

Abstract
Blocking quorum sensing (QS) pathways has attracted considerable interest as an approach to suppress virulence in bacterial pathogens. Toward this goal, we recently developed analogues of a native autoinducing peptide (AIP-III) signal that can inhibit AgrC-type QS receptors and attenuate virulence phenotypes in Staphylococcus aureus. Application of these compounds is limited, however, as they contain hydrolytically unstable thioester linkages and have only low aqueous solubilities. Herein, we report amide-linked AIP analogues with greatly enhanced hydrolytic stabilities and solubilities relative to our prior analogues, whilst maintaining strong potencies as AgrC receptor inhibitors in S. aureus. These compounds represent powerful new tools for the study of QS.
Keywords: autoinducing peptides, cyclic peptides, quorum sensing, Staphylococcus aureus, virulence
Graphical abstract
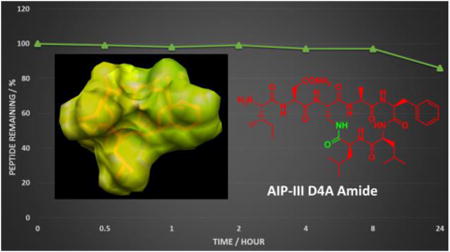
New lactam analogues of peptidic quorum sensing inhibitors in Staphylococcus aureus are reported with significantly enhanced physical prosperities, whilst maintaining their strong biological activities. Structural analyses reveal that they adopt conformations similar to their thioester precursors, and corroborate their activities in cell-based assays for AgrC-type receptor modulation.
Quorum sensing (QS) is a cell-cell signaling mechanism that is intimately connected to virulence in many common pathogens,[1] including Staphylococcus aureus.[2] QS enables bacteria to assess their population density and synchronize group behaviors at high cell number, allowing pathogens to overwhelm their host.[1] Bacteria with non-functional QS systems have been shown to be less virulent in numerous infection models.[3] Accordingly, inhibition of QS has garnered attention as a potential target to combat infection,[1, 4] and substantial recent effort has been focused on QS in S. aureus.[2b, 3a, 3b, 4b]
QS in bacteria, at the most basic level, is regulated by small molecule or peptide signals and their cognate receptors.[1-2] Signal concentration increases with cell density, and once a threshold concentration is reached, productive signal: receptor binding alters gene expression levels to permit the initiation of group behaviors, such as toxin production, motility, or biofilm formation.[1] S. aureus uses an accessory gene regulator (agr) system for QS that is centered on the interaction between the autoinducing peptide (AIP) signal and its target transmembrane receptor, AgrC.[2] To date, four distinct AIP: AgrC pairs have been characterized in S. aureus strains, forming four QS specificity groups (I–IV). The S. aureus AIP signals share two main structural features: (i) a macrocyclic thioester linked via a conserved cysteine and the C-terminus, and (ii) an N-terminal exocyclic tail (Figure 1).[2b]
Figure 1.

Structures of a native AIP (AIP-III), an inhibitor derived therefrom (AIP-III D4A), and a scaffold studied herein (AIP-III D4A Amide). Single letter codes used for amino acids; Dap = L-diamino propionic acid.
So far, most efforts to block QS in S. aureus have focused on the AIP ligand, either with the intent to competitively inhibit AIP:AgrC binding[5] or, to a lesser extent, sequester the AIP ligand away from its target receptor.[4b] Blocking downstream transcriptional activation by AgrC has also been accomplished.[3b] These chemical strategies provide means to modulate the agr system with both temporal and spatial control, and gain insights into its role in S. aureus infections. Our laboratory has focused on blocking AIP:AgrC binding, and we recently reported analogues of AIP-III (Figure 1) that inhibit AgrC receptors and associated toxin production in S. aureus.[6] Despite their high potencies, these compounds (and many other reported AIP analogues)[5] have significant limitations, most notably the instability of the native thioester bridge and their relatively low solubilities in aqueous media.[7] Herein, we report an approach toward addressing these shortcomings through the development of new AIP-III analogues containing non-native amide bridges that are highly potent, stable, and soluble pan-group inhibitors of agr-type QS in S. aureus.
Our earlier studies identified AIP-III as a superior peptidic scaffold for the design of pan-group AgrC inhibitors.[6a] A seemingly simple modification to the native AIP-III structure, replacing Asp4 with Ala, converted the peptide into an extremely potent inhibitor, AIP-III D4A (Figure 1). Subsequent NMR studies revealed that this replacement drastically altered the structure of AIP-III.[6b] While AIP-III displayed a distinctive hydrophobic, triangular “knob” composed of three endocyclic side chains (Phe5, Leu6, and Leu7) and a separately positioned N-terminal tail domain presenting an “anchor point” necessary for activation, the D4A modification caused the peptide to fold up into a compact, amphipathic spheroid where the hydrophobic knob effectively sequestered the N-terminal tail.[6b] We propose that this compact, knob-like structure allows AIP-III D4A to compete with the native ligand for AgrC receptors and, in the absence of a correctly positioned tail, inhibit their function.
In the current study, we sought to leverage our past results and redesign AIP-III D4A to increase its utility as a probe to study QS in S. aureus. We reasoned that replacement of the thioester with an amide linkage (Figure 1) would increase its hydrolytic stability, and potentially its solubility, in aqueous media. Surprisingly, very few non-thioester AIP analogues have been studied to date.[5] We previously examined AIP-I peptoid-peptide analogues with lactam bridges,[8] but these compounds had limited activity. Muir,[3c, 9] Williams,[10] and Otto[11] have studied lactam replacements in the native S. aureus AIP-I and AIP-II signals and the S. epidermidis AIP-I signal, and identified peptides capable of low to moderate non-cognate (i.e., non-self) AgrC receptor inhibition and very weak cognate AgrC receptor activation. No other lactam-derived AIP analogues have been reported, to our knowledge.
To start, we developed a solid-phase synthetic route to amide-bridged AIP analogues in which the conserved Cys was replaced with L-diamino propionic acid (Dap; see SI). We applied this route for the construction of three new AIP-III analogues (listed in Table 1): the amide versions of native AIP-III and our two most potent AgrC inhibitors, AIP-III D4A and its truncated analogue, tAIP-III D2A (lacking the N-terminal tail).[6a] Simple logP calculations on these peptides (see SI) indicated they were less hydrophobic than their thioester precursors, suggestive that they could have enhanced aqueous solubilities.
Table 1.
Peptides evaluated in this study. [a]Dap =L-diamino propionic acid.
| Peptide name | Sequence |
|---|---|
| AIP-III Amide | Ile-Asn-(Dap-Asp-Phe-Leu-Leu) |
| AIP-III D4A Amide | Ile-Asn-(Dap-Ala-Phe-Leu-Leu) |
| tAIP-III D2A Amide | Ac-(Dap-Ala-Phe-Leu-Leu) |
| AIP-III[b] | Ile-Asn-(Cys-Asp-Phe-Leu-Leu) |
| AIP-III D4A[b] | Ile-Asn-(Cys-Ala-Phe-Leu-Leu) |
| tAIP-III D2A[b] | Ac-(Cys-Ala-Phe-Leu-Leu) |
See Table S-1 in the SI for MS and HPLC characterization data.
Control peptides; reported in ref. [6a].
We evaluated the amide-bridged peptides for their abilities to modulate AgrC receptor activity in S. aureus groups-I–IV using reporter gene assays. For AgrC activation, we used a set of S. aureus agr-null strains each harbouring a P3-blaZ reporter and agrCA from groups-I, -II, -III, or -IV.[12] For AgrC inhibition, we used S. aureus strains (groups-I–IV) with functional agr circuits and P3-gfp reporter plasmids.[13] In these strains, AgrC activation or inhibition was quantified by measuring β-lactamase activity or GFP fluorescence, respectively.
None of the amide-bridged AIPs were capable of AgrC activation in any of the groups (at concentrations up to 40 μM). This result for AIP-III Amide is congruent with previous studies of the AIP-I and AIP-II lactams, both of which are only very weak cognate receptor agonists.[9-10] Turning to the inhibition data (Table 2), AIP-III Amide was found to be a very weak to moderate inhibitor of the four AgrC receptors; interestingly, it was most potent against AgrC-III, which contrasts with the lack of self-inhibition observed previously for the AIP-I and AIP-II lactams.[3c, 9-10] These results, when compared to the relatively potent non-cognate receptor antagonism displayed by the native AIP-III signal (Table 2), underscore the importance of the thioester linkage for strong non-cognate AgrC receptor inhibition by AIP-III.
Table 2.
IC50 values of the amide-bridged AIP analogues against AgrCs I–IV determined using S. aureus fluorescence reporter strains.[a]
Gratifyingly, and in contrast to AIP-III Amide, the amide-bridged analogues of our lead inhibitor, AIP-III D4A, had largely comparable inhibitory activities as their parent thioester counterparts (Table 2). With the exception of tAIP-III D2A Amide against AgrC-II (15-fold reduction in activity compared to tAIP-III D2A), the activity differences between the amide-bridged analogues and their thioester counterparts did not exceed a 5-fold reduction, and, in most cases, the activity differences were within error. These results suggest that the AIP-III D4A and tAIP-III D2A scaffolds are more tolerant to linkage modifications relative to the native AIP-III signal. Notably, these compounds represent the first lactam-bridged AIP analogues with strong, pan-group inhibitory activities (picomolar to low nanomolar IC50 values) against S. aureus AgrC-type receptors.
We next examined the 3D structures of the lactam AIP-III analogues to gain insights into the origins of their biological activities. Specifically, we were interested to determine if the consensus structural features that we previously identified as critical for AgrC-III inhibition and activation (see above) also applied to these amide-bridged analogues.[6b, 6d] We used NMR spectroscopy to elucidate the 3D solution structures of the peptides, utilizing conditions analogous to those in our past report (see SI).[6b] Starting with AIP-III Amide, as this analogue was incapable of AgrC-III activation and instead moderately inhibited AgrC-III, we expected it to possess a hydrophobic, knob-like motif to enable interaction with AgrC-III, but to lack an appropriate fourth anchor point in the correct orientation. The NMR data revealed that this analogue did possess a knob motif analogous to AIP-III (Figure 2A); however, unlike AIP-III where the exocyclic tail projects from the top of the triangular knob to presumably correctly orient the activating anchor (Figure 2B), the exocyclic tail in AIP-III Amide projects from the left side of the triangular knob (Figure S-3A) and the Ile1 side chain is located in close proximity to the Leu7 side chain (Figure S-4A). We propose that this alternate tail orientation for AIP-III Amide hinders AgrC-III activation; it may also present a steric blockade that prevents the knob from productively binding to AgrC receptors in general, thus providing an explanation for the relatively low to moderate activity of this peptide overall in any of the S. aureus groups.
Figure 2.
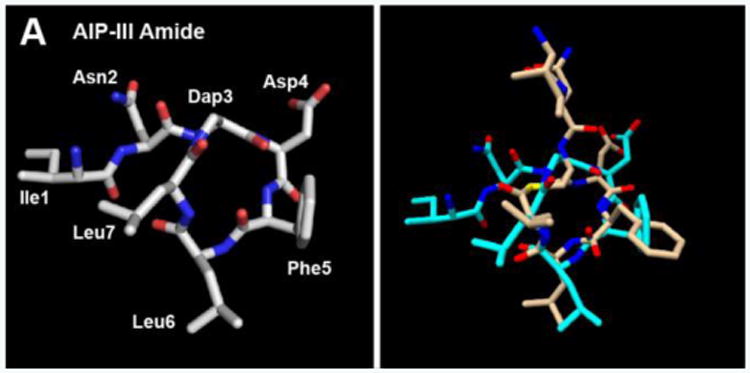
A) Heavy atom lowest energy structure of AIP-III Amide. B) Overlay of AIP-III Amide (cyan) and AIP-III (tan) structures. AIP-III structure from ref. [6b].
As AIP-III D4A Amide exhibited activities comparable to those of its parent thioester (AIP-III D4A) in the reporter assays, we expected this analogue to possess a largely similar structure in solution. We observed just this. Although the conformation of the AIP-III D4A Amide macrocycle was somewhat different than that of AIP-III D4A (Figure 3), the overall structure of AIP-III D4A Amide revealed an obvious endocyclic knob motif (Figures S-3B and S-5), with the tail residing behind the macrocycle and projecting to the left side, and the Ile1 side chain distanced from Leu7 (relative to AIP-III Amide; Figure S-4B). We reason that this orientation of the AIP-III D4A Amide tail disallows activation, but does not restrict competitive AgrC binding (and thereby the observed strong inhibition), in contrast to the AIP-III Amide tail (see Figure S-4A vs. S4B).
Figure 3.
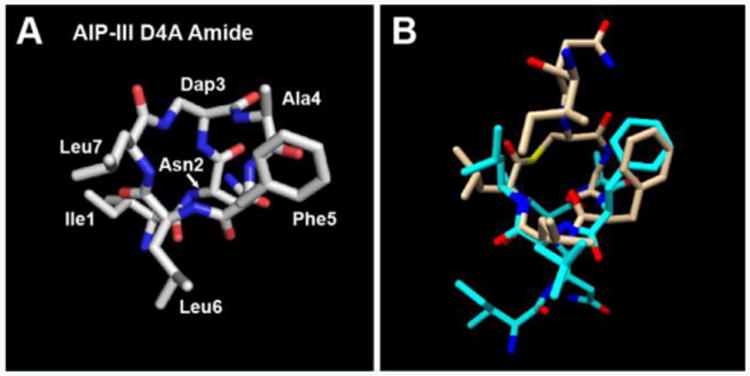
A) Heavy atom lowest energy structure of AIP-III D4A Amide. B) Overlay of AIP-III D4A Amide (cyan) and AIP-III D4A (tan) structures. AIP-III D4A structure from ref. [6b].
Turning to tAIP-III D2A Amide, this peptide had similar inhibitory activities as its thioester counterpart (tAIP-III D2A) against AgrC-III and -IV, and was only slightly less potent against AgrC-I and -II. We thus expected to observe a triangular knob motif for this analogue as well, similar to that we had previously observed for tAIP-III D2A.[6b] The NMR analyses of tAIP-III D2A Amide revealed a very structured conformation (Figures 4A and S-3C; backbone atoms RMSD of 0.01 Å for the 10-structure ensemble). Interestingly, this amide analogue displayed a “reversed” triangular knob motif when compared to tAIP-III D2A (see Figures 4B and C); namely, when viewed with the Phe3 and Leu5 residues to the right and left, respectively, the Leu4 side chain in tAIP-III D2A Amide is positioned higher than those for Phe3 and Leu5 (Figure 4C), as opposed to being lower as in tAIP-III D2A (Figure 4B). It is tempting to speculate that tAIP-III D2A Amide binds AgrC receptors in an opposite way relative to other AIP-III derivatives; that is, the Phe3 residue interacts with the AgrC binding pocket usually occupied by Leu5, and vice versa (see Figures 4C vs. 4D). In our prior study, we previously hypothesized that the roles of the hydrophobic side chains of AIP-III (as part of the knob or the anchor) may be interchangeable.[6b] Additional studies are needed to fully explore these possibilities. Nevertheless, the 3D structures of the three amide-bridged AIP-III analogues described herein, when coupled with their biological activity profiles in the AgrC reporter assays, support the general structural trends that we have previously identified for AgrC inhibition and activation.
Figure 4.
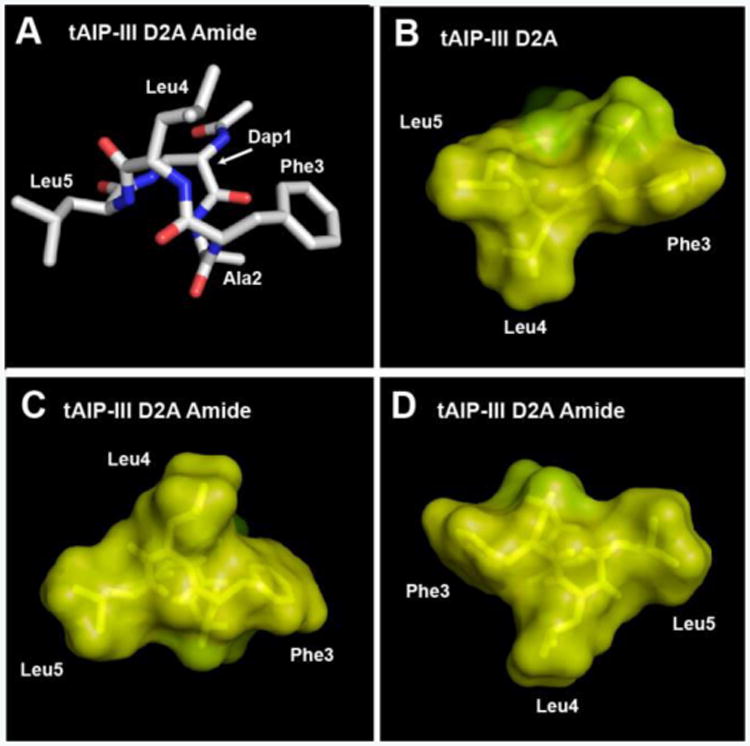
A) Heavy atom lowest energy structure of tAIP-III D2A Amide. Space-filling models of B) tAIP-III D2A (from ref. [6b]), C) tAIP-III D2A Amide, and D) tAIP-III D2A Amide (showing the alternate triangular knob) displaying hydrophobic (yellow) and hydrophilic (green) surfaces.
Lastly, we sought to determine whether the replacement of the thioester with an amide confers increased overall stability to the peptides. We had observed empirically that all three amide-bridged AIPs were more soluble in aqueous media relative to the parent thioesters, congruent with the logP calculations. To probe stability, we initially chose to compare the stability of AIP-III D4A to its amide analogue, AIP-III D4A Amide. However, AIP-III D4A could not be solubilized at the concentrations necessary for the stability studies, further highlighting the solubility limitations of this compound. We thus compared the stability of the native AIP-III to AIP-III D4A Amide instead.
To monitor hydrolytic stability, solutions of the two peptides (0.33 mM) were prepared in different buffers (PBS, pH 7.4 and ammonium carbonate, pH 8.0) and incubated at 37°C. To monitor proteolytic stability, a mixture of Trypsin and Chymotrypsin was added to the peptides in ammonium carbonate prior to incubation. HPLC was used to quantitate the amount of intact peptide over time. In PBS, 30% of the AIP-III thioester was degraded after 72 h, while AIP-III D4A Amide remained intact (Figure 5A). In the more alkaline ammonium carbonate solution, the stability of the thioester-linked peptide dramatically decreased, and AIP-III was fully degraded in ∼8 h. In contrast, AIP-III D4A Amide was found to be significantly more stable at pH 8.0, with >90% intact peptide remaining after 24 h (and >60% remaining after 72 h).[14] Treatment of the peptides with Trypsin/Chymotrypsin in ammonium carbonate yielded similar results as in the buffer alone (Figure 5B). This result is not unexpected, as cyclization is a common method to confer proteolytic stability to linear peptides. Nonetheless, the replacement of the thioester with an amide linkage markedly increased the overall hydrolytic stability of AIP-III D4A Amide. Further, the amide replacement increased the aqueous solubilities of all the amide-bridged AIP analogues and significantly facilitated their purification and handling overall.[15]
Figure 5.
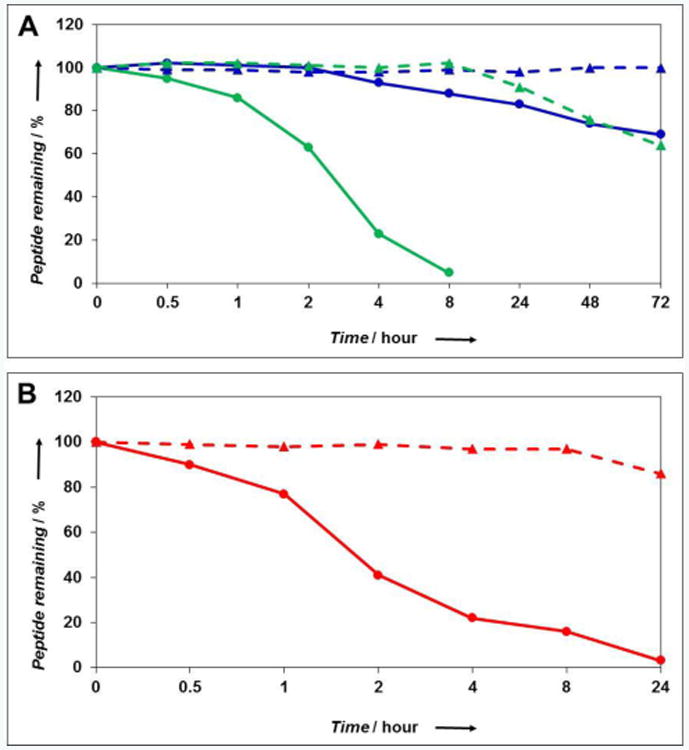
Peptide stability studies as monitored by HPLC. AIP-III and AIP-III D4A Amide data are shown as circles with solid lines or triangles with dashed lines, respectively. A) Stability in aqueous solutions (blue is PBS, pH 7.4; green is ammonium carbonate, pH 8.0). B) Stability towards enzymatic degradation by Trypsin/Chymotrypsin (red). See SI for methods.
As such, the AIP-III analogues reported here constitute robust new tools to study the role of agr-type QS in S. aureus virulence, especially in biologically relevant environments. Their activity profiles also further underscore the utility of the native AIP-III scaffold for the generation of potent AgrC inhibitors.
Supplementary Material
Acknowledgments
This research was supported by the Office of Naval Research (N00014-16-1-2185), Kimberly-Clark Corporation, and Burroughs Welcome Fund. T. Y. was supported by the UW–Madison NIH Biotechnology Training Program (T32 GM08349). The National Magnetic Resonance Facility at Madison is supported by the NIH (P41 GM103399). NMR facilities in the Department of Chemistry are supported in part by the NIH (1 S10 RR13866-01). We thank Prof. Richard Novick and Prof. Alexander Horswill for providing S. aureus strains, and Joseph Vasquez for technical assistance.
Footnotes
Supporting Information for this article is given via a link at the end of the document.
References
- 1.Rutherford ST, Bassler BL. Cold Spring Harb Perspect Med. 2012;2:a012427. doi: 10.1101/cshperspect.a012427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Wang B, Muir TW. Cell Chem Biol. 2016;23:214–224. doi: 10.1016/j.chembiol.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Thoendel M, Kavanaugh JS, Flack CE, Horswill AR. Chem Rev. 2011;111:117–151. doi: 10.1021/cr100370n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Khan BA, Yeh AJ, Cheung GYC, Otto M. Expert Opin Investig Drugs. 2015;24:689–704. doi: 10.1517/13543784.2015.1019062. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sully EK, Malachowa N, Elmore BO, Alexander SM, Femling JK, Gray BM, DeLeo FR, Otto M, Cheung AL, Edwards BS, Sklar LA, Horswill AR, Hall PR, Gresham HD. PLoS Pathog. 2014;10:e1004174. doi: 10.1371/journal.ppat.1004174. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Mayville P, Ji G, Beavis R, Yang H, Goger M, Novick RP, Muir TW. Proc Natl Acad Sci U S A. 1999;96:1218–1223. doi: 10.1073/pnas.96.4.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Galloway WRJD, Hodgkinson JT, Bowden SD, Welch M, Spring DR. Chem Rev. 2011;111:28–67. doi: 10.1021/cr100109t. [DOI] [PubMed] [Google Scholar]; b) Amara N, Krom BP, Kaufmann GF, Meijler MM. Chem Rev. 2011;111:195–208. doi: 10.1021/cr100101c. [DOI] [PubMed] [Google Scholar]; c) O'Connell KM, Hodgkinson JT, Sore HF, Welch M, Salmond GP, Spring DR. Angew Chem Int Ed. 2013;52:10706–10733. doi: 10.1002/anie.201209979. [DOI] [PubMed] [Google Scholar]
- 5.Gordon CP, Williams P, Chan WC. J Med Chem. 2013;56:1389–1404. doi: 10.1021/jm3014635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Tal-Gan Y, Stacy DM, Foegen MK, Koenig DW, Blackwell HE. J Am Chem Soc. 2013;135:7869–7882. doi: 10.1021/ja3112115. [DOI] [PubMed] [Google Scholar]; b) Tal-Gan Y, Ivancic M, Cornilescu G, Cornilescu CC, Blackwell HE. J Am Chem Soc. 2013;135:18436–18444. doi: 10.1021/ja407533e. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Tal-Gan Y, Stacy DM, Blackwell HE. Chem Commun. 2014;50:3000–3003. doi: 10.1039/c4cc00117f. [DOI] [PubMed] [Google Scholar]; d) Tal-Gan Y, Ivancic M, Cornilescu G, Blackwell HE. Org Biomol Chem. 2016;14:113–121. doi: 10.1039/c5ob01735a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kratochvil MJ, Tal-Gan Y, Yang T, Blackwell HE, Lynn DM. ACS Biomat Sci Eng. 2015;1:1039–1049. doi: 10.1021/acsbiomaterials.5b00313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fowler SA, Stacy DM, Blackwell HE. Org Lett. 2008;10:2329–2332. doi: 10.1021/ol800908h. [DOI] [PubMed] [Google Scholar]
- 9.Lyon GJ, Wright JS, Christopoulos A, Novick RP, Muir TW. J Biol Chem. 2002;277:6247–6253. doi: 10.1074/jbc.M109989200. [DOI] [PubMed] [Google Scholar]
- 10.McDowell P, Affas Z, Reynolds C, Holden MT, Wood SJ, Saint S, Cockayne A, Hill PJ, Dodd CE, Bycroft BW, Chan WC, Williams P. Mol Microbiol. 2001;41:503–512. doi: 10.1046/j.1365-2958.2001.02539.x. [DOI] [PubMed] [Google Scholar]
- 11.Otto M, Echner H, Voelter W, Gotz F. Infect Immun. 2001;69:1957–1960. doi: 10.1128/IAI.69.3.1957-1960.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lyon GJ, Wright JS, Muir TW, Novick RP. Biochemistry. 2002;41:10095–10104. doi: 10.1021/bi026049u. [DOI] [PubMed] [Google Scholar]
- 13.Kirchdoerfer RN, Garner AL, Flack CE, Mee JM, Horswill AR, Janda KD, Kaufmann GF, Wilson IA. J Biol Chem. 2011;286:17351–17358. doi: 10.1074/jbc.M111.231258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.The major degradation product for AIP-III D4A Amide in ammonium carbonate was a result of Asn2 deamidation. See SI for additional discussion.
- 15.Increased hydrophilicity may also reduce their propensity for binding serum lipoproteins, which are known to sequester native AIP ligands. See: Manifold-Wheeler BC, Elmore BO, Triplett KD, Castleman MJ, Otto M, Hall PR. J Immunol. 2016;196:328–335. doi: 10.4049/jimmunol.1501835.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.