

Abstract
Bioluminescence imaging is a powerful approach to visualize specific events occurring inside live mice. Animals can be made to glow in response to the expression of a gene, the activity of an enzyme, or the growth of a tumor. But bioluminescence requires the interaction of a luciferase enzyme with a small molecule luciferin, and its scope has been limited by the mere handful of natural combinations. Here we show that mutants of firefly luciferase can discriminate between natural and synthetic substrates in the brains of live mice. Using adeno-associated viral (AAV) vectors to express luciferases in the brain, we find that mutant luciferases that are inactive or weakly active with D-luciferin can light up brightly when treated with the aminoluciferins CycLuc1 and CycLuc2 or their respective FAAH-sensitive luciferin amides. Further development of selective luciferases promises to expand the power of bioluminescence and allow multiple events to be imaged in the same live animal.
Keywords: imaging agents, luminescence, protein engineering, neurochemistry, drug delivery
Graphical Abstract
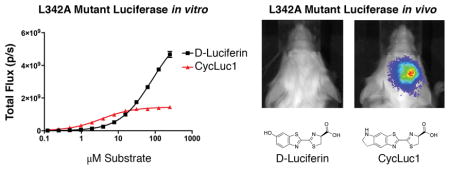
Mice do not ordinarily glow in the dark. But if you introduce the enzyme firefly luciferase and the small molecule D-luciferin, they will glow in much the same way as a firefly does. Detection of the light emitted from these animals can be used to spy on specific events that would otherwise be invisible, such as gene expression and enzyme activity.[1,2] One limitation of this powerful technique has been the paucity of distinguishable luciferins and luciferases, restricting our ability to visualize multiple events in the same live animal using bioluminescence.
Firefly luciferase is particularly well suited to in vivo imaging.[2] The wavelength of the emitted light extends well into the red and near-infrared wavelengths that pass most easily through living tissues. Furthermore, the luciferin substrate is stable, easily administered, and offers several strategies to create luminescent probes.[2–5] In efforts to expand upon the bioluminescent palette of the firefly while retaining its inherent benefits, we have synthesized new luciferin analogs and mutated the luciferase enzyme.[6–8] We found that mutant luciferases can discriminate between D-luciferin and aminoluciferin analogs in vitro and in live cells.[7,8] However, part of the basis for this discrimination is a loss of substrate affinity, and it has been unclear how this discrimination would manifest in the context of live animals.[9]
Here we perform bioluminescence imaging in the brain, and show that in this challenging environment, firefly luciferase mutants can discriminate between the natural substrate D-luciferin and synthetic luciferin analogs. We find that a balance of substrate selectivity and substrate affinity must be achieved to maintain high photon flux in vivo, more restrictive conditions than exist in vitro. Nonetheless, simple point mutants of luciferase are capable of discriminating between substrates. This work will help guide the development of additional luciferase-luciferin pairs that can be used to shed light on the molecular basis of disease and drug action in live animals.
We recently reported that the combination of three active-site mutations (R218K, L286M, and S347A) yielded a luciferase with 10,000-fold selectivity for a synthetic aminoluciferin substrate over the natural substrate D-luciferin (Figure 1).[8] However, this selectivity came at the cost of an increased apparent Km compared to the individual mutants and the wild-type luciferase. A higher Km is not an issue for bioluminescence assays performed in vitro, but it could pose a problem for in vivo imaging, where substrate access is limiting.[9]
Figure 1.
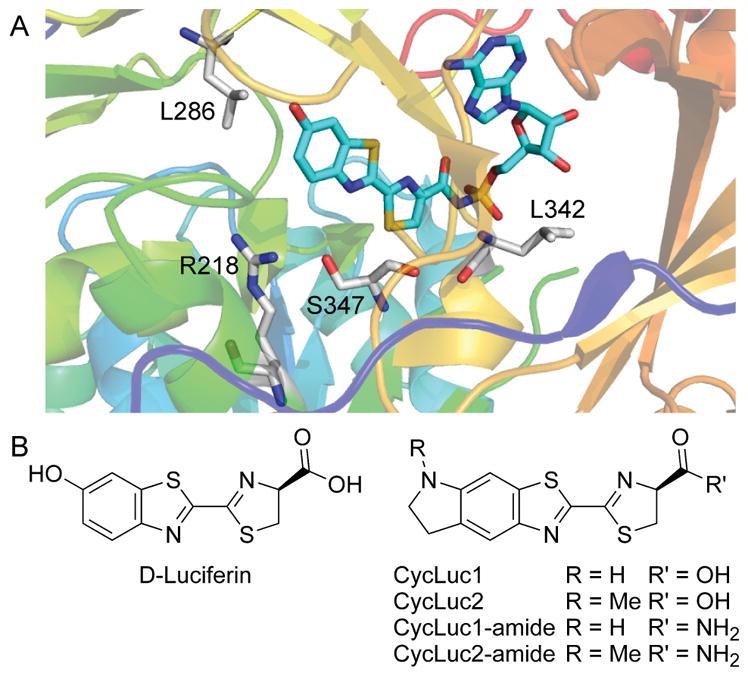
A) Crystal structure of firefly luciferase bound to a luciferyl-adenylate analog[10] (PDB 4G36, rendered in PyMOL). Mutations to R218, L286, and S347 in the luciferin binding pocket have allowed tuning of substrate specificity.[7,8] L342 does not contact the core luciferin structure and is distal from the structural differences between D-luciferin, CycLuc1 and CycLuc2 (B).
Light emission from a luciferin substrate requires activation by ATP to form an adenylated intermediate (Figure 1, Figure S1). Although residues in the ATP binding site make no direct contact with the core luciferin scaffold, the ATP binding site can influence (and be influenced by) luciferin binding. High ATP concentration exacerbates the product inhibition that is observed with aminoluciferins (Figure S2). The apparent Km value for ATP depends on the luciferin substrate and is much lower for CycLuc1 and CycLuc2 compared to D-luciferin (Table 1, Figure S2). Taking advantage of this behavior can thus conceivably be used to distinguish between luciferins. Mutation of leucine 342, part of the GYGLTE motif found in many adenylate-forming superfamily members,[11] can greatly increase the apparent Km values for both D-luciferin and ATP yet retain high maximal luciferase activity.[12] We found that the L342A mutation confers substrate selectivity which compares favorably to the previously reported R218K mutant (Figure 2, Figures S2–S3). The change in apparent Km values for aminoluciferins is muted compared to D-luciferin (Table 1). Combination of the two mutations into a single luciferase results in further selectivity over D-luciferin (Table 1, Figure 2). Although not as selective as the R218K/L286M/S347A luciferase,[8] the R218K/L342A double mutant yields the highest maximal sustained photon flux with a synthetic luciferin to date, and no longer suffers product inhibition (Figure 2, Figures S2–S3).
Table 1.
Apparent Km values for luciferins and ATP with wild-type and mutant luciferases. NA = poor fit of data to a sigmoidal dose-response curve.
| Km(app) (μM) | D-Luciferin | CycLuc1 | CycLuc2 | |||
|---|---|---|---|---|---|---|
| Luciferin | ATP | Luciferin | ATP | Luciferin | ATP | |
| WT[a] | 6.2 ± 0.18 | 68 ± 3.0 | NA | NA | 0.69 ± 0.08 | NA |
| R218K[a] | 157 ± 7.7 | 408 ± 19 | 2.0 ± 0.22 | 50 ± 5.3 | NA | 16 ± 3.0 |
| L342A[a] | 232 ± 13 | 1320 ± 52 | 4.2 ± 0.39 | 96 ± 15 | 0.81 ± 0.06 | 22 ± 2.3 |
| R218K+ L342A[b] | > 250 | > 4000 | 100 ± 4.1 | 398 ± 51 | 40 ± 2.1 | 238 ± 17 |
ATP titrations were performed at 100 μM D-luciferin, 10 μM CycLuc1/2; or
1 mM D-luciferin, 100 μM CycLuc1/2.
Figure 2.

Photon flux from purified wild-type (WT) and mutant luciferases treated with high and low concentration of luciferin substrate, imaged as described in the Methods.
To evaluate the performance of these three luciferase mutants in vivo, each codon-optimized mutant was cloned into an adeno-associated viral (AAV) plasmid under the control of a CMV promoter,[9] and packaged into AAV9 vectors.[13,14] The AAV9-luciferase viruses were then used to transduce FVB mice in the brain striatum.[9] Unlike the use of luciferase-expressing tumor cells, this viral gene delivery method allows luciferase expression in endogenous mouse cells, for the life of the mouse (well over a year). As we have previously described for AAV9 transduction with the WT luciferase, each luciferase-expressing mouse was imaged after i.p. injection with a phosphate-buffered saline (PBS) solution of each luciferin.[4,9] These experiments were performed in the exact same set of mice, imaged with each substrate on sequential days.
For all of the mutant luciferases, brain bioluminescence was dramatically reduced when using D-luciferin compared to CycLuc1 and CycLuc2 (Figure 3). With the L342A luciferase, D-luciferin gave no signal, while both aminoluciferins gave signal that was >50-fold over background. For the R218K luciferase, D- luciferin signal was ~3-fold over background, but CycLuc1 and CycLuc2 were 40–50 fold brighter still. However, combining the R218K and L342A mutations did not yield synergism in vivo.
Figure 3.

Mutant luciferase performance in the brains of live mice. A) Photon flux from a single AAV-transduced mouse compared after imaging with the indicated luciferase and luciferin (D-luciferin, 400 μmol/kg; CycLuc1, 20 μmol/kg; CycLuc2, 10 μmol/kg; CycLuc1-amide, 1 μmol/kg; CycLuc2-amide, 0.4 μmol/kg); B) Photon flux from the region of interest (ROI), n = 3 mice for each mutant luciferase. * p <0.05; ** p < 0.01; ns = not significant by t-test.
Although no photon flux above background was observed with D-luciferin for the double mutant, the photon flux was also substantially lower for CycLuc1 and CycLuc2 than for the individual point mutants (Figure S4). Thus, at least in the brain, the double mutant was less useful as a selective luciferase for these substrates compared to L342A or R218K alone.
Luciferin amides, sensors for the enzyme fatty acid amide hydrolase (FAAH), potentially offer a way to deliver luciferins into the brain more effectively.[4,15] Because of high FAAH activity in the brain, the amides are converted into their respective luciferins, and imaging can be performed with very low doses of the luciferin amide.[4] We therefore tested whether the amides of CycLuc1 and CycLuc2 (Figure 1) could be used to detect AAV transduction in the brain with the mutant luciferases (Figure 3). Remarkably, i.p. injection of just 0.4 μmol/kg CycLuc2-amide allowed imaging of L342A luciferase in the brain with a signal that was >400-fold over background, equivalent or better than its performance with WT luciferase (Figure S5). By contrast, 400 μmol/kg D-luciferin failed to yield any detectable signal (Figure 3). Luciferin comparison at low equimolar doses further revealed the relative potency of each analogue (Figure S6).
Firefly luciferase exhibits a great deal of structural plasticity, and accepts an almost bewildering array of molecules as substrates.[2,8] Although we demonstrate here that mutant luciferases can strongly discriminate against D-luciferin in vivo, the inherent promiscuity of firefly luciferase has posed a challenge for the construction of truly orthogonal luciferases. That is, we can identify mutated enzymes that will emit light with CycLuc2 but not D-luciferin, but as of yet the converse has not been achieved. For most of the selective luciferases described thus far, we do not believe that the changes to the luciferin and luciferase are mirroring the classic “bump-hole” approach used for engineering ligand-receptor interactions.[16,17] Rather, the primary basis for substrate selectivity is likely a loss of affinity for the core features of D-luciferin. Because aminoluciferins bind with higher affinity, they remain viable substrates. Going forward, one approach to more selective luciferases is to take advantage of homologous enzymes that are inherently more selective than firefly luciferase, and may therefore offer guidance as to how more precise substrate selectivity can be achieved.[18] Here we also show that the interplay between the luciferin and ATP binding sites can play a role in discriminating between luciferins. Although leucine 342 is located far from the 6′-hydroxyl of D-luciferin (i.e., the site of the CycLuc1 and CycLuc2 modifications), mutation of this residue allows substrate selectivity in the brain. Future mechanistic work in vitro will be aimed at establishing the molecular basis for this interplay and uncovering how ATP binding affects the product inhibition seen with many luciferin analogs (Figure S2). Perhaps ATP can rebind luciferase prior to release of the oxyluciferin product, resulting in a trapped complex.
Using AAV-delivered reporters, we have a platform on which to evaluate luciferases, luciferins, and luciferin-based sensors in different animal models and tissues.[4,19–22] Despite the ability of the L342A luciferase to emit light given sufficient quantities of D-luciferin and ATP in vitro, in the context of the live mouse brain we do not detect a signal. On the other hand, strong brain bioluminescence is observed from the L342A luciferase when using the aminoluciferins, but not with the double mutant R218K/L342A, despite its higher peak performance in vitro. Based on these results, a prudent design consideration (and proxy) for in vivo activity is luciferase performance at low substrate concentration, rather than the maximal rate. And in the development of new luciferins and luciferin-based sensors for in vivo use, it is also important to keep in mind the role substrate modification has on the ability of the molecule to access particular tissues in the live animal.[15] Together, we expect that the judicious application of mutant luciferases and modified luciferins will greatly expand the repertoire of noninvasive optical imaging tools.
Experimental Section
Experimental Details are provided in the Supporting Information.
Supplementary Material
Acknowledgments
We thank the Aronin Lab (UMMS) for the use of a stereotactic frame and luminometer, and the Viral Vector Core for packaging of AAV9. This work was supported by the US National Institutes of Health (R01 EB013270 and R01 DA039961).
References
- 1.Prescher JA, Contag CH. Curr Op Chem Biol. 2010;14:80–89. doi: 10.1016/j.cbpa.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 2.Adams ST, Miller SC. Curr Op Chem Biol. 2014;21:112–120. doi: 10.1016/j.cbpa.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takakura H, Kojima R, Kamiya M, Kobayashi E, Komatsu T, Ueno T, Terai T, Hanaoka K, Nagano T, Urano Y. J Am Chem Soc. 2015;137:4010–4013. doi: 10.1021/ja511014w. [DOI] [PubMed] [Google Scholar]
- 4.Mofford DM, Adams ST, Reddy GSKK, Reddy GR, Miller SC. J Am Chem Soc. 2015;137:8684–8687. doi: 10.1021/jacs.5b04357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Porterfield WB, Jones KA, McCutcheon DC, Prescher JA. J Am Chem Soc. 2015;137:8656–8659. doi: 10.1021/jacs.5b02774. [DOI] [PubMed] [Google Scholar]
- 6.Reddy GR, Thompson WC, Miller SC. J Am Chem Soc. 2010;132:13586–13587. doi: 10.1021/ja104525m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harwood KR, Mofford DM, Reddy GR, Miller SC. Chem Biol. 2011;18:1649–1657. doi: 10.1016/j.chembiol.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mofford DM, Reddy GR, Miller SC. J Am Chem Soc. 2014;136:13277–13282. doi: 10.1021/ja505795s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Evans MS, Chaurette JP, Adams ST, Jr, Reddy GR, Paley MA, Aronin N, Prescher JA, Miller SC. Nat Methods. 2014;11:393–395. doi: 10.1038/nmeth.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sundlov JA, Fontaine DM, Southworth TL, Branchini BR, Gulick AM. Biochemistry. 2012;51:6493–6495. doi: 10.1021/bi300934s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weimar JD, DiRusso CC, Delio R, Black PN. J Biol Chem. 2002;277:29369–29376. doi: 10.1074/jbc.M107022200. [DOI] [PubMed] [Google Scholar]
- 12.Branchini BR, Southworth TL, Murtiashaw MH, Boije H, Fleet SE. Biochemistry. 2003;42:10429–10436. doi: 10.1021/bi030099x. [DOI] [PubMed] [Google Scholar]
- 13.Gao G, Vandenberghe LH, Alvira MR, Lu Y, Calcedo R, Zhou X, Wilson JM. J Virol. 2004;78:6381–6388. doi: 10.1128/JVI.78.12.6381-6388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cearley CN, Wolfe JH. Mol Ther. 2006;13:528–537. doi: 10.1016/j.ymthe.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 15.Mofford DM, Miller SC. ACS Chem Neurosci. 2015;6:1273–1275. doi: 10.1021/acschemneuro.5b00195. [DOI] [PubMed] [Google Scholar]
- 16.Belshaw PJ, Schoepfer JG, Liu KQ, Morrison KL, Schreiber SL. Angew Chem Int Ed Engl. 1995;34:2129–2132. [Google Scholar]
- 17.Shah K, Liu Y, Deirmengian C, Shokat KM. Proc Natl Acad Sci USA. 1997;94:3565–3570. doi: 10.1073/pnas.94.8.3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mofford DM, Reddy GR, Miller SC. Proc Natl Acad Sci USA. 2014;111:4443–4448. doi: 10.1073/pnas.1319300111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Limberis MP, Wilson JM. Proc Natl Acad Sci USA. 2006;103:12993–12998. doi: 10.1073/pnas.0601433103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Mol Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- 21.Tarantal AF, Lee CCI. Hum Gene Ther. 2010;21:143–148. doi: 10.1089/hum.2009.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Orabi AI, Sah S, Javed TA, Lemon KL, Good ML, Guo P, Xiao X, Prasadan K, Gittes GK, Jin S, et al. J Biol Chem. 2015;290:11309–11320. doi: 10.1074/jbc.M115.647933. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.