

Abstract
Aims
The aim of this study was to use data from an experimental induced blood stage malaria clinical trial to characterize the antimalarial activity of the new compound Actelion‐451840 using pharmacokinetic/pharmacodynamic (PK/PD) modelling. Then, using simulations from the model, the dose and dosing regimen necessary to achieve cure of infection were derived.
Methods
Eight healthy male subjects were infected with blood stage P. falciparum. After 7 days, a single dose of 500 mg of Actelion‐451840 was administered under fed conditions. Parasite and drug concentrations were sampled frequently. Parasite growth and the relation to drug exposure were estimated using PK/PD modelling. Simulations were then undertaken to derive estimates of the likelihood of achieving cure in different scenarios.
Results
Actelion‐451840 was safe and well tolerated. Single dose treatment markedly reduced the level of P. falciparum parasitaemia, with a weighted average parasite reduction rate of 73.6 (95% CI 56.1, 96.5) and parasite clearance half‐life of 7.7 h (95% CI 7.3, 8.3). A two compartment PK/PD model with a steep concentration−kill effect predicted maximum effect with a sustained concentration of 10–15 ng ml−1 and cure achieved in 90% of subjects with six once daily doses of 300 mg once daily.
Conclusions
Actelion‐451840 shows clinical efficacy against P. falciparum infections. The PK/PD model developed from a single proof‐of‐concept study with eight healthy subjects enabled prediction of therapeutic effects, with cure rates with seven daily doses predicted to be equivalent to artesunate monotherapy. Larger doses or more frequent dosing are not predicted to achieve more rapid cure.
Keywords: challenge model, malaria, pharmacodynamics, pharmacokinetics, Plasmodium falciparum, proof of concept
What is Already Known about this Subject
A challenge model provides a novel approach of assessing the efficacy of antimalarial drugs under controlled conditions. Healthy subjects are infected with the blood stage P. falciparum parasite.
The experimental infection under tightly controlled conditions, as opposed to patient populations, allows for close monitoring of parasite growth and drug effects, including controlling the dose of parasites administered compared with malaria infections induced by a mosquito bite.
P. falciparum strain 3D7 has a well‐characterised drug sensitivity profile, allowing for comparison with other drugs.
What this Study Adds
The present study in eight subjects provides the results of the proof‐of‐concept study of Actelion‐451840, showing that the new compound is active against blood stage P. falciparum infection.
The implementation of the challenge model in the controlled environment of clinical studies generated robust PK and PD data. Model‐based predictions based on a large simulated population of patients allow estimation of curative doses or dosing regimens in patients with clinical malaria.
The model predicted that 90% of subjects would be cured with six daily doses of Actelion‐451840. Thus, a small study coupled with population modelling provided sufficient information for decision‐making.
Introduction
Malaria is one of the most important infectious diseases, threatening half of the world's population. In accordance to the latest estimates by the World Health Organization (WHO), in 2014, there were 198 million cases of this parasitic disease (infection with P. falciparum) out of the estimated 3.3 billion people at risk and with an estimated 584000 deaths. 90% of the mortality was in sub‐Saharan Africa, mostly among children under 5 years of age, attributing 78% of deaths 1. The WHO has declared malaria control a global development priority and has changed its recommendation from control programmes to eradication programmes. New drugs will be needed to make this possible 2.
The widespread resistance of P. falciparum to conventional monotherapies such as chloroquine, amodiaquine and sulfadoxine/pyrimethamine led to an urgent need for new therapies to combat increasing levels of resistance. The WHO currently recommends artemisinin‐based combination therapy (ACT) as the first line therapy in areas with high prevalence of resistance 3. Even though the artemisinins are the most potent and rapidly acting antimalarial agents available to date 4, they are associated with high recrudescence rates when used as monotherapy. However, worrying evidence of its decreasing efficacy is now widespread through south east Asia 5. Because of the concern for resistance development, new drugs with new mechanisms of action are needed. The development of these drugs requires fast efficient processes.
Actelion‐451840 is a new chemical entity 6 and potent inhibitor of both multidrug‐resistant and sensitive P. falciparum asexual blood stage parasites, with in vitro IC 50 values in the sub‐nanomolar range (7G8: 0.3 nm; Dd2: 0.7 nm; NF54: 0.6 nm) 7. The proposed mechanism of action of 451840 is the blocking of the MDR1 pump on the parasitophorous digestive vacuole membrane in addition to gametocidal activity 7. Similar to artemisinins, 451840 targets the blood stages of infection and has a rapid onset of action. Furthermore, 451840 is efficacious in animal models and has a good safety profile. Its mechanism of action is under investigation, with in vitro evidence suggesting that its activity is mediated by blocking a parasite‐digestive vacuole membrane‐resident transporter termed PfMDR1 7. These properties make 451840 a potential new drug for the treatment of P. falciparum malaria.
The safety and pharmacokinetic profile (PK) of single doses of 10, 50, 200 and 500 mg in the fasted state and 200 and 500 mg in the fed state in healthy subjects have been reported previously 6. No serious adverse events or adverse events leading to discontinuation of the study were observed in 30 subjects treated with 451840. The relatively low exposure in the fasted state increased approximately 14‐fold when the drug was administered in the fed state, with slightly less than dose‐proportional exposure 6.
Historically, antiparasitic activity of candidate antimalarial drugs was determined in large phase 2 trials in patients. These require large amounts of resources and long timelines for execution. The benefits of obtaining such data using a human blood stage challenge model as previously described 8 include the ability to obtain critical data earlier in the drug development process, in a controlled setting with lower potential for variability in efficacy and safety, not confounded by previously acquired protective immune responses 9, different sites, investigators, etc., and that it has simpler logistics, i.e. participant recruitment, drug supply, data management, etc.
The current study served as the proof‐of‐concept study to characterize the antimalarial effect of a single dose of 500 mg 451840 on P. falciparum parasite clearance from the blood of experimentally infected healthy subjects.
Methods
Induced blood stage malaria infection was used to characterize the activity of 451840 against early blood stage P. falciparum infection. The procedure is described in 8.
Subjects
Eight healthy male subjects agreeing to use a double barrier method of contraception for at least 14 days up to the time of dosing through 90 days after dosing were recruited.
Study design
The study was a proof‐of‐concept, single centre, open label phase 1b study. Each participant in the cohort was intravenously inoculated on day 0 with ~1800 viable P. falciparum (strain 3D7)‐infected human erythrocytes (blood stage P. falciparum challenge inoculum, BSPC). Subjects were monitored daily (a.m.) on an outpatient basis from day 4 until quantitative real‐time polymerase chain reaction (PCR) was positive for the presence of malaria parasites. Once PCR‐positive, subjects were monitored twice daily for adverse events and the unexpected early onset of symptoms, signs or parasitological evidence of malaria. When parasite counts reached ≥1000 counts ml−1 (or ≥5,000 counts ml−1 in a particular subject), subjects were admitted to the study unit and treatment with 451840 was started. A 25 ml suspension containing 500 mg 451840 was administered as a single dose orally under fed conditions followed by 175 ml of tap water.
Compulsory commencement of treatment with Riamet® (artemether/lumefantrine) to ensure complete clearance of any gametocytes present occurred on the 16th day post 451840 treatment or earlier (in case of PCR evidence of recrudescence, parasite counts exceeding 500 parasites ml−1). The regimen consisted of four tablets per os twice a day at 0, 12, 24, 36, 48 and 60 h, making a total dose of 24 tablets in six doses. The drug was administered with fatty food, following e FDA guidance 10. Subjects consumed a high fat (approximately 50% of total caloric content of the meal) and high calorie (approximately 800 to 1000 calories) breakfast on the dosing day, completed over a 20 min period and finished no longer than 10 min before study drug administration. The high fat/high calorie breakfast had the same composition for all subjects, two eggs fried in butter, two strips of bacon, two slices of toast with butter, 100 g of hash brown potatoes and 250 ml of whole milk.
Pharmacokinetic assessments
Blood for PK assessment of 451840 was sampled pre‐dose, at 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 14, 16, 20, 24, 48, 72, 96 and 144 h after drug administration. The bioanalytical methods were the same as previously reported 6. Drug concentrations were determined by liquid chromatography with tandem mass spectrometry (LC–MS/MS). The lower limit of quantification (LLOQ) was 0.1 ng ml−1. The inter‐batch precision was between 2.4% and 5.3%, whereas the inter‐batch accuracy was in the range of 95.3% to 103.6% of nominal concentration.
An ex vivo bioassay to assess the inhibitory effect of treated subjects' serum on the growth of P. falciparum (laboratory strain NF54) was undertaken by quantification of [3H]‐hypoxanthine incorporated in post‐dose samples relative to pre‐dose controls as previously described 11, 12. The active drug concentrations in the bioassay are expressed in equivalents of 451840 6. The LLOQ was 0.2 ng ml−1. Samples were collected at 0, 2, 4, 8, 12, 16, 20, 24 and 48 h. Samples taken at 72, 96 and 144 h after drug intake concentrations were almost entirely below the LLOQ and not used for data analysis.
Parasitological assessments
Assessment of parasitaemia was undertaken by PCR as previously described 13. Blood samples were collected at baseline, at 08.00 h on day 4, and then at 08.00 h and 20.00 h from day 5 to confinement. During confinement, blood samples were collected for PCR at 0, 2, 4, 8, 12, 16, 20, 24, 30, 36 and 48 h (exit from unit) as well as at 60, 72, 84, 96, 120 and 144 h after 451840 intake. Further samples were taken on an individual basis until the last artemether/lumefantrine dosing day and at the final visit (day 28/end of study). Previous studies had indicated that following treatment with some licensed antimalarial drugs, such as sulfadoxine/pyrimethamine 14 and piperaquine 15, gametocytaemia becomes apparent after drug treatment. As an exploratory analysis, to test if 451840 blocks the appearance of gametocytaemia, we undertook PCR analysis for the mature gametocyte‐specific transcript pfs25 as previously described 15.
Statistical considerations
The number of subjects was determined based on previous studies that had determined that a cohort size of eight subjects provided 80% power to identify a difference of 25% in the rate of decline of parasite kinetics as statistically significant in a two‐sided test at the 5% level 8.
Statistical summary of PK and PD data
Individual plasma and serum concentration–time data as well as derived PK parameters were summarised descriptively. PK parameters were evaluated from the plasma concentrations using non‐compartmental methods in the Phoenix WinNonlin® software version 6.1 16. The other analyses were carried out using SAS software version 9.3 17.
Pharmacodynamics and statistical assessments
Efficacy parameters were analysed descriptively. The subject specific parasite reduction rate over a 48 h period (PRR48, i.e. the fold‐reduction in parasitaemia over a 48 h period (the asexual life cycle of the parasite) after removing potential lag and tail phases), denoting the drug‐specific killing rate 8, its 95% confidence interval (CI) and the parasite clearance half‐life (time to 50% reduction in parasite concentration) were derived from the slopes of log‐linear regression models of subjects with appropriate overall fit (P value of overall model F test <0.001) as detailed in 14. The drug‐specific slope was derived as weighted average with weights proportional to the inverse variance, from which drug specific PRR48 and parasite clearance half‐life were derived 14.
The lower limit of detection (LOD) was 64 parasites ml−1 of blood 13. Values below the LOD (but still detectable parasite counts) were substituted by LOD/2 and values that were non‐detectable were substituted by 1 11. An analysis of previous studies indicated that if parasitaemia falls below a calculated threshold of 0.003 parasites ml−1, parasites would not rebound, denoting eradication from the system. The cure threshold was therefore set to 0.003 parasites ml−1.
PK/PD modelling
A PK/PD model was developed to characterize the relation between drug concentration and parasite clearance. The PK model characterises absorption, distribution and elimination of the drug. Parasite concentration was modelled using an exponential growth model. The change in parasite concentration was modelled as the joint effects of parasite growth and drug concentration using the model in 8:
The initial condition is P0, the parasite concentration at the time of inoculation. P denotes the parasite count ml−1, t time in h, G the first order parasite growth rate in absence of drug, D the maximum parasite reduction rate (drug specific), c the drug concentration in ng ml−1 and IC 50 the drug concentration required to achieve half the maximum parasite reduction rate. The parameter γ denotes an optional non‐linearity parameter defining the steepness of the concentration−effect curve (also known as the Hill coefficient).
The model describes exponential growth and killing of the parasites. When no drug is present (concentration c = 0), the parasite concentration changes by the growth factor G per unit of time, h. For example, if G = 0.1, the parasite concentration increases by 0.1 or 10% per h. The drug effect manifests by a kill rate that depends on the drug concentration, increasing with higher concentrations c.
The resulting parasite count change h−1 is given as the difference between the growth rate and the drug effect, characterized by D, IC 50 and the current drug concentration, c. For example, if G = 0.1, D = 0.2, and c = IC 50, then G − D*(c/(c + IC 50)) = 0.1–0.2*(1/2) = 0 and thus the drug effect counterbalances the parasite growth and there is no change in parasite concentration. For higher concentrations, the parasite concentration reduces over time.
R 18 was used for the programming of the modelling data sets and data exploration. Fitting of the population PK/PD model, the determination of the model parameters that best fitted the observed data, was carried out using Monolix 19 and the stochastic approximation of expectation maximization (SAEM) algorithm. Data below the LLOQ (PK) and the LOD (PD) were tagged as BLQ and estimated, corresponding to the method M3 20. Visualization of the model and simulation of alternative doses and dosing regimens were performed using Mlxplore 21, including inter‐individual and stochastic variability.
Results
Subject demographics
All eight subjects were male with a mean (range) age of 24 (19–38) years, body weight of 77 (59–98) kg, height of 179 (167–191) cm and a BMI of 24 (20–28) kg m−2. The races were Caucasian (4), Indian (2), Asian (1) and Native Hawaiian (1). One subject was an ex‐smoker and all others never smoked.
Tolerability
451840 was well tolerated with no AEs attributed to the investigational drug. Mild AEs attributable to malaria, which the subjects were told to expect, were common. These typically occurred at the time of parasite recrudescence (see below). Except for one AE of nausea that was considered related to the administration of artemether/lumefantrine, all AEs were attributed to malaria. The most frequently reported AE attributed to malaria was headache (87.5%), followed by pyrexia (62.5%) and myalgia (62.5%). All AEs were mild to moderate and resolved without sequelae by the end of the study. No clinically significant changes in vital signs were documented. No clinically significant abnormalities in haematology, clinical chemistry, coagulation variables or electrocardiogram recordings were identified.
Pharmacokinetics
Following single dose administration of 500 mg 451840, the geometric mean (95% CI) AUC(0,∞) and C max was 1284.4 ng ml−1 h (919.6, 1794.1) and 121.7 ng ml−1 (90.6, 163.5), respectively. Median t max was 4 h (minimum–maximum: 3–6 h). The geometric mean t 1/2 was 36.4 h (30.2–43.9). The apparent values based on antiparasitic activity in the bioassay, following single dose administration of 500 mg 451840 were the following: the geometric mean (95% CI) AUC(0,∞) and C max were 6081.5 ng ml−1 h (4040.1–9590.9) and 547.0 ng ml−1 (390.7–792.7), respectively. Median t max was 4.0 h (minimum–maximum: 4–8 h). The geometric mean t 1/2 was 6.8 h (5.1–9.2). Individual PK profiles are shown in Figure 1.
Figure 1.
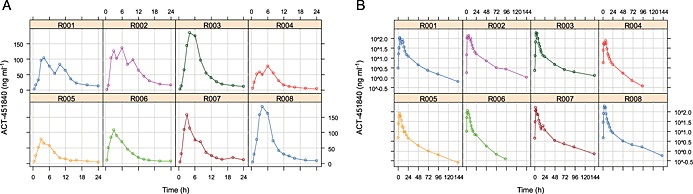
Pharmacokinetics: individual drug concentration–time profiles, linear (A) and semi‐logarithmic scale (B). (R001  R002
R002  R003
R003  R004
R004  R005
R005  R006
R006  R007
R007  R008
R008  )
)
Pharmacodynamics
All subjects reached the threshold for initiation of treatment, 1000 parasites ml−1, on the same day (day 7) with the exception of one subject, R002, who had a parasite concentration just below the threshold. Treatment was started simultaneously for all subjects (168 h after parasite inoculation). After an initial reduction in parasitaemia, recrudescent parasite growth was observed in all subjects after 100 h and in one subject after more than 200 h (Figure S2, subject R002). This required per‐protocol rescue treatment with artemether/lumefantrine (Figure 2).
Figure 2.
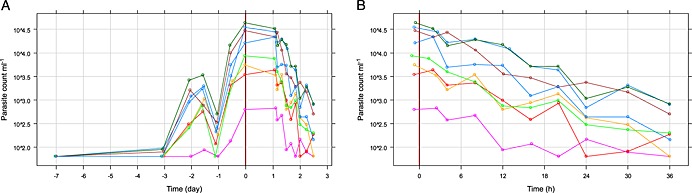
Pharmacodynamics: parasite concentration over time after inoculum (A) and after treatment start, semi‐logarithmic scale (B). (R001 R002 R003 R004 R005 R006 R007 R008 )
The drug specific PRR48 and parasite clearance half‐life were calculated as 73.6/48 h (95% CI 56.1, 96.5, n = 8) and 7.7 h (95% CI 7.3, 8.3, n = 8), respectively. All individuals had significant regression estimates (P < 0.001) and contributed to the drug specific estimates (Figure S1). For one subject (12.5%), non‐detectable parasite density at 48 h (parasite clearance) was observed.
Gametocytaemia
No evidence of the appearance of gametocytes as determined by PCR for the gametocyte specific transcript pfs25 was observed.
PK/PD modelling
Exploratory data analysis, visualisations of individual drug concentration over time (PK) and parasite concentration over time (PD) profiles, shows variability between subjects, but generally a consistent pattern. Drug elimination showed a biphasic profile on the semi‐logarithmic scale, suggesting a two compartment PK model (Figure 1).
Parasite growth showed a similar pattern between subjects. The drug effect, parasite reduction over time, showed very similar effects with respect to the decline (slope), starting from different baseline levels (Figure 2).
Starting with a one compartment model without absorption lag time, introducing a second compartment and absorption lag time improved the model fit significantly. Therefore, the PK model was identified as a two compartment model with absorption lag time and first order absorption and elimination.
Fitting a PK model with Hill coefficient showed large standard errors in the parameter estimate for the Hill coefficient. Fixing the Hill coefficient to 1 provided stable PK parameter estimates. A joint estimation of PK and PD allowed estimation of the Hill coefficient with reasonable accuracy. The initial aim to first fit the PD data prior to start of treatment, i.e. only the parasite growth phase, proved numerically unstable while fitting the entire PD data (before and after drug administration) provided parameter estimates that varied only slightly between different models (e.g. a model estimating the PD and keeping the PK fixed vs. a joint estimation of PK and PD). The final model is therefore a joint estimation of PK and PD parameters of all data, before and after drug administration.
The drug effect was estimated to be very steep, similar to an on/off phenomenon where the maximum drug effect is reached at relatively low concentrations of 10–15 ng ml−1 (the concentration was mostly above that level over 24 h). Variations of the model, with and without Hill coefficient, with some of the parameters fixed to particular values, all estimated a low IC 50 parameter. Fixing IC 50 to higher values resulted in very large estimates of the Hill coefficient, counterbalancing the (too) large IC 50. The final model included a steepness parameter, the Hill coefficient γ.
The final parameter estimates are given in Table 1. The variability relative to the parameter estimates (%RSE) is generally low with the exception of the parameter P0, the initial parasite concentration at time of inoculation, −168 h relative to drug administration. These concentrations were not available and the large variability reflects the observed inter‐individual differences in parasite count and one subject with unusually low parasite growth. The parasite growth, G, as well as the drug specific parameters D, IC 50 and γ, show little variability, reflecting the consistent patterns of parasite growth and drug effect (slope of decline). The inter‐individual differences are assigned to different initial parasite counts, confirmed by the data (Figure 2).
Table 1.
Final parameter estimates
| Parameter | Estimate | %RSE |
|---|---|---|
| Population parameters | ||
| k a (1 h −1 ) | 0.23 | 6 |
| t lag (h) | 0.45 | 2 |
| k (1 h −1 ) | 0.31 | 12 |
| V/F (l) | 1337.89 | 9 |
| k 12 (1 h −1 ) | 0.11 | 7 |
| k 21 (1 h −1 ) | 0.03 | 7 |
| P 0 (count ml −1 ) | 0.11 | 58 |
| G (unitless) | 6.67 | 4 |
| D (unitless) | 18.09 | 5 |
| IC 50 (ng ml −1 ) | 2.70 | 10 |
| γ (unitless) | 2.46 | 7 |
| Random effects (standard deviations) | ||
| k a | 0.14 | 38 |
| t lag | 0.05 | 33 |
| k | 0.29 | 26 |
| V | 0.20 | 34 |
| k 12 | 0.06 | 157 |
| k 21 | 0.08 | 129 |
| P 0 | 1.18 | 26 |
| G | 0.03 | 183 |
| D | 0.04 | 105 |
| IC 50 | 0.25 | 27 |
| γ | 0.08 | 89 |
| Error terms | ||
| Additive error PK | 4.38E‐09 | 3.72E + 06 |
| Multiplicative error PK | 0.265 | 7 |
| Additive error PD | 26.1 | 31 |
| Multiplicative error PD | 0.558 | 7 |
The small additive error term in PK and the associated large %RSE indicated that there might be no additive error for the PK. The low IC 50 value in combination with a Hill coefficient above 2 suggested a steep effect curve. Even low drug concentrations achieved effects close to the maximum. In fact, almost all concentrations measured over a 24 h period after dosing were above the IC 50 (Figure 1), suggesting that the drug effect was basically constant above a certain threshold similar to an on–off effect (Figure 3).
Figure 3.
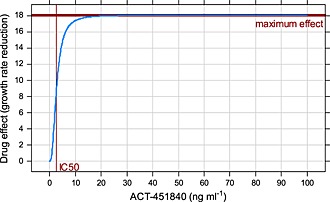
Concentration–effect curve over the observed range of drug concentration
Figure 4 shows the model‐predicted parasite count for a population typical subject and the 80% range together with the observed data on a semi‐logarithmic scale. The initial parasite growth phase becomes apparent as well as the drug effect with a minimum parasite count predicted after approximately 72 h (population typical) and varying between 1 to 5 days.
Figure 4.
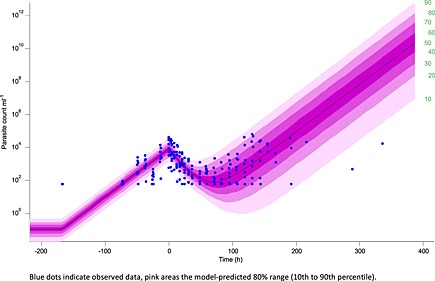
Model visualization PD: observed and model‐predicted parasite count with 10th to 90th percentile
Further model diagnostics are provided in the online supplement. They indicate that the PK and the PD model both fit the data well on population and individual levels.
Predictions
The PK/PD relationship suggested that the maximum effect was reached over almost the entire 24 h dosing interval such that higher doses or more frequent dosing would not yield larger effects on the parasite count. Further simulations showed that the effect of 300 mg doses once daily would achieve similar efficacy.
Figure 5 shows the population typical model‐predicted PK (A) and PD (B) for 1, 2, 3 and 4 doses of 500 mg 451840. It becomes apparent that four (or more) doses are predicted to be needed to achieve cure in a population typical subject.
Figure 5.
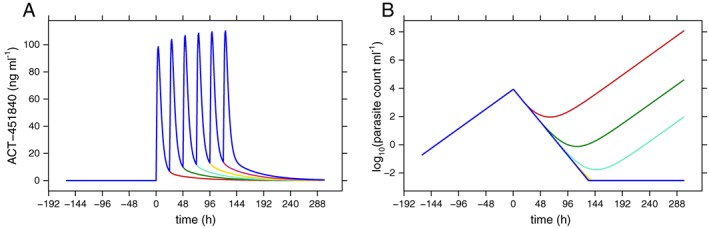
Model‐predicted population typical PK (A) and PD (B) for one to four doses of 500 mg once daily
To assess which number of doses would be required to not only lead to a cure for a typical or ‘average’ subject but to estimate how many subjects in a population would be cured, larger populations were simulated from the PK/PD model and the percentages of cure were assessed.
Figure 6 shows a simulation of parasite concentration in 500 subjects for one to six doses of 500 mg once daily. The line shows the population typical response and the coloured areas the corresponding 10 to 90% range for one to six doses of 500 mg once daily of 451840. The figure suggests that less than 10% of subjects are predicted to achieve cure with a single dose of 500 mg 451840. For four doses, more than half of the subjects are predicted to reach cure and six doses are required to achieve cure in more than 90% of subjects, typically achieved after approximately 1 week.
Figure 6.
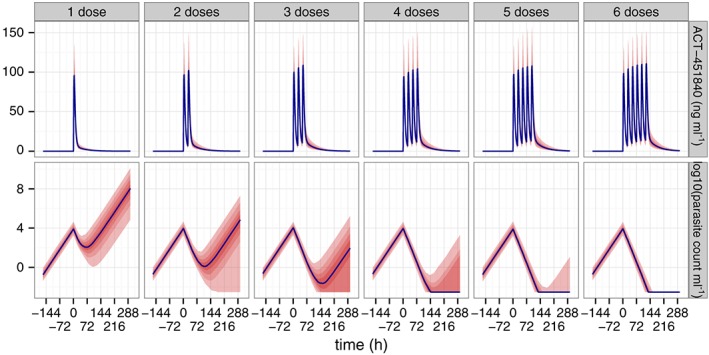
Simulated parasite concentration, population typical and 80% range for one to six doses of 500 mg once daily
Discussion
This small clinical trial including eight healthy volunteers experimentally infected with blood stage P. falciparum proved to be a highly efficient means of obtaining informative data on the clinical activity of an investigational antimalarial, 451840, never before tested in patients with levels of parasitaemia that cause clinical illness of significant severity. The data obtained enabled modelling of the activity of the drug to derive informative prediction of its curative potential. The experimental conditions of this proof‐of‐concept study following the induced blood stage infection generated high quality data, with the observed PK and PD profiles generally consistent between the different subjects.
There was, however, some inter‐individual variability, including one subject in whom parasite growth did not reach the pre‐defined treatment threshold of 1000 parasites ml−1 for treatment. In previous studies 22 significant inter‐individual variability in parasite growth has been observed. This may have an underlying genetic basis, as previously reported 23. This subject with the parasitaemia below the specified threshold was, however, included in the study, as logistic considerations necessitate that treatment be started simultaneously in all subjects. The decision to include or not include data from this subject in the PK/PD analysis would possibly have an effect on the estimated parasite growth. However, of note, parasite clearance as observed in the slope of the parasite count (supplemental data) was similar in this subject to that of the other study subjects.
A single 500 mg dose of 451840 administered with fatty food demonstrated a good antiparasitic effect in all subjects, with a parasite clearance half‐life of 7.7 h. Food effects are not uncommon for antimalarial drugs, e.g. halofantrine 24. When drug development continues, a new formulation may mimic food effect. The variability observed here is not unusually large.
However, the antimalarial effect was insufficiently potent or prolonged to achieve complete parasite clearance in any of the eight subjects. The PK profile of 451840 resembled that previously reported in the phase 1 study 6, with exposure following a single dose administration of 500 mg of 451840 similar to that observed in non‐infected healthy subjects under fed conditions. Similarly, the PD parameters were estimated with low variability (up to 10%) with the exception of the initial parasite count, P0, reflecting inter‐individual differences in parasite growth. The PK/PD model developed from the study data provided a robust characterization of PK and PD with low variability in parameter estimates, well capturing drug concentrations, parasite growth rate (in the absence of drug), and parasite reduction (drug effect).
No severe or serious adverse events were reported at the tested dose of 500 mg of 451840. No AEs were attributed to the drug, and except for nausea attributed to the administration of artemether/lumefantrine, all AEs were attributed to malaria. As has been typically observed in previous human challenge studies 8, 15, 25, AEs attributable to malaria were common. These are generally mild and reversible, responding to antipyretics such as paracetamol.
The model‐based predictions suggested that six daily doses 451840 dosed at 500 mg daily would be sufficient to achieve cure in more than 90% of subjects. This is similar to cure rates reported with seven daily doses of artesunate monotherapy 17. The model also suggested that a daily dose of 500 mg elicits close to the maximally achievable effect PD effect, indicating that higher doses or more frequent dosing would not change the overall antiparasitic effect substantially and, further, that lower doses down to 300 mg could achieve similar effects.
Potential limitations in the extrapolation to patients include higher parasite concentrations in patients and possibly altered PK and/or PD in presence of malaria. These challenge studies are undertaken in subjects with no history of malaria exposure (past travel to a malarious area was an exclusion criterion). The data were from subjects where no baseline antiparasitic immunity was present. Parasite specific antibodies were first detected about 3 weeks after drug exposure. An advantage of this model was that it provided an estimate of the minimum antiparasitic effect in a non‐immune population and clinical use in a malaria‐endemic area would likely result in a response of a greater magnitude.
The bioassay results confirmed those obtained in the previous study 6, so that the concentration−time profiles of 451840 estimated by bioassay were approximately four times higher compared with those determined by LC–MS/MS. Although the metabolism of 451840 is not well understood, this result suggests the presence of circulating active metabolites in humans 6.
It is notable that, despite the recrudescent parasitaemia observed, treatment with 451840 did not lead to induction of gametocytes, as determined by PCR for the gametocyte‐specific transcript, pfs25. This is in contrast to observations made in a previous study with piperaquine 15 in which recrudescent parasitaemia following subcurative therapy was followed by the appearance of gametocytaemia. These observations suggest that 451840 may be active against early stage gametocytes and corroborate in vitro data indicating that the drug is active against gametocytes 7. Activity against transmission stages of the parasite is a desirable characteristic in selection of new antimalarial drugs under consideration for development.
Antimalarial drugs with novel mechanisms of action are urgently required to combat the emergence of artemisinin‐resistant parasites. This study based on eight healthy subjects demonstrates that a challenge study can support decision making very early in the development process of a new drug, saving several clinical studies that would have been conducted otherwise. Although single dose treatment with 451840 did not fully provide the desired results, its use in combination deserves further investigation since it represents a class of drugs with a promising new mode of action and has a good tolerability and safety profile.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: AK, JD and AM had support from Actelion Pharmaceuticals Ltd for the submitted work. AK, JD and AM were employed by and held stock options for Actelion Pharmaceuticals Ltd in the previous 3 years.
Principal investigator of this study was Dr James S. McCarthy, QIMR Berghofer Medical Research Institute (Brisbane, Australia), whereas the clinical part of the study was conducted at the premises of Q‐Pharm Pty Ltd (Herston, QLD, Australia). Both QIMR Berghofer and Q‐Pharm Pty Ltd received financial compensation from Medicines for Malaria Venture (Geneva, Switzerland) and Actelion Pharmaceuticals Ltd (Allschwil, Switzerland).
LM and JSM were employed by QIMR Berghofer Medical Research Institute in the previous 3 years, JJM had support from the Medicines for Malaria Venture for the submitted work and was employed by Medicines for Malaria Venture in the previous 3 years and there were no other relationships or activities that could appear to have influenced the submitted work.
This study was sponsored by Actelion Pharmaceuticals Ltd (Allschwil, Switzerland), the Medicines for Malaria Venture (MMV, Geneva) and the Wellcome Trust (London UK, grant 095909/Z/11/Z). The following collaborators are gratefully acknowledged: The subjects participating in this study and the staff from Q‐Pharm Pty Ltd (Brisbane, Australia) for the study conduct, Dr Olaf Mackenroth and Dr Christoph Siethoff (Swiss BioQuant AG, Reinach, Switzerland) for the drug concentration measurements, Dr Sergio Wittlin (bioanalytical laboratory of the Swiss Tropical and Public Health Institute, Basel, Switzerland) for the 451840 bioassay, Dr Paul Brian (Actelion Preclinical Project Management, Allschwil, Switzerland) and Michelle Lee (Actelion Pharmaceuticals Australia Pty., Sydney, Australia) for study coordination.
Supporting information
Figure S1 PD regression: observed data and individual regression fits
Figure S2 Model diagnostics PK: observed concentration and model fits by subject
Figure S3 Model diagnostics PD: observed parasite count and model fits
Figure S4 Model diagnostics PK: prediction‐corrected VPC (linear and logarithmic scale)
Figure S5 Model diagnostics PD: prediction‐corrected VPC (linear and logarithmic scale)
Figure S6 Model diagnostics PK: Observed vs. predicted drug concentration (linear and logarithmic scale)
Figure S7 Model diagnostics PD: Observed vs. predicted parasite count (linear and logarithmic scale)
Figure S8 Model diagnostics PK: Residual diagnostics. Individual‐weighted residuals (IWRES) and normalised prediction distribution errors (NPDE) vs. time and predicted concentration, residual distribution (theoretical distribution line and empirical histogram), quantile−quantile plot of empirical residual quantiles vs. normal distribution
Figure S9 Model diagnostics PD: Residual diagnostics. Residual diagnostics. Individual‐weighted residuals (IWRES) and normalized prediction distribution errors (NPDE) vs. time and predicted concentration, residual distribution (theoretical distribution line and empirical histogram), quantile−quantile plot of empirical residual quantiles −. normal distribution
Supporting info item
Krause, A. , Dingemanse, J. , Mathis, A. , Marquart, L. , Möhrle, J. J. , and McCarthy, J. S. (2016) Pharmacokinetic/pharmacodynamic modelling of the antimalarial effect of Actelion‐451840 in an induced blood stage malaria study in healthy subjects. Br J Clin Pharmacol, 82: 412–421. doi: 10.1111/bcp.12962.
Footnotes
The compound's actual name is ACT‐451840. To avoid confusion with artemisinin combination therapy (ACT), we refer to it as Actelion‐451840 in the summary and as 451840 in the main manuscript.
References
- 1. World Health Organization . World Malaria Report 2014, 2014.
- 2. The malERA Consultative Group on Drugs . A research agenda for malaria eradication: Drugs. PLoS Med 2011; 8: e1000402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Roll Back Malaria Partnership . The global malaria action plan. 2008. http://www.rollbackmalaria.org/
- 4. Ashley EA, White NJ. Artemisinin‐based combinations. Curr Opin Infect Dis 2005; 18: 531–6. [DOI] [PubMed] [Google Scholar]
- 5. Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, et al. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 2014; 371: 411–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bruderer S, Hurst N, de Kanter R, Miraval T, Pfeifer T, Donazzolo Y, et al. First‐in‐humans study of the safety, tolerability, and pharmacokinetics of ACT‐451840, a new chemical entity with antimalarial activity. Antimicrob Agents Chemother 2015; 59: 935–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ng C, Siciliano G, Lee M, de Almeida MJ, Corey VC, Bopp SE, et al. CRISPR-Cas9-modified pfmdr1 protects Plasmodium falciparum asexual blood stages and gametocytes against a class of piperazine-containing compounds but potentiates artemisinin-based combination therapy partner drugs. Molecular Microbiology 2016. doi: 10.1111/mmi.13397. [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- 8. McCarthy JS, Sekuloski S, Griffin PM, Elliott S, Douglas N, Peatey C, et al. A pilot randomised trial of induced blood‐stage Plasmodium falciparum infections in healthy volunteers for testing efficacy of new antimalarial drugs. PLoS One 2011; 6: e21914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rogerson SJ, Wijesinghe RS, Meshnick SR. Host immunity as a determinant of treatment outcome in plasmodium falciparum malaria. Lancet Infect Dis 2010; 10: 51–9. [DOI] [PubMed] [Google Scholar]
- 10. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER) . Guidance for Industry: Food‐effect bioavailability and fed bioequivalence studies, 2002.
- 11. Teja‐Isavadharm P, Peggins JO, Brewer TG, White NJ, Webster HK, Kyle DE. Plasmodium falciparum‐based bioassay for measurement of artemisinin derivatives in plasma or serum. Antimicrob Agents Chemother 2004; 48: 954–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moehrle JJ, Duparc S, Siethoff C, van Giersbergen PL, Craft JC, Arbe‐Barnes S, et al. First‐in‐man safety and pharmacokinetics of synthetic ozonide OZ439 demonstrates an improved exposure profile relative to other peroxide antimalarials. Br J Clin Pharmacol 2013; 75: 524–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rockett RJ, Tozer SJ, Peatey C, Bialasiewicz S, Whiley DM, Nissen MD, et al. A real‐time, quantitative PCR method using hydrolysis probes for the monitoring of Plasmodium falciparum load in experimentally infected human volunteers. Malar J 2011; 10: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Marquart L, Baker M, O'Rourke P, McCarthy JS. Evaluating the pharmacodynamic effect of antimalarial drugs in clinical trials by quantitative PCR. Antimicrob Agents Chemother 2015; 59: 4249–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pasay C, Rockett R, Sekuloski S, Griffin P, Marquart L, Peatey C, et al. Piperaquine monotherapy of drug sensitive P. falciparum infection results in rapid clearance of parasitemia but is followed by the appearance of gametocytemia. J Infect Dis 2016; pii: jiw128. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pharsight Corporation . Phoenix WinNonlin: Pharsight Corporation, 1999.
- 17. SAS Institute Inc . Base SAS 9.1.3 Procedures Guide. Cary, NC: SAS Institute Inc, 2006. [Google Scholar]
- 18. R Development Core Team . R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing, 2009. [Google Scholar]
- 19. Lixoft‐Incuballiance . Monolix User Guide. In, 4.2.0 Edition, Orsay, France, 2012.
- 20. Beal SL. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn 2001; 28: 481–504. [DOI] [PubMed] [Google Scholar]
- 21. Lixoft‐Incuballiance . MLXPlore: Monolix Model Explorer Version 1.0.0. In, 4.2.0 Edition, Orsay, France, 2014.
- 22. Khoury D, Cromer D, Gobeau N, Möhrle J, McCarthy J, Davenport M. Determining drug effectiveness and implications for drug screening. Submitted.
- 23. Duncan CJ, Draper SJ. Controlled human blood stage malaria infection: current status and potential applications. Am J Trop Med Hyg 2012; 86: 561–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Milton K, Edwards G, Ward S, Orme M, Breckenridge A. Pharmacokinetics of halofantrine in man: effects of food and dose size. Br J Clin Pharmacol 1989; 28: 71–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McCarthy J, Marquart L, Sekuloski S, Trenholme KR, Elliott S, Griffin P, et al. Linking murine and human Plasmodium falciparum challenge models in a translational path for antimalarial drug development. Antimicrobial Agents and Chemotherapy. Accepted manuscript posted online 4 April 2006, doi: 10.1128/AAC.02883-15 AAC.02883-15 [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 PD regression: observed data and individual regression fits
Figure S2 Model diagnostics PK: observed concentration and model fits by subject
Figure S3 Model diagnostics PD: observed parasite count and model fits
Figure S4 Model diagnostics PK: prediction‐corrected VPC (linear and logarithmic scale)
Figure S5 Model diagnostics PD: prediction‐corrected VPC (linear and logarithmic scale)
Figure S6 Model diagnostics PK: Observed vs. predicted drug concentration (linear and logarithmic scale)
Figure S7 Model diagnostics PD: Observed vs. predicted parasite count (linear and logarithmic scale)
Figure S8 Model diagnostics PK: Residual diagnostics. Individual‐weighted residuals (IWRES) and normalised prediction distribution errors (NPDE) vs. time and predicted concentration, residual distribution (theoretical distribution line and empirical histogram), quantile−quantile plot of empirical residual quantiles vs. normal distribution
Figure S9 Model diagnostics PD: Residual diagnostics. Residual diagnostics. Individual‐weighted residuals (IWRES) and normalized prediction distribution errors (NPDE) vs. time and predicted concentration, residual distribution (theoretical distribution line and empirical histogram), quantile−quantile plot of empirical residual quantiles −. normal distribution
Supporting info item