trial. Epilepsia 2007; 48: 484–9. [DOI] [PubMed] [Google Scholar]
- 45. Kim HJ, Kim TE, Joo EY, Seo DW, Lee SY, Hong SB. Effect of comedication on lamotrigine clearance in Korean epilepsy patients. Clin Chim Acta 2015; 438: 269–73. [DOI] [PubMed] [Google Scholar]
- 46. Chan V, Morris RG, Ilett KF, Tett SE. Population pharmacokinetics of lamotrigine. Ther Drug Monit 2001; 23: 630–5. [DOI] [PubMed] [Google Scholar]
- 47. Yamamoto Y, Inoue Y, Matsuda K, Takahashi Y, Kagawa Y. Influence of concomitant antiepileptic drugs on plasma lamotrigine concentration in adult Japanese epilepsy patients. Biol Pharm Bull 2012; 35: 487–93. [DOI] [PubMed] [Google Scholar]
- 48. Zevin S, Benowitz NL. Drug interactions with tobacco smoking. An update. Clin Pharmacokinet 1999; 36: 425–38. [DOI] [PubMed] [Google Scholar]
- 49. Yuen AW, Land G, Weatherley BC, Peck AW. Sodium valproate acutely inhibits lamotrigine metabolism. Br J Clin Pharmacol 1992; 33: 511–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kaufman KR, Gerner R. Lamotrigine toxicity secondary to sertraline. Seizure 1998; 7: 163–5. [DOI] [PubMed] [Google Scholar]
- 51. Christensen J, Sandgaard AP, Sidenius P, Linnet K, Licht RW. Lack of interaction between sertraline and lamotrigine in psychiatric patients: a retrospective study. Pharmacopsychiatry 2012; 45: 119–21. [DOI] [PubMed] [Google Scholar]
- 52. Prescribing information for Lamictal® . [online]. Available at: https://www.medicines.org.uk/emc/medicine/4228 (last accessed 23 April 2015).
- 53. Wootton R, Soul‐Lawton J, Rolan PE, Sheung CT, Cooper JD, Posner J. Comparison of the pharmacokinetics of lamotrigine in patients with chronic renal failure and healthy volunteers. Br J Clin Pharmacol 1997; 43: 23–7. [DOI] [PubMed] [Google Scholar]
- 54. Grant M. The Genetic Determinants of Lamotrigine Dosing in Epilepsy [Master thesis]. Liverpool: University of Liverpool, 2010.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S1 Pharmacokinetic priors used for the analysis
S2 Graphical summary of the meta‐analysis. Parameter estimates (symbols) with 95% CI (error bars); Relative study weight is presented by horizontal bars
S3 nonmem control stream
S4 Influence of smoking on post‐hoc estimates of individual patient's lamotrigine clearance (CL) with the base population model across various genotype groups
S5 Influence of co‐treatment with inhibitors of metabolism on post‐hoc estimates of individual patient's lamotrigine clearance (CL) with the base population model across various genotype groups
S6 Multiple imputation of missing smoking status
S7 Conditional density of smoking given lamotrigine oral clearance (left) and ROC curve for logistic regression (right)
Supporting info item