

Abstract
Aim
To estimate whether laxatives prescribed for constipation in Parkinson's disease (PD) could moderate rigidity. Constipation predates diagnosis of PD by decades. Deposition of misfolded protein may begin in the gut, driven by dysbiosis. Successive antimicrobial exposures are associated with cumulative increase in rigidity, and rigidity has biological gradients on circulating leukocyte‐subset counts.
Methods
Retrospective service evaluation, in a gut/brain axis clinic, yielded an interrupted time series, relating maintenance laxative and other medication to rigidity, in consecutive outpatients identified by inclusion and exclusion criteria. Objective assessment of rigidity was used to bring greater sensitivity to change, validated against subjective gold standard (UPDRS).
Results
There were 1493 measurements of torque required to extend (flexor rigidity) and flex (extensor rigidity) the forearm in 79 PD patients over 374 person‐years. Both were strongly associated with UPDRS (P < 0.001 and P = 0.008, respectively). Before exhibition of laxative, flexor rigidity increased by 6% (95% CI 1, 10) per year, plateauing at −2% (−4, 1) per year after, with no shift at initiation. Change in slope was significant (P = 0.002), and manifest in those naïve to antiparkinsonian medication. The change was replicated for individual laxative classes (bulk, osmotic, enterokinetic). There was no temporal change in extensor rigidity. Limited experience with a quanylate cyclase‐C receptor agonist (17 patients, 6 person‐years) indicated a large and significant step down in flexor and extensor rigidity, of 19% (1, 34) and 16% (6, 24) respectively (P = 0.04 and <0.001).
Conclusions
Maintenance laxative usage was associated with apparent stemming of the temporal increase in rigidity in PD, adding to indicative evidence of a continuing role of gastrointestinal dysbiosis in pathogenesis.
Keywords: bulk, enterokinetic and quanylate cyclase‐C receptor agonist, dysbiosis of Parkinson's disease, gut/brain axis clinic, laxatives for constipation, objective quantification rigidity
What is Already Known about this Subject
Constipation markedly affects the quality of life in Parkinson's disease (PD), and predates diagnosis by decades.
The majority of PD patients have small intestinal bacterial overgrowth at presentation, probably a consequence of caeco‐ileal reflux.
There are biological gradients of quantified rigidity on circulating inflammatory markers: improving transit may protect.
What this Study Adds
Flexor rigidity increased by a clinically significant amount year‐on‐year before exhibition of maintenance laxatives, plateauing thereafter.
A similar pattern was seen with bulk, osmotic and enterokinetic laxatives, pointing to a common mechanism.
In contrast, linaclotide, which increases gastrointestinal secretion, was associated with a large step down in rigidity.
Introduction
Parkinson's disease (PD) is the second most common neurodegenerative disorder, with an estimated prevalence of 1% in people aged over 60 years in industrialized countries 1. James Parkinson noted the co‐morbidity of constipation 2. Constipation is more prevalent than PD in the general population and, like PD, its prevalence increases with age 3, 4. Prospectively, infrequent bowel movements are associated with developing PD 5. Retrospectively, frequency diverges from that of controls three decades before the median age of PD diagnosis 6. Morbidity which predates diagnosis is more likely to be closely linked to its aetiology, and to have drivers and perpetuators in common.
In PD, aggregates of misfolded α‐synuclein (Lewy bodies) are found throughout the enteric nervous system from upper oesophagus to rectum, in coeliac and para‐vertebral sympathetic ganglia, and in the dorsal vagal nuclei (which serve the gastrointestinal tract) 7, 8. Enteric deposition occurs early in PD, even predating diagnosis 9, 10, 11, 12. In the brain, aggregation ‘spreads’ through closely interconnected regions, from brain stem to substantia nigra, areas of midbrain and basal forebrain, eventually reaching the neocortex 13. It has been postulated that there is analogous spread from gut to brain, via the vagus, through an uninterrupted series of susceptible neurons. Indeed, a sub‐population of α‐synuclein expressing myenteric neurons, synaptically connected to vagal efferents, has been described in rodents 14.
It is not known what initiates or ‘seeds’ misfolding of α‐synuclein, converts containment to progression, or drives progression in PD. Although purified monomeric α‐synuclein can aggregate to form fibrils in vitro, it required introduction of fibrils to produce insoluble aggregates in cultures of neurons overexpressing α‐synuclein 15. Enteric α‐synuclein aggregates are described in clinical and experimental colitis 16, that is with local inflammation in the gut. Lewy bodies are not the first manifestation of neurodegeneration: reduced tyrosine hydroxylase immunoreactivity in the substantia nigra is present before ‘spread’ of Lewy bodies from caudal brain stem 17. In early PD, inflammation in the affected nigro‐striatal pathway (measured by imaging microglial activation) accompanies loss of presynaptic dopamine transporter 18, becoming more widespread on follow‐up 19. In advanced disease, histopathology shows persistent microglial activation despite marked neuronal loss 20. Thus, inflammation may be a primary process, not just secondary to neuronal death. Indeed, a systemic process would better explain the synucleinopathy, not just in cholinergic but also in vasoactive intestinal peptide and dopaminergic neurons of the enteric nervous system, and in sympathetic ganglia, including cardiac innervation 7, 8. Moreover, circulating inflammatory markers may represent peripheral drivers of central neuroinflammation, since they are associated with severity of cardinal facets of established PD 21, 22, 23, 24 and predict incident PD. There is a higher concentration of interleukin‐6 in blood collected 4 years prior to PD diagnosis 25. Hence emerging therapeutic strategies to prevent, contain and clear α‐synuclein deposition may be jeopardized by persistence of a peripheral driver. This is likely to emanate from the gastrointestinal tract, or a stimulus related to it in the immuno‐inflammatory or metabolic milieu 26.
Bradford Hill 27 wrote ‘The clear dose–response curve admits of a simple explanation’: implication causality. We have found biological gradients of immuno‐inflammatory milieu and of exposome (i.e. environmental exposures) on objectively measured rigidity in PD. Small intestinal bacterial overgrowth in PD, which is a common accompaniment of the slow gastrointestinal transit, is associated with higher circulating natural killer cell and total CD4+ counts, and there is a biological gradient between flexor rigidity and natural killer count, modulated by total CD4+ count 24. (CD4+ subset includes regulatory T‐cells which inhibit natural killer effector mechanisms.) Moreover, successive courses of antimicrobials are associated with a cumulative increase in flexor rigidity, over and above the effect of time, and irrespective of indication 28. Repeated antimicrobial interventions might progressively increase dysbiosis in the ‘fermentation bioreactor’ of the right colon, with adverse immunological, immune‐regulatory and/or metabolomic consequences 29.
Conversely, improving intestinal transit by maintenance laxatives may, by moderating dysbiosis, protect against rigidity. Understanding the development of disease‐specific pathophysiology clearly requires long‐term observation. Service evaluation of laxative usage is a potential tool, provided documentation of rigidity is adequate. This condition obtains in our gut/brain axis clinic.
Methods
Service evaluation setting and design
The setting is a gut/brain axis clinic, with a focus on PD and an emphasis on quantifying disease facets to document temporal change and response to treatment. Consecutive patients exhibiting parkinsonism, attending between August 2002 and July 2014, were potential candidates for the retrospective service evaluation (audit). Number of patients entered and time over which each was evaluated were determined by availability of objective measurement of rigidity and the inclusion and exclusion criteria. For inclusion, diagnosis of idiopathic PD was made according to UK Brain Bank Criteria, with at least three supportive criteria, and exclusion of other causes of parkinsonism 30. Responsiveness to large doses of levodopa was not a requirement. Presentation to the clinic with moderate or severe levodopa‐associated motor fluctuations 31 was an exclusion, so that longer term motor effects were not confounded by within‐day swings. Any data obtained after onset of such fluctuations were censored. Patients using sedatives or anti‐dopaminergic medication were excluded. Those using stimulant laxatives were excluded, because of the risk of colonic damage with long‐term use 32 and the need, where small intestinal bacterial overgrowth is common, to speed small intestinal, as well as colonic, transit. Since there is no standard checklist for service evaluation, reference was made, where relevant, to the TREND checklist for nonrandomized evaluations 33.
Criteria for use of laxatives and treatment protocol
All PD patients were encouraged to have adequate fluid intake and a high residue diet. At each visit, they were questioned about frequency of defecation, form and consistency of stool, and common intestinal symptoms (abdominal discomfort and pain, bloating, distension and flatus, straining, feeling of incomplete evacuation and sudden urge to defecate). Physical abdominal examination was made. Where the history suggested spurious diarrhoea from colonic faecal overload, abdominal X‐ray was requested.
The decision to assign to laxative treatment was based on severity of clinical picture, not time‐span as used in diagnosing idiopathic functional constipation (three months' symptoms within preceding year criterion) 34. Initial maintenance treatment was with bulk or osmotic laxative. Where the picture did not improve, this was followed by both combined, and then (since it became licensed) by adding the enterokinetic, serotonin 5‐HT4 receptor agonist, prucalopride (1 mg daily for 2 weeks, then 2 mg daily if tolerated). Laxative class, individual agent/formulation within bulk and osmotic classes, and dosage were adjusted to achieve maximum efficacy with tolerability, taking account of patient preference. More recently, the quanylate cyclase‐C receptor agonist, linaclotide, was prescribed, alone or as an adjuvant, only if irritable bowel syndrome with constipation (IBS‐C) had been diagnosed by Rome II criteria 34. (Linaclotide is currently approved in the US to treat either chronic idiopathic constipation or IBS‐C, restricted to latter indication in Europe.) Guanylate cyclase‐C activation stimulates chloride and bicarbonate secretion and inhibits sodium absorption, thereby increasing water flow into the intestine and speeding transit.
Strategy for use of antiparkinsonian and other relevant medication
Need to introduce or modify antiparkinsonian medication was according to doctor's and patient's perception of the impact and progression of the disease, with reference to NICE guidelines. We sought to avoid motor complications of levodopa by using longer t 1/2 medication for as long as this did not impair quality of life. In feedback to the general practitioner, maintenance laxatives were tabulated, alongside any antiparkinsonian, β‐adrenoceptor antagonist (for tremor) or antidepressant medication. Patients were questioned as to what medication they were actually taking at each visit. Antimicrobial courses initiated between visits were not recorded routinely throughout, unless relevant to the discussion or spontaneously reported.
Outcomes
Usual practice in clinic is to measure upper limb rigidity objectively, on the side judged clinically to be more rigid at presentation 35. Torque, required by an electric motor, to move the relaxed supported forearm horizontally, through a 40° arc about the elbow at a controlled velocity, is measured. Greater resistance to passive movement in flexor than in extensor muscles is characteristic of parkinsonism, that is ‘flexor rigidity’ is greater than ‘extensor rigidity’. The former is represented by the grand mean for torque required to extend, the latter by that to flex. Swing duration is 1.3 s, with sampling every 25 ms, the pause between swings varying (1–3 s) to reduce the effect of anticipation. One minute's acclimatization precedes the 2½ min recording. There was no temporal drift in calibration values. Unshod weight and height (potential between‐patient covariates) are measured at each visit. For the service evaluation, raw torque/time outputs were reprocessed by an observer blind to date and medication.
Although an objective continuous measure of rigidity should be more sensitive to change than a categorical clinical predictor, the United Parkinson's Disease Rating Scale (UPDRS) scores are the current gold standards. UPDRS scores (0–4) for rigidity in the upper limbs and neck and for pronation‐supination movements of hands are recorded on the great majority of visits.
Outcomes are discussed with the patient, alongside the fuller clinical picture, to inform management.
Statistical analysis
Association between paired objective and subjective measures was tested using Gaussian linear mixed regression models. Objective rigidity measurements are positively skewed and, thus, a natural logarithmic transformation was applied prior to their use as an outcome variable. Patient‐specific intercepts were included, as a random effect, to account for dependencies due to repeated measurements over time in each patient. The fixed‐effect predictor variables, ‘subjective measurement’ and ‘patient‐specific time since first measurement’, were included. The model was fitted using maximum likelihood, assuming an independent variance–covariance structure. The Wald test was used to assess the null hypothesis of no association between the subjective and objective assessment, after adjusting for any temporal trend in the objective.
Gaussian linear mixed regression models were used to estimate the impact of any maintenance laxative medication on rigidity, as assessed objectively. Linear splines, corresponding to the time periods before and after laxative initiation, with a knot at initiation, were used to estimate change in temporal disease progression. To allow for a potential ‘shift’ in rigidity at initiation, an indicator variable of pre/post initiation was included in the model. Individual classes of laxative were investigated, allowing for any confounding by inclusion of the other classes as covariates. Random intercepts for each patient and independent random coefficients for temporal trends, prior to and post initiation, were included. Antiparkinsonian and other relevant medications were incorporated as time‐varying covariates to allow for any effect that they may have on rigidity. Models were applied also to the restricted subset of data from patients not receiving these medications, where no such confounding effects could occur. Model predictions and standard errors for fixed effects were obtained directly from the model's parameter and variance–covariance matrix estimates. Mixed effects ordered logit models were used to assess any change in temporal trend in subjective rigidity measurements after initiating laxatives.
Insufficient time post‐initiation of the quanylate cyclase‐C receptor agonist precluded an assessment of its impact on the temporal trend in rigidity. An estimate of the relative change post‐initiation was obtained in a random coefficient model with a single temporal trend for each patient.
All statistical analyses were performed in Stata/MP 13.0.
Results
Characteristics of patients included
Table 1 gives the characteristics of 79 PD patients meeting the selection criteria, at the start of their period of evaluation.
Table 1.
Characteristics at start of period evaluated in 79 patients
| Characteristic | Description | |
|---|---|---|
| Demographic | Age (years)* | 66 (55 to 69) |
| Gender (male)† | 45 | |
| Height (m)‡ | 1.69 (0.10) | |
| Weight (kg)‡ | 75.2 (13.6) | |
| Time since diagnosis (years)* | 2.8 (0.8 to 5.4) | |
| Hoehn and Yahr staging (1/2/3/4/5)† | 17/50/6/5/1 | |
| Antiparkinsonian medication | Any (yes)† | 36 |
| Dopaminergic agonist | 26 | |
| Monoamine oxidase‐B inhibitor | 25 | |
| Amantadine | 8 | |
| Levodopa combination | 15 | |
| Anticholinergic | 18 | |
| Laxative | Any (yes)† | 10 |
| Bulk | 9 | |
| Osmotic | 5 | |
| Other | 0 | |
| β‐blocker | Propranolol† | 3 |
| Antidepressant | Citalopram† | 4 |
Median (interquartile range).
Count.
Mean (SD).
During their evaluation period, the 79 had 1493 measurements [mean 18.9 (SD 13.5) per person] of each flexor and extensor rigidity over a median of 4.4 (interquartile range 2.4–7.0) years, a total of 374 person‐years. Table 2 gives exposure to bulk‐forming (ispaghula husk/methylcellulose/sterculia), osmotic (macrogols) and enterokinetic (prucalopride) laxatives during the evaluation period. The corresponding pre‐ and post‐intervention measurements are summarized in Figure 1. The relatively short exposure to a guanylate cylase‐C receptor agonist (linaclotide) and related measurements are summarized in Figure 2.
Table 2.
Exposure to laxative, antiparkinsonian, antidepressant and β‐adrenergic during period evaluated in 79 patients
| Data | Number of patients | Median time interval (interquartile range) years | Person‐years |
|---|---|---|---|
| Maintenance laxative(s): | 60 | 3.2 (1.1, 5.1) | 209 |
| bulk‐forming | 59 | 2.9 (1.0, 5.0) | 199 |
| osmotic | 56 | 2.8 (1.1, 4.4) | 176 |
| enterokinetic | 25 | 1.9 (0.7, 2.6) | 43 |
| Antiparkinsonian agent(s): | |||
| dopaminergic agonists | 48 | 3.3 (2.1, 6.8) | 206 |
| monoamine oxidase‐B inhibitor | 48 | 3.4 (1.1, 6.5) | 195 |
| anticholinergic | 31 | 4.5 (2.4, 7.6) | 151 |
| levodopa combination | 31 | 3.5 (2.3, 6.6) | 127 |
| amantadine | 24 | 3.0 (1.8, 6.9) | 95 |
| Antidepressant | 13 | 3.6 (0.7, 9.7) | 66 |
| Β‐adrenergic receptor antagonist | 11 | 4.0 (0.6, 4.6) | 38 |
Figure 1.
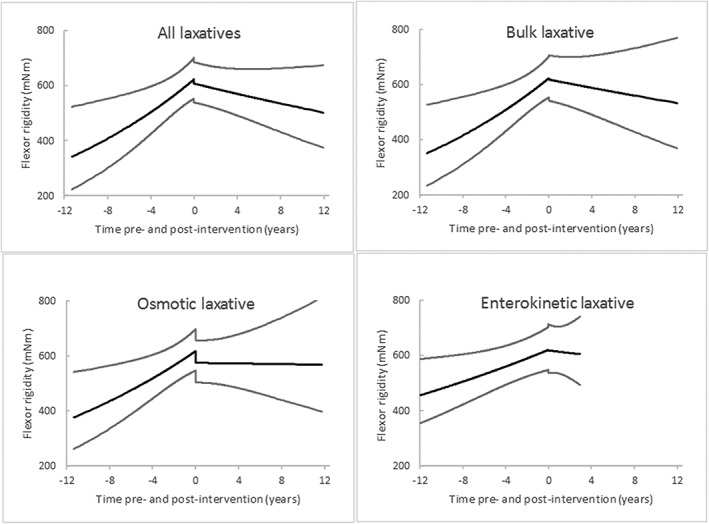
Time trends and 95% CI in flexor rigidity in relation to initiation of laxative classes at time 0. Of 1493 measurements in 79 patients, post‐intervention time trends are based on 811 measurements (mean 13.5 (SD 10.3) per person) of flexor‐rigidity in 60 receiving any laxative(s), 781 (13.2 (10.3)) in 59 receiving bulk laxative, 686 (12.3 (9.09)) in 56 receiving osmotic laxative, and 191 (7.6 (4.9)) in 25 receiving enterokinetic laxative. Time trends pre‐intervention with any laxative and with bulk, osmotic and enterokinetic laxatives are based on the residual 682, 712, 807 and 1302 measurements, respectively
Figure 2.
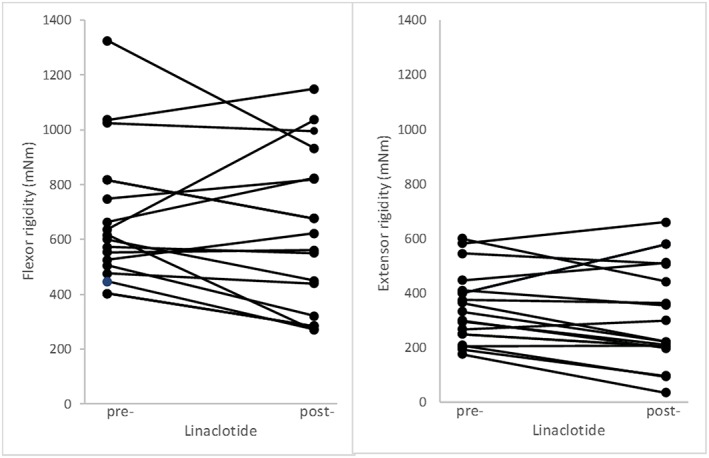
Changes in flexor‐ and extensor‐rigidity (in milli‐Newton metres) after initiation of linaclotide in 17 patients. Pre‐initiation, there were 329 measurements of flexor‐ and extensor‐rigidity (mean 19 (SD 14) per person) over a median of 232 (interquartile range 100 to 346) weeks, and post‐initiation 48 sets (2 (1) per person) over 18 (6, 30) weeks
Exposure to antiparkinsonian medication [dopaminergic agonists (pramipexole, cabergoline), monoamine oxidase‐B inhibitor (selegiline), anticholinergic (trihexyphenidyl, with a maximum total daily dose, 6 mg, at lower end of usual range), levodopa combination (with extracerebral dopa‐decarboxylase inhibitor ± catechol‐O‐methyl transferase inhibitor), antidepressants (citalopram) and β‐adrenergic receptor antagonist (propranolol) during this period is also shown in Table 2. A total of 104 antimicrobial courses were recorded in 58 patients: one course in 28 patients, two in 14 and three in 16.
Validation of objective against subjective assessment
Table 3 summarizes overall tests of significance of association between objective measures of rigidity and paired‐observations on the categorical predictors, UPDRS scores. All subjective assessments of rigidity (except on less‐affected upper limb) were indicative of measured flexor and extensor rigidity. For example, there was a 7.5% (95% CI 2.5–12.7) relative increase in flexor rigidity between slight (score 1) and none (0), a 48.2% (20.9–81.8) between severe (score 4) and none (0). Similarly, there were 6.4% (1.0–12.1) and 41.4% (13.0–76.9) relative increases in extensor rigidity between scores 1 and 0, and 4 and 0, respectively. The pronation‐supination score on the more affected side was indicative of measured flexor and extensor rigidity, on the less affected side only of flexor rigidity.
Table 3.
Cross‐referencing objectively measured rigidity against United Parkinson's Disease Rating Scale (UPDRS) scores using 1242 paired‐observations* in 79 patients
| UPDRS item | Objectively measured rigidity on worse side (n = 1240 paired measurements) | |
|---|---|---|
| Flexor | Extensor | |
| P‐value | P‐value | |
| Rigidity upper limb worse side | <0.0001 | 0.008 |
| Rigidity upper limb better side | 0.1 | 0.4 |
| Rigidity neck | 0.004 | 0.02 |
| Pronation‐supination upper limb worse side | 0.006 | 0.04 |
| Pronation‐supination upper limb better side | <0.0001 | 0.97 |
UPDRS rating not applied, or applied by a different observer, on 27% of 1493 occasions when rigidity measured objectively.
Temporal trend in rigidity before and after the initiation of laxatives
Table 4a shows the 5.5% per year increase in flexor rigidity before maintenance laxatives were introduced, and plateauing after. The difference in slopes was statistically significant (P = 0.002). There was no shift in rigidity at the time of intervention. In contrast, there was no change in temporal trend for extensor rigidity. The ratio of objectively measured flexor to extensor rigidity reflected the pattern for flexor rigidity. The time‐varying covariates, class of antiparkinsonian medication, β‐blocker and antidepressant medication and antimicrobial courses, had no significant effect on flexor rigidity at the 0.05 level. No important interactions between laxative and antiparkinsonian medication on flexor rigidity were observed. The UPDRS categorical rating of ‘rigidity’ proved insensitive to the change.
Table 4.
Change in rigidity in relation to initiating maintenance laxative, based on 1493 measurements in 79 patients
| (a) Effect of laxatives overall on flexor and extensor rigidity and their ratio | |||
|---|---|---|---|
| Measurement | Change (% per year) * mean (95% CI) | Shift (%) mean (95% CI) | |
| Pre‐intervention | Post‐intervention | At intervention | |
| Flexor rigidity | 5.5 (1.4–9.7) | −1.6 (−3.8–0.7) | −2.5 (−8.6–4.0) |
| P‐value | 0.008 | 0.2 | 0.4 |
| Extensor rigidity | 1.1 (−2.6–5.0) | 0.9 (−1.8–3.6) | 2.4 (−4.9–10.3) |
| P‐value | 0.6 | 0.5 | 0.5 |
| Flexor/Extensor ratio | 4.9 (0.8–9.2) | −2.3 (−4.9–0.3) | −4.6 (−11.3–2.7) |
| P‐value | 0.02 | 0.08 | 0.2 |
| (b) Comparison of effect of different laxative classes on flexor rigidity | |||
|---|---|---|---|
| Class of laxative | Change in flexor rigidity (% per year) * mean (95% CI) | Shift (%) mean (95% CI) | |
| Pre‐intervention | Post‐intervention | At intervention | |
| Bulk | 5.2 (1.4–9.2) | −1.2 (−3.9–1.5) | −0.7 (−8.2–7.4) |
| P‐value | 0.007 | 0.4 | 0.9 |
| Osmotic | 4.5 (1.0–8.0) | −0.1 (−3.0–2.9) | −6.8 (−13.5–0.3) |
| P‐value | 0.01 | 0.9 | 0.06 |
| Enterokinetic | 2.6 (0.3–4.9) | −0.7 (−7.1–6.0) | −0.4 (−8.6–8.5) |
| P‐value | 0.02 | 0.8 | 0.9 |
Adjusted for effects of time‐varying covariates: antiparkinsonian medication by class, β‐blocker and antidepressant medication and antimicrobial courses.
Table 4b shows that the temporal increase in flexor rigidity of PD was no longer observed following introduction of maintenance laxative. This apparent stemming of progression was similar for bulk, osmotic and enterokinetic laxative classes. On exhibition of osmotic laxative, there appeared to be a downward shift in rigidity (7%, P = 0.06). Figure 1 shows these findings in time perspective: profiles for these individual laxative classes were similar to those for any maintenance laxatives.
The findings were replicated in data obtained whilst patients were naïve to antiparkinsonian, β‐blocker and antidepressant medication and without antimicrobial courses. Over a third of rigidity measurements had been made prior to receipt of such medication (558 measurements in 44 patients [mean 12.7 (SD 11.6) per person] over a median period without laxative therapy of 1.4 (interquartile range 0.5–4.5) years, 119 person‐years). The core analysis showed a 7.8% (95% CI 3.2–12.5) per year increase in flexor rigidity (P < 0.001) before laxatives were introduced, no increase (2.7% (−3.4–9.2), P = 0.4) after, no shift on initiation. Again, there was no change in temporal trend for extensor rigidity.
Figure 2 shows the change in rigidity in response to exhibition of linaclotide over a median of 4.5 months, a total of 6 person‐years. The relative change was large and seen in extensor as well as flexor rigidity: −18.7% (−0.6–−33.6) (P = 0.04) for flexor and −15.7% (−6.4–−24.0) (P < 0.001) for extensor (after adjustment for time and relative changes with other medicinal interventions). Rigidity was no higher when linaclotide was introduced than at introduction of other laxatives: the scope to respond was not greater for linaclotide.
Discussion
Synopsis of key findings
We report an unyielding increase in flexor rigidity, of on average 5.5% per year, prior to introduction of laxatives. This was despite capture of patients early in PD (when over half were untreated) and intensive structured review of treatment (average follow‐up interval 2½ months, when on‐demand visits are included). Extensor rigidity was stable, so the flexor/extensor ratio increased: this is compatible with worsening of flexed posture. Exhibition of maintenance laxatives was associated with loss of temporal increase in flexor rigidity, even in those naïve to antiparkinsonian medication.
The similar pattern occurring with bulk, osmotic and enterokinetic laxatives pointed to a common mechanism of action on rigidity, dependent on increasing intestinal transit and/or reducing faecal overload. The enterokinetic agent would have also promoted gastric emptying 36, but there was no evidence that this was important. Early experience with introducing maintenance quanylate cyclase‐C receptor agonist points to a different pattern of response: a large downward shift in both flexor and extensor rigidity, 17.2% (on average) with a median of 4½ months' exposure.
Strengths and limitations in context of literature
Controlled trials of laxatives in idiopathic chronic constipation tend to be of short‐term monotherapy. This service evaluation is analogous to an intention‐to‐treat, open intervention study over years, albeit that the laxatives were intended to treat constipation not rigidity. That is, the expected outcome was on a comorbidity not a cardinal sign. A limitation is the lack of a randomized comparator. However, the gold standard of a long‐term prospective, placebo‐controlled, randomized comparison presents ethical and practical difficulties. Since slow gastrointestinal transit is intrinsic to PD, a parallel arm without laxatives is not a long‐term option. Using a placebo to evaluate laxative treatment in PD, even over weeks, requires a ‘rescue medication’ protocol 37, 38. Where the primary outcome is stemming the increase in rigidity over years, a more realistic comparison may be between ‘standard clinical care with pro re nata (prn) laxatives’ and ‘actively and meticulously managed, tailored maintenance schedules within fixed protocols’.
Objective quantification of rigidity allows titration of maintenance therapy 35, 39, at successive visits. It has the advantage of lack of observer bias, clearer definition of magnitude, greater sensitivity to change, and separating flexor and extensor rigidity to give a physiological explanation of posture. A placebo effect is extremely unlikely. This is not a performance test, where subjects could ‘pull themselves together’ after, say, introducing maintenance laxative. (Moreover, it is difficult to envisage physiologically how subjects could counteract, over years, the temporal increment in resistance to passive movement as PD progresses.) We suggest that long‐term objective monitoring of rigidity should be adopted widely, and to this end present the validation of our methodology against relevant categorical predictors (UPDRS).
Although the current statistical analysis takes account of potential confounding by treatments relating to PD, information on antimicrobial courses from general practice is incomplete, and no information on other modalities (e.g. physiotherapy), judged unlikely to affect rigidity per se, is included.
Comparative studies with focus on objective quantification of rigidity would help establish commonality and differences in laxative effects. There has been no prospective study of prescription of linaclotide for constipation in PD, setting the scene for discovery of its wider effects on the syndrome. No change in rigidity was noted in a placebo‐controlled trial of lubiprostone (a chloride channel activator which also increases gastrointestinal fluid secretion) for constipation in 52 PD patients 40. However, only UPDRS ratings were used and exposure was for just 4 weeks.
Proposed mechanism and crucial future research directions
Our work indicates a potential new approach to the management of rigidity, opening the door to pragmatic and exploratory cause/effect studies. It uses on‐the‐shelf‐medication. Given that constipation occurs early in the natural history, maintenance laxative usage has potential to be an initial therapeutic strategy for rigidity, as well as an adjuvant to conventional symptomatic treatment.
Since the loss of temporal increment in rigidity was observed in patients naïve to antiparkinsonian and other relevant medications, a mechanism of laxative influence on drug pharmacokinetics 41 finds no material support here. Laxatives may reduce rigidity by moderating a gastrointestinal dysbiosis, intrinsic to PD, or secondary to/exacerbated by the comorbidity of constipation. The systemic immuno‐inflammatory response may be primarily mediated by the gut‐associated lymphoid tissue of the small intestine, but it is the colonic microbiota which contribute most to the metabolomic output 29. The metabolome may have a direct toxic effect or influence immune‐regulation. Moderating the dysbiosis may lessen neuroinflammation and this has a direct effect on rigidity, as suggested by the biological gradient with circulating natural killer count 24. It may also downregulate a vicious cycle of intraneuronal aggregation of misfolded α‐synuclein, neuronal death from cytotoxicity, its leakage from dead cells into the extracellular space, and uptake into neighbouring cells with consequent cytotoxic effect 8.
The gut microbiota has recently been described as ‘a world once believed irrelevant but now looming large on the Parkinson's disease horizon’ 42, but interest goes back a long way. For example, the path to a Helicobacter aetio‐pathological hypothesis 43 was paved by observation of an excess of previously documented peptic ulcers in PD in 1965 44 and the proposition that an infectious agent was involved in both in 1979 45. This was well before discovery of Helicobacter pylori‐associated gastritis in 1983 46. In a randomized controlled trial in PD, biopsy‐proven H. pylori eradication reduced hypokinesia on gait analysis 47 (level of evidence is 1b as an individual trial). Hypokinesia improved over the year post‐eradication, with overall clinical benefit, but objective measurement revealed that rigidity worsened. Both hypokinesia and rigidity plateaued over the next year. Improved hypokinesia was independent of any (stable, long t 1/2) antiparkinsonian medication (receipt of levodopa an exclusion). Tight surveillance of antimicrobial usage in PD showed the hypokinesia effect to be indication‐specific, the rigidity effect non‐specific 28. Lack of integrity of commensal microbiota may predispose to failure of tolerance to particular foreign and self antigens 48. Indeed, there is evidence that adaptive immunity/autoimmunity, as well as innate immunity with bystander damage, has a role in the pathogenesis 20, 26. Since H. pylori and hydrogen breath test positivity are inversely related in PD 24, increased rigidity may flag acquisition of small intestinal bacterial overgrowth. However, hydrogen breath test per se did not account for leukocyte associations with rigidity 24. Other measures of overgrowth, a component or non‐bacterial associate of overgrowth, or underlying colonic dysbiosis might.
Two approaches to defining the gut microbiome in PD, using 16S ribosomal RNA gene sequencing (studying 72 patients/72 controls and 38 patients/34 controls, respectively) have been reported 49, 50, but with no consensus. Definitive work is needed using whole genome sequencing to give greater taxonomic resolution (species, and even strain, rather than family/genus) and greater precision 51. Sample size needs to be adequate to characterize the microbiota of PD within a spectrum of neuropsychiatric and gastrointestinal disease, and in terms of its facets and comorbidities. The control group needs to be robust and well‐characterized in these respects. Metagenomic exploration should be set in context of the immuno‐inflammatory and metabolic milieu. Changes in exposome and milieu associated with amelioration of rigidity by laxative and other intervention need definition.
Conclusions
The immediate therapeutic implication is that maintenance laxatives are disease‐modifying in PD. The aetio‐pathogenic implication is that change in the gut environment has a continuing and profound, but at least in part reversible, adverse effect. Actively and meticulously managed, tailored maintenance laxative schedules within fixed protocols, compared with standard clinical care of prn laxatives, is a potential tool for exploring the effect of exposome drivers and immuno‐inflammatory and metabolomic mediators on objectively measured flexor rigidity.
Like other chronic diseases, PD is likely to be multistep, multifactorial, but this does not preclude a systematic explanation and interim clinical solutions. Targeting environmental influences has potential for cost‐effective screening, prophylaxis and amelioration. Indeed, the misfolded protein theory of PD pathogenesis need not relegate any environmental theory to being ‘hit‐and‐run’ and ‘remote’ in time.
Competing Interests
All authors have completed the Unified Competing Interest form at www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: RJD and SMD had support from the Psychiatry Research Trust, London, for the submitted work; no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years; no other relationships or activities that could appear to have influenced the submitted work.
We thank the Psychiatry Research Trust, London for financial support. It received grants from and the Cyril Corden Trust, and the Cecil Pilkington Charitable Trust, and donations from Brian Newman & Louise Barton and Richard & Diana Gloyn. Barclays Corporate Social Responsibility Ambassador, Nicholas Smith, coordinated a fundraising programme for the Psychiatry Research Trust. Open access for this article was funded by King's College London. We also thank staff of The Maudsley Hospital and Pharmacy for their help in the care of the patients, in particular Glynis Ivin and Neville Desouza.
Augustin, A. D. , Charlett, A. , Weller, C. , Dobbs, S. M. , Taylor, D. , Bjarnason, I. , and Dobbs, R. J. (2016) Quantifying rigidity of Parkinson's disease in relation to laxative treatment: a service evaluation. Br J Clin Pharmacol, 82: 441–450. doi: 10.1111/bcp.12967.
References
- 1. de Lau LML, Breteler MMB. Epidemiology of Parkinson's disease. Lancet Neurol 2006; 5: 525–35. [DOI] [PubMed] [Google Scholar]
- 2. Parkinson J. An Essay on the Shaking Palsy. London: Sherwood Neely and Jones, 1817. [Google Scholar]
- 3. Sonnenberg A, Koch TR. Epidemiology of constipation in the United States. Dis Colon Rectum 1989; 32: 1–8. [DOI] [PubMed] [Google Scholar]
- 4. Locke GR, Pemberton JH, Phillips SF. American Gastroenterological Association Medical Position Statement: guidelines on constipation. Gastroenterology 2000; 119: 1761–6. [DOI] [PubMed] [Google Scholar]
- 5. Chen H, Burton EA, Ross GW, Huang X, Savica R, Abbott RD, et al. Research on the premotor symptoms of Parkinson's disease: clinical and etiological implications. Environ Health Perspect 2013; 121: 1245–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Charlett A, Dobbs RJ, Weller C, Dobbs SM. Stasis in the gut: the source of xenobiotic in idiopathic parkinsonism. Eur J Clin Pharmacol 1997; 52 (suppl): 168. [Google Scholar]
- 7. Braak H, Del Tredici K. Nervous system pathology in sporadic Parkinson disease. Neurology 2008; 70: 1916–25. [DOI] [PubMed] [Google Scholar]
- 8. Lebouvier T, Chaumette T, Paillusson S, Duyckaerts C, Bruley des Varannes S, Neunlist M, et al. The second brain and Parkinson's disease. Eur J Neurosci 2009; 30: 735–41. [DOI] [PubMed] [Google Scholar]
- 9. Shannon KM, Keshavarzian A, Dodiya HB, Jakate S, Kordower JH. Is alpha‐synuclein in the colon a biomarker for premotor Parkinson's disease? Evidence from three cases. Mov Disord 2012; 27: 716–9. [DOI] [PubMed] [Google Scholar]
- 10. Shannon KM, Keshavarzian A, Mutlu E, Dodiya HB, Daian D, Jaglin JA, et al. Alpha‐synuclein in colonic submucosa in early untreated Parkinson's disease. Mov Disord 2012; 27: 709–15. [DOI] [PubMed] [Google Scholar]
- 11. Hilton D, Stephens M, Kirk L, Edwards P, Potter R, Zajicek J, et al. Accumulation of α‐synuclein in the bowel of patients in the pre‐clinical phase of Parkinson's disease. Acta Neuropathol 2014; 127: 235–41. [DOI] [PubMed] [Google Scholar]
- 12. Sánchez‐Ferro Á, Rábano A, Catalán MJ, Rodríguez‐Valcárcel FC, Fernández Díez S, Herreros‐Rodríguez J, et al. In vivo gastric detection of α‐synuclein inclusions in Parkinson's disease. Mov Disord 2015; 30: 517–24. [DOI] [PubMed] [Google Scholar]
- 13. Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 2003; 24: 197–211. [DOI] [PubMed] [Google Scholar]
- 14. Phillips RJ, Walter GC, Wilder SL, Baronowsky EA, Powley TL. Alpha‐synuclein‐immunopositive myenteric neurons and vagal preganglionic terminals: autonomic pathway implicated in Parkinson's disease? Neurosci 2008; 153: 733–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Luk KC, Song C, O'Brien P, Stieber A, Branch JR, Brunden KR, et al. Exogenous α‐synuclein fibrils seed the formation of Lewy body‐like intracellular inclusions in cultured cells. Proc Natl Acad Sci USA 2009; 106: 20051–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Grathwohl SA, Steiner JA, Britschgi M, Brundin P. Mind the gut: secretion of α‐synuclein by enteric neurons. J Neurochem 2013; 125: 487–90. [DOI] [PubMed] [Google Scholar]
- 17. Milber JM, Noorigian JV, Morley JF, Petrovitch H, White L, Ross GW, et al. Lewy pathology is not the first sign of degeneration in vulnerable neurons in Parkinson's disease. Neurology 2012; 79: 2307–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ouchi Y, Yoshikawa E, Sekine Y, Futatsubashi M, Kanno T, Ogusu T, et al. Microglial activation and dopamine terminal loss in early Parkinson's disease. Ann Neurol 2005; 57: 168–75. [DOI] [PubMed] [Google Scholar]
- 19. Ouchi Y, Yagi S, Yokokura M, Sakamoto M. Neuroinflammation in the living brain of Parkinson's disease. Parkinsonism Relat Disord 2009; 15S3: S200–4. [DOI] [PubMed] [Google Scholar]
- 20. Orr CF, Rowe DB, Mizuno Y, Mori H, Halliday GM. A possible role for humoral immunity in the pathogenesis of Parkinson's disease. Brain 2005; 28: 2665–74. [DOI] [PubMed] [Google Scholar]
- 21. Charlett A, Dobbs RJ, Purkiss AG, Wright DJ, Peterson DW, Weller C, et al. Cortisol is higher in parkinsonism and associated with gait deficit. Acta Neurol Scand 1998; 97: 77–85. [DOI] [PubMed] [Google Scholar]
- 22. Dobbs RJ, Charlett A, Purkiss AG, Dobbs SM, Weller C, Peterson DW. Association of circulating TNF‐a and IL‐6 with ageing and parkinsonism. Acta Neurol Scand 1999; 100: 34–41. [DOI] [PubMed] [Google Scholar]
- 23. Reale M, Larlori C, Thomas A, Gambi D, Perfetti B, Di Nicola M, et al. Peripheral cytokines profile in Parkinson's disease. Brain Behav Immun 2009; 23: 55–63. [DOI] [PubMed] [Google Scholar]
- 24. Dobbs RJ, Charlett A, Dobbs SM, Weller C, Ibrahim MAA, Iguodala O, et al. Leukocyte‐subset counts in idiopathic parkinsonism provide clues to a pathogenic pathway involving small intestinal bacterial overgrowth: a surveillance study. Gut Pathogens 2012; 4: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen H, O'Reilly EJ, Schwarzschild MA, Ascherio A. Peripheral inflammatory biomarkers and risk of Parkinson's disease. Am J Epidemiol 2008; 167: 90–5. [DOI] [PubMed] [Google Scholar]
- 26. Dobbs SM, Dobbs RJ, Weller C, Charlett A, Augustin A, Taylor D, et al. Peripheral aetiopathogenic drivers and mediators of Parkinson's disease and co‐morbidities: role of gastrointestinal microbiota. J Neurovirol 2016; 22: 22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hill AB. The environment and disease: association or causation? Proc R Soc Med 1965; 58: 295–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dobbs SM, Charlett A, Dobbs RJ, Weller C, Iguodala O, Smee C, et al. Antimicrobial surveillance in idiopathic parkinsonism: indication‐specific improvement in hypokinesia following Helicobacter pylori eradication and nonspecific effect of antimicrobials for other indications in worsening rigidity. Helicobacter 2013; 18: 187–96. [DOI] [PubMed] [Google Scholar]
- 29. Neish AS. Microbes in gastrointestinal health and disease. Gastroenterology 2009; 136: 65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico‐pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992; 55: 181–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Goetz CG, Tilley BC, Shaftman SR, Stebbins GT, Fahn S, Martinez‐Martin P, et al. Movement Disorder Society‐sponsored revision of the Unified Parkinson's Disease Rating Scale (MDS‐UPDRS): scale presentation and clinimetric testing results. Mov Disord 2008; 23: 2129–70. [DOI] [PubMed] [Google Scholar]
- 32. Joo JS, Ehrenpreis ED, Gonzalez L, Kaye M, Breno S, Wexner SD, et al. Alterations in colonic anatomy induced by chronic stimulant laxatives: the cathartic colon revisited. J Clin Gastroenterol 1998; 26: 283–6. [DOI] [PubMed] [Google Scholar]
- 33. Des Jarlais DC, Lyles C, Crepaz N, TREND Group . Improving the reporting quality of nonrandomized evaluations of behavioral and public health interventions: the TREND statement. Am J Public Health 2004; 94: 361–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Thompson WG, Longstreth GF, Drossman DA, Heaton KW, Irvine EJ, Müller‐Lissner SA. Functional bowel disorders and functional abdominal pain. Gut 1999; 45 (Suppl II): II43–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kirollos C, Charlett A, Bowes SG, Purkiss AG, O'Neill CJ, Weller C, et al. Time course of physical and psychological responses to selegiline monotherapy in newly diagnosed, idiopathic parkinsonism. Eur J Clin Pharmacol 1996; 50: 7–18. [DOI] [PubMed] [Google Scholar]
- 36. Bouras EP, Camilleri M, Burton DD, Thomforde G, McKinzie S, Zinsmeister AR. Prucalopride accelerates gastrointestinal and colonic transit in patients with constipation without a rectal evacuation disorder. Gastroenterology 2001; 120: 354–60. [DOI] [PubMed] [Google Scholar]
- 37. Camilleri M, Kerstens R, Rykx A, Vandeplassche L. A placebo‐controlled trial of prucalopride for severe chronic constipation. N Engl J Med 2008; 358: 2344–54. [DOI] [PubMed] [Google Scholar]
- 38. Lembo AJ, Schneier HA, Shiff SJ, Kurtz CB, MacDougall JE, Jia XD, et al. Two randomized trials of linaclotide for chronic constipation. N Engl J Med 2011; 365: 527–36. [DOI] [PubMed] [Google Scholar]
- 39. Kirollos C, Charlett A, O'Neill CJA, Kosik R, Mozol K, Purkiss AG, et al. Objective measurement of activation of rigidity: diagnostic, pathogenetic and therapeutic implications in parkinsonism. Brit J Clin Pharmacol 1996; 41: 557–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ondo W, Kenney C, Sullivan K, Davidson A, Hunter C, Jahan I, et al. Placebo‐controlled trial of lubiprostone for constipation associated with Parkinson disease. Neurology 2012; 78: 1650–4. [DOI] [PubMed] [Google Scholar]
- 41. Neira WD, Sanchez V, Mena MA, de Yebenes JG. The effects of cisapride on plasma L‐dopa levels and clinical response in Parkinson's disease. Mov Disord 1995; 10: 66–70. [DOI] [PubMed] [Google Scholar]
- 42. Vizcarra JA, Wilson‐Perez HE, Espay AJ. The power in numbers: gut microbiota in Parkinson's disease. Mov Disord 2015; 30: 296–8. [DOI] [PubMed] [Google Scholar]
- 43. Dobbs SM, Dobbs RJ, Weller C, Charlett A. Link between Helicobacter pylori infection and idiopathic parkinsonism. Med Hypotheses 2000; 55: 93–8. [DOI] [PubMed] [Google Scholar]
- 44. Strang RR. The association of gastro‐duodenal ulceration with Parkinson's disease. Med J Aust 1965; 52: 842–3. [DOI] [PubMed] [Google Scholar]
- 45. Szabo S. Dopamine disorder in duodenal ulceration. Lancet 1979; ii: 880–2. [DOI] [PubMed] [Google Scholar]
- 46. Warren JR, Marshall B. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet 1983; 1: 273–5. [PubMed] [Google Scholar]
- 47. Dobbs SM, Dobbs RJ, Weller C, Charlett A, Bjarnason IT, Lawson AJ, et al. Differential effect of Helicobacter pylori eradication on time trends in brady/hypokinesia and rigidity in idiopathic parkinsonism. Helicobacter 2010; 15: 279–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio CW, Santacruz N, et al. Peripheral education of the immune system by colonic commensal microbiota. Nature 2011; 478: 250–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Scheperjans F, Aho V, Pereira PA, Koskinen K, Paulin L, Pekkonen E, et al. Gut microbiota are related to Parkinson's disease and clinical phenotype. Mov Disord 2015; 30: 350–8. [DOI] [PubMed] [Google Scholar]
- 50. Keshavarzian A, Green SJ, Engen PA, Voigt RM, Naqib A, Forsyth CB, et al. Colonic bacterial composition in Parkinson's disease. Mov Disord 2015; 30: 1351–60. [DOI] [PubMed] [Google Scholar]
- 51. O'Sullivan DM, Laver T, Temisak S, Redshaw N, Harris KA, Foy CA, et al. Assessing the accuracy of quantitative molecular microbial profiling. Int J Mol Sci 2014; 15: 21476–91. [DOI] [PMC free article] [PubMed] [Google Scholar]