

Abstract
Manipulating single molecules and systems of molecules with mechanical force is a powerful technique to examine their physical properties. Applying force requires attachment of the target molecule to larger objects using some sort of molecular tether, such as a strand of DNA. DNA handle attachment often requires difficult manipulations of the target molecule, which can preclude attachment to unstable, hard to obtain, and/or large, complex targets. Here we describe a method for covalent DNA handle attachment to proteins that simply requires the addition of a preprepared reagent to the protein and a short incubation. The handle attachment method developed here provides a facile approach for studying the biomechanics of biological systems.
Keywords: forced unfolding, magnetic tweezers, optical tweezers, SpyTag, SpyCatcher, protein folding, membrane protein
Introduction
Single‐molecule mechanical manipulation techniques have been widely utilized for studying mechanical properties of proteins in diverse areas such as protein folding, binding, translocation, degradation, and enzymatic catalysis.1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 Polyprotein or DNA handles are typically affixed to the target of interest so that the molecule can be readily attached to chamber surface or beads/tips so that force can be applied. The molecular handles prevent undesirable nonspecific adhesion of target proteins to the surfaces, and their well‐characterized force responses validate the single molecule conditions. Although polyprotein handles can be conveniently encoded, they are often not suitable for more complex targets like membrane proteins or ribosomes where they may prevent protein expression or assembly.13, 14, 15, 16 Oftentimes, DNA handles are more convenient because they can be attached after expression and assembly. Moreover, DNA handles can provide longer spacers between surfaces or beads and are therefore more appropriate for larger complexes.
A simple approach for coupling DNA handles to proteins is to introduce cysteine residues at the point of attachment, allowing covalent linkage using thiol chemistry.17, 18, 19, 20, 21, 22, 23, 24, 25 Thiol coupling imposes limitations, however, because all the other reactive cysteines must be removed, which is not always possible, and it becomes increasingly difficult with larger proteins that have more cysteines. HaloTag has been used, but requires the addition of a large protein domain to the target, which can limit its utility.16, 26, 27, 28, 29 A clever ybbR tag method was devised to attach DNA handles to green fluorescent protein or the enormous ribosome.30, 31, 32, 33, 34 In this method, a short ybbR peptide tag was inserted into the target proteins, and Sfp phosphopantetheinyl transferase was then used to connect an ∼20 to 30 base oligonucleotide linked to the thiol on the pantothenate moiety of Coenzyme A. Once the oligonucleotide was attached, longer handles could then be appended by ligation. While this method can clearly be used for handle attachment to complex molecules, all the manipulations and incubations required for attachment must be performed on the target itself, which is fine for abundant and highly stable targets like ribosomes, but can be difficult if the target is hard to obtain in large quantities or is unstable as is the case for many membrane proteins.
Here we describe an approach using a SpyTag/SpyCatcher system in which all the difficult DNA attachment chemistry is performed on the easy to obtain SpyCatcher protein. Once the SpyCatcher‐DNA conjugate is made, attachment to the target only requires a short incubation at room temperature. Thus, the method should be suitable for DNA handle attachment to a wide variety of complex and sensitive target molecules. We tested the approach on a previously characterized membrane protein GlpG22 and show that the attachment method does not significantly alter the mechanical properties of the system.
Results
The handle attachment method is outlined in Figure 1. First SpyCatcher‐DNA conjugate reagents are prepared, bearing either biotin or digoxigenin (dig) affinity tags that can be attached to the chamber surface or beads. The target protein encoding the 13 amino acid SpyTag peptide sequence is then mixed with the SpyCatcher‐DNA conjugate allowing covalent attachment to the SpyTag via an isopeptide bond between a Lys side chain in SpyCatcher and an Asp side chain in the SpyTag.35, 36
Figure 1.

Handle attachment procedure for single molecule manipulations. The DNA handles are first linked to the MBP‐SpyCatcher fusion using the traditional thiol chemistry approach. Separately the target protein (in this case SpyTag‐GlpG) is prepared with added SpyTag peptides (genetically encoded). Upon simple mixing, SpyTag‐GlpG becomes covalently linked to the MBP‐SpyCatcher‐DNA via an isopeptide bond (see box inset). For unfolding experiments with GlpG, we employ bicelles to provide bilayer conditions, which are fully compatible with the attachment method. The DNA‐conjugated GlpG can then be tethered between the glass support and magnetic bead via the biotin/neutravidin and dig/antidig linkages, and subject to mechanical force.
For convenient purification of the protein and the DNA conjugate, we expressed SpyCatcher as a fusion to maltose binding protein (MBP‐SpyCatcher). We introduced a unique Cys into the MBP‐SpyCatcher protein to which 512 bp DNA handles could be attached using maleimide chemistry. The MBP‐SpyCatcher‐DNA conjugate could be conveniently purified away from unconjugated DNA or protein by employing anion exchange chromatography to select for molecules that possess DNA moiety, and then by amylose affinity chromatography to select for molecules that possess MBP moiety. Although MBP is a relatively large affinity tag, we found that DNA conjugation prevented affinity purification using a simple 6xHis tag. Moreover, because both the target protein and DNA attachment points are within the SpyTag/SpyCatcher domain (i.e., MBP is not on the mechanical pulling axis), the MBP purification tag will not experience a force during the pulling experiments, so it is not necessary to remove it. Note that the complex manipulations needed to prepare and purify the MBP‐SpyCatcher‐DNA conjugate are all performed on the simple and easy to prepare MBP‐SpyCatcher protein. Once the conjugate is prepared, it can simply be used as an added reagent to attach to any target possessing a SpyTag sequence.
To test the method, we employed the rhomboid protease GlpG as a target protein. GlpG is a membrane protein with six transmembrane helices. Previously, we had extensively characterized the mechanical unfolding of GlpG using DNA handles coupled using traditional disulfide attachment to introduced Cys residues at the N‐ and C‐termini,22 allowing us to test whether the new method affected the observed mechanical properties.
SpyTag peptides (AHIVMVDAYKPTK)35, 36 were appended to the N‐ and C‐ termini of GlpG (SpyTag‐GlpG) to allow for linkage to the MBP‐SpyCatcher‐DNA conjugate. Figure 2(a) shows the progress of the linkage reaction when we added a nominal 13:1 molar ratio of SpyTag‐GlpG to MBP‐SpyCatcher‐DNA (1.2 µM SpyTag‐GlpG, 0.09 µM MBP‐SpyCatcher‐DNA). We obtained ∼80% linkage in only 5 min and nearly quantitative linkage after 1 h at 22°C. Both singly and doubly conjugated SpyTag‐GlpG were observed in an approximately 1:1 ratio [Fig. 2(b)]. We were surprised that we needed to add such a large excess of SpyTag‐GlpG relative to MBP‐SpyCatcher‐DNA, but it is possible that at these low concentrations SpyTag‐GlpG could stick to microcentrifuge tube surfaces so that the effective concentration is much lower than the nominal concentration. Regardless of the cause, it is a simple matter to perform a titration to determine the appropriate concentration ratios.
Figure 2.
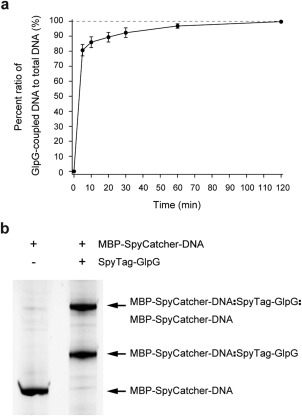
Kinetics and gel analysis for DNA handle attachment to GlpG. (a) Percent ratio of GlpG‐coupled DNA to total DNA as a function of time. The percent ratios are estimated from triplicate gels. The reaction was performed at 22°C and pH 7.5. (b) SDS‐PAGE gel showing the attachment of DNA handles to GlpG after 1 h incubation. The upward shifts from the MBP‐SpyCatcher‐DNA band indicate the coupling of GlpG to one or two DNA handles. The gel is stained with nucleic acid gel stain.
There are many possible products from the conjugation reaction. In addition to singly conjugated forms, there are three possible doubly conjugated forms (two biotin tags, two dig tags, or a mixture), yet all are invisible to the magnetic tweezer experiment except the form with both a biotin tag and a dig tag, which can bind to both a neutravidin treated glass surface and an antidig treated magnetic bead (Fig. 1, bottom).
The tethered GlpG proteins were subjected to a slow force ramp (∼0.3 pN/s) by approaching a pair of magnets to magnetic beads. As a response to the force, the extension, i.e., the end‐to‐end distance of the GlpG‐DNA conjugate increased in a gradual manner as the DNA stretches and around 25 pN abrupt jumps in extension occurred, indicating the highly cooperative unfolding of the GlpG protein (Fig. 3).
Figure 3.
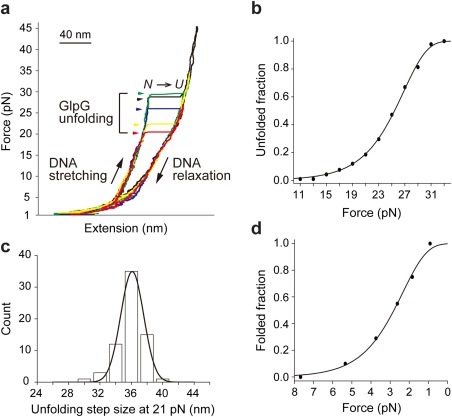
Single‐molecule forced unfolding experiments for tethered GlpG. (a) Representative force‐extension curves showing the repetitive unfolding events of a single GlpG linked using the SpyTag/SpyCatcher system described here. The symbols N and U denote the folded and unfolded states respectively. (b) Unfolded fraction as a function of force (n = 91). Fitting yields unfolding rate at zero force. (c) Unfolding step size distribution at a constant 21 pN (n = 67). (d) Folded fraction as a function of force (n = 88). Fitting yields folding rate at zero force.
To test whether the new tethering method altered the observed mechanical unfolding/folding properties of GlpG, we measured the folding and unfolding rates and step size for the unfolding events as described previously22 (Fig. 3 and Table 1). As shown in Table 1, the results were similar to those found previously with a direct disulfide tethering method (P > 0.05). These results indicate the MBP‐SpyCatcher‐DNA handles do not significantly alter the observed mechanical unfolding events. Moreover, the SpyTag/SpyCatcher complex has high mechanical stability35 and did not provide any additional rupture events at forces up to ∼45 pN, a typical force regime in protein mechanical responses (Fig. 3).
Table 1.
Step Size and Rates for GlpG Unfolding/Folding from Two Linkage Methods
| Unfolding size (nm) | Unfolding rate (×10−5 s−1) | Folding rate (×10−2 s−1) | |
|---|---|---|---|
| SpyTag | 36.10 (0.05) | 8.53 (1.43) | 3.56 (0.53) |
| Disulfide | 35.63 (0.09) | 5.64 (0.91) | 3.91 (0.54) |
| P | 0.1385 | 0.0900 | 0.6442 |
Numbers in parentheses indicate standard deviation for the step size and standard error for the rate constants. The P values were assessed by Welch's t‐test.
Discussion
Our approach for protein/DNA conjugation has several advantages for the mechanical manipulation of proteins. (1) The hard protein chemistry of DNA handle coupling and purification steps can be done on the well‐behaved and less valuable SpyCatcher protein rather than a precious target protein. (2) The rapid, spontaneous attachment reaction is a major advantage for proteins sensitive to thermal stress and vulnerable to aggregation. (3) The rapid, spontaneous reaction also allows us to use low concentration (tens of nM) of DNA conjugate, which is hard to obtain in large amounts. (4) The binding of SpyTag and SpyCatcher is not a thiol chemistry, and thus we can study native proteins without engineering out the native cysteines of the target proteins. (5) The high mechanical rigidity of the SpyTag/SpyCatcher complex allows for observations of unfolding transitions without complications from additional rupture events from the complex. A limitation of the method is that it would be difficult to attach handles in the middle of a protein for pulling with different geometries. It is not impossible, however, as the SpyTag peptide can also be internally encoded.35 The new handle attachment method established here should be generally applicable for the mechanical manipulation of highly complex and sensitive systems.
Materials and Methods
Cloning, expression, and purification of SpyTag‐GlpG
The GlpG protein employed here corresponds to residues 87 to 276 of E. coli GlpG.22 The SpyTag peptide (AHIVMVDAYKPTK)35, 36 and a linker (GSGESG) were added to both N‐ and C‐termini by two sequential PCR amplifications. The prior GlpG construct in a pTrcHisB vector22 was used as a template for the first PCR reaction. We employed the following primers that include the SpyTag and a linker (annealing regions underlined): FWD: 5’‐AATGGTCGA TGCGTATAAACCGACGAAAGGTTCAGGAGAGTCAGGCGCCGCCTGTTTGCG−3’, REV: 5’‐GGCGTCCACCATCACGATGTGGGCACCACTTTCACCACTACCACATTTTCGTTTTCGCGC−3’. The gel‐purified product was then used as the template for the second PCR reaction. The second primers completed the SpyTag sequence and include a 24 to 29 bp overlap with SacI/HindIII digested pTrcHisB. Second PCR primers were (annealing regions underlined): FWD: 5’‐ATGGGGCATCATCATCATCATCATGAGCTCGCTCATATTGTAATGGTCGATGCGTATAAACC−3’, REV: 5’‐CTCATCCGCCAAAACAGCCAAGCTTACTCCTTCGTCGGCTTGTAGGCGTCCACCATCACG−3’. The gel‐purified PCR product was cloned into pTrcHisB vector at the SacI and HindIII sites, preserving the N‐terminal 6xHis‐tag to generate the SpyTag‐GlpG construct shown in Figure 4(a). The SpyTag‐GlpG protein was expressed in E. coli BL21‐Gold (DE3) and purified as previously described.22 Aliquots of the purified GlpG (∼15 µM) in 50 mM Tris (pH 7.5), 150 mM NaCl, 0.1% DDM were flash frozen in liquid nitrogen, and stored at −80°C.
Figure 4.

Amino acid sequences for (a) SpyTag‐GlpG and (b) MBP‐SpyCatcher. The SpyTag and SpyCatcher are shown in red, GlpG and MBP in blue, 6xHis‐tag in green, and TEV protease site in yellow. The unique cysteine residue in MBP‐SpyCatcher for DNA handle conjugation is underlined.
Cloning, expression, and purification of MBP‐SpyCatcher
A DNA segment encoding the maltose binding protein (MBP) tag was PCR‐amplified from the pMAL‐c2X vector (New England Biolabs) using the primers, FWD: 5’‐TTTAACTTTAAGAAGGAGATATACATATGAAAATCGAAGAAGGTAAACTGGTAATC and REV: 5’‐ATGGTGATGGTAGTACGACATGCCATTAGTCTGCGCGTC. The gel‐extracted DNA fragment was inserted into NdeI‐linearized pDEST14‐SpyCatcher vector35 using the Gibson ISO assembly procedure.37 The resulting plasmid was then modified using a PCR‐based mutagenesis strategy, which replaced the three amino acids (Ala‐Met‐Val) following the TEV protease site with the sequence Gly‐Cys‐Gly to allow for DNA conjugation. Briefly, the MBP‐containing SpyCatcher plasmid was used as the template for PCR‐amplification with the primers, FWD: 5’‐GGTGGTTGTGGTGATACCTTATCAGGTTTATCAAGTGAGCAAG and REV: 5’‐CTGAAAATACAGGTTTTCGGTCGTTG, the reaction was then treated with DpnI restriction enzyme and subsequently with polynucleotide kinase and T4 DNA ligase, before transformation into competent E. coli DH5α. Putative positive transformants were mini‐prepped and their sequence confirmed by DNA sequencing (Genewiz) to ensure that the final expression plasmid, pMBP‐SpyCatcher, encodes a fusion protein with the following organization of MBP, 6xHis, TEV protease site, Cys‐containing linker, and SpyCatcher shown as in Figure 4(b).
The expression plasmid was transformed into E. coli BL21(DE3) and an overnight culture was used to inoculate four Ultra Yield flasks (Thomson Instrument Company) each containing 1 L of Terrific broth supplemented with ampicillin (100 µg/mL). The cells were grown with shaking at 37°C until they reached an OD600 of ∼1.0 at which point the temperature was lowered to 30°C and protein expression induced with 0.75 mM IPTG. Growth was continued for an additional 4 to 5 h and the cells harvested by centrifugation. The cell pellet was frozen pending cell lysis. After thawing, the cell pellet was resuspended in lysis buffer (50 mM Tris (pH 8.0), 200 mM NaCl, 1 mM EDTA, 1 mM DTT, 1 mM PMSF) supplemented with a cocktail of protease inhibitors (Sigma‐Aldrich). The cells were lysed using an EmulsiFlex C3 cell homogenizer (Avestin) and the lysate was centrifuged at 39,000g for 40 min at 4°C. The supernatant was decanted, filtered using a 1.0 µm polyethersulfone membrane, and loaded on a 5 mL MBPTrap column (GE Healthcare Bio‐Sciences) equilibrated in 50 mM Tris (pH 8.0), 200 mM NaCl, 1 mM TCEP (Buffer A) at 2 mL/min. The column was washed extensively with Buffer A and then bound proteins were eluted with a step gradient to 10 mM maltose in Buffer A. The fractions containing the fusion protein were pooled and concentrated, and the fusion protein was further purified by size exclusion chromatography using a Sephacryl S‐100 column (GE Healthcare Bio‐Sciences) equilibrated in 50 mM Tris (pH 8.0), 150 mM NaCl, 1 mM TCEP at a flow rate of 1 mL/min. Fractions containing pure MBP‐SpyCatcher proteins were pooled, concentrated to ∼150 µM, aliquots flash‐frozen with liquid nitrogen, and stored at −80°C pending use.
Conjugation of DNA to MBP‐SpyCatcher
A 512 bp DNA possessing a distinct binding tag (biotin or digoxigenin), was PCR‐amplified from a λ DNA template (final 2.5 μg/mL; New England Biolabs, N3011S) using forward primer CATGTGGGTGACGCGAAA modified with 5′ amine group, and reverse primer TCGCCACCATCATTTCCA modified with either 5′ biotin or digoxigenin (final 1 μM each; Integrated DNA Technologies). 4 mL of each PCR product was purified using a HiSpeed Plasmid Maxi Kit (Qiagen, 12662), eluted into ∼1 mL of 0.1M sodium bicarbonate (pH 8.4), and then mixed to total ∼2 mL (each ∼1.5 μM final concentration).
To attach a maleimide functional group to the amine‐modified end, we used the SM(PEG)2 reagent (Thermo Fisher Scientific, 22102), which is a heterobifunctional crosslinker with N‐hydroxysuccinimide (NHS) ester and the desired maleimide group [Fig. 5(a)]. 8 μL of 250 mM SM(PEG)2 crosslinker (dissolved in dimethyl sulfoxide) was added to the ∼2 mL DNA mixture and incubated for 20 min at room temperature (RT). The sample was then divided into two ∼1 mL samples for purification with the HiSpeed Plasmid Maxi Kit, each eluted into ∼1 mL of 0.1M sodium phosphate (pH 7.3), 150 mM NaCl, and then combined to yield ∼2 mL of ∼1.1 μM DNA conjugate mixture. To attach the DNA handles to the MBP‐SpyCatcher protein, we mixed 200 μL of the purified MBP‐SpyCatcher protein to the ∼2 mL DNA handle with final concentration of ∼14 μM and ∼1 μM each, and incubated the mixture for 2 h at RT [Fig. 5(a)].
Figure 5.
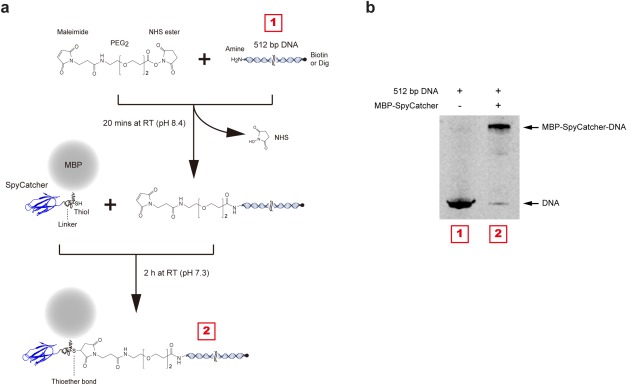
Linking DNA to MBP‐SpyCatcher. (a) Schematic diagram of the conjugation process. The amine‐modified 512 bp DNA were activated with a maleimide group and then conjugated to MBP‐SpyCatcher protein by maleimide‐cysteine crosslinking chemistry. The purification of MBP‐SpyCatcher‐DNA construct was performed sequentially by ion exchange and MBP‐tag affinity chromatography. (b) SDS‐PAGE gel showing successful conjugation. The upward shift from the DNA band indicates the MBP‐SpyCatcher coupling to DNA. The gel was stained with a nucleic acid gel stain.
To separate unconjugated SpyCatcher proteins and DNA in the SpyCatcher/DNA mixture, we first employed an anion exchange column. The mixture (DNA and MBP‐SpyCatcher‐DNA) was bound to a 1 mL HiTrap Q HP column (GE Healthcare Bio‐Sciences, 29‐0513‐25) equilibrated with 20 mM Tris (pH 7.5), and then eluted using a gradient to 1M NaCl (gradient volume = 25 mL). The eluted sample was collected in 1 mL fractions, and peak fractions of the DNA constructs were pooled. Unreacted DNA was further removed by employing an amylose affinity resin that only captures the construct with MBP tag, i.e., MBP‐SpyCatcher‐DNA. We loaded the pooled sample onto a column charged with ∼1 mL amylose resin (New England Biolabs, E8021S) and incubated for 2 h at RT with slow tilt rotation. We washed the column with ∼60 mL of 50 mM Tris (pH 7.4), 200 mM NaCl, 20 mM imidazole (Buffer B), and then eluted the MBP‐SpyCatcher‐DNA from the amylose resin by applying 10 mM maltose in Buffer B. Using 30K Amicon Centrifugal Filter Unit (EMD Millipore, UFC803024), the MBP‐SpyCatcher‐DNA construct was concentrated to ∼100 nM of ∼300 μL and stored at −80°C in 10 μL aliquots. The covalent conjugation of MBP‐SpyCatcher and DNA was confirmed by separation by 4 to 20% SDS‐PAGE (GenScript, M42015) with staining for DNA molecules with GelRed nucleic acid gel stain (Biotium, 41003) [Fig. 5(b)].
DNA‐SpyTag‐GlpG sample preparation
1 μL of ∼15 µM SpyTag‐GlpG and 11 μL of ∼100 nM MBP‐SpyCatcher‐DNA were mixed so that the final solution contained 45 mM Tris (pH 7.4), 150 mM NaCl, 2 mM TCEP, 15 mM imidazole, 7 mM maltose, and 0.1% DDM. After 1 h incubation at RT, 9 μL of the mixture was diluted to ∼800 μL with 50 mM Tris (pH 7.5), 150 mM NaCl, 2 mM TCEP, 1.3% bicelle to make the final DNA concentration ∼1.0 nM, and stored at −80°C in 10 μL aliquots. The bicelle mixture consisted of DMPC lipid (Avanti Polar Lipids, 850345P) and CHAPSO detergent (Affymetrix, C317) at a 2.5:1 molar ratio as previously described.22
Coating of magnetic bead with antidigoxigenin
We used N‐hydroxysuccinimide (NHS) ester crosslinking chemistry to coat magnetic beads with antidigoxigenin (antidig). With a magnetic concentrator (Thermo Fisher Scientific, 12321D), 34 μL of 2.8 μm carboxylated magnetic beads (Thermo Fisher Scientific, 14305D) were equilibrated in 1 mL of 0.1M MES (pH 6.0), 0.5M NaCl, 0.1% Tween 20. To activate the carboxylic acids with NHS esters, we added 100 μL of a solution of 50 mM 1‐ethyl‐3‐(3‐dimethylaminopropyl)carbodiimide and 100 mM sulfo‐NHS and incubated for 15 min at RT with gentle mixing on a rotator. The activated magnetic beads were equilibrated in 1 mL of 0.1M sodium phosphate (pH 7.3), 150 mM NaCl, 0.1% Tween 20 using the magnetic concentrator. We then added 60 μL of 1.1 μM antidig (Sigma‐Aldrich, 11333089001) and incubated for 3 h at RT with gentle mixing on a rotator. To quench unreacted NHS esters, we equilibrated the beads in 1 mL of 0.1M Tris (pH 8.5), 0.1% Tween 20 and incubated for 10 min at RT with gentle mixing on a rotator. The antidig coated magnetic beads were washed with 0.1M sodium phosphate (pH 7.3), 150 mM NaCl, 0.01% Tween 20, resuspended in 34 μL of the same buffer, and then stored at 4°C.
PEG/biotin coating of glass coverslip
We coated the surface of glass coverslips (25 × 50 mm, No 1.5; VWR) with a combination of two different NHS ester‐functionalized polyethylene glycols (PEG‐NHS),38, 39 i.e., 125:1 molar ratio of methylated PEG‐NHS (mPEG; Laysan Bio, MPEG‐SVA‐5000) and biotin‐conjugated PEG‐NHS (biotin‐PEG; Laysan Bio, Biotin‐PEG‐SVA‐5000) using amine‐NHS ester crosslinking chemistry. The mPEG molecules prevent nonspecific binding events of beads and proteins whereas the biotin‐PEGs specifically bind DNA‐conjugated GlpGs via biotin‐neutravidin linkage. The coverslips were first cleaned with 1M KOH in a sonication bath for 20 min, washed with distilled water, and then functionalized with amine groups (silanization reaction) using a solution of N‐(2‐aminoethyl)−3‐aminopropyltrimethoxysilane (UCT, A0700), acetic acid and methanol in a 1:5:100 volume ratio. The coverslips were incubated with the silanization solution for 12 min at RT, sonicated for 1 min in the sonication bath, and incubated for an additional 20 min at RT. The coverslips were washed with methanol, then distilled water, and then dried with nitrogen gas. PEGs were coupled by incubating 60 μL of a PEG‐NHS solution (40 mM PEG mixture in 0.1M sodium bicarbonate, pH 8.4) between two coverslips for 4 to 5 h at RT in a humidity chamber. The coverslips were washed with distilled water, dried with nitrogen gas, and stored at −20°C.
Single‐molecule microscope sample chamber
A sample chamber of ∼15 μL channel volume (≡1 CV) was constructed by putting together a PEG/biotin‐coated coverslip (see above) and a KOH‐cleaned coverslip (24 × 40 mm, No 1.5; VWR) with double‐sided tape. 1 μL of 1.0 μm sized polystyrene beads coated with streptavidin (Polysciences, 24162) were washed and equilibrated in 300 μL of 0.1M sodium phosphate (pH 7.4), 150 mM NaCl, 0.1% Tween 20 by repeated centrifugation and resuspension. We injected 1 CV of the bead solution into the ∼90 μm thick channel by capillary action and incubated for 90 s at RT. The nonmagnetic polystyrene beads bind to PEG/biotin‐coated surface via biotin‐streptavidin linkage and are used to correct vertical and lateral drift of sample stage. The chamber surfaces were further passivated by flowing through three CVs of 100 mg/mL bovine serum albumin, and then washed with 50 mM Tris (pH 7.5), 150 mM NaCl. The sample chamber was then equilibrated in 50 mM Tris (pH 7.5), 150 mM NaCl, 2 mM TCEP, 1.3% bicelle (Buffer C). The bicelle mixture consisted of DMPC lipid and CHAPSO detergent at a 2.5:1 molar ratio as described above.
To bind the DNA‐linked GlpG constructs to the PEG/biotin‐coated surface, we first attached neutravidin (NTV) molecules to the biotin‐modified DNA handles. We added 1 μL of 167 nM NTV (Thermo Fisher Scientific, A‐2666) to 10 μL of the DNA‐linked GlpG stock described above and incubated for 20 min at RT (NTV: biotin‐DNA = 30: 1). The NTV‐bound GlpG sample was then diluted to 22 μL total volume with Buffer C (i.e., ∼100 pM of NTV‐biotin‐DNA‐GlpG‐DNA‐digoxigenin), and 1 CV of the diluted sample was injected into the chamber and incubated for 10 min at RT to allow binding to the PEG/biotin‐coated glass surface. The vacant biotin binding sites of the neutravidin/streptavidin molecules were blocked by washing with three CVs of short 30 nt DNA oligonucleotides modified by biotin (10 μM in Buffer C) and incubating for 5 min at RT. The chamber was then equilibrated in 50 mM Tris‐HCl (pH 7.5), 150 mM NaCl, 1.3% bicelle (Buffer D). With the magnetic concentrator (the same one used above), 1 μL of antidig‐coated magnetic beads were washed with 300 μL of 50 mM Tris (pH 7.5), 150 mM NaCl once and with 50 μL of Buffer D twice. One CV of the magnetic beads, resuspended in 50 μL Buffer D, were introduced into the chamber, and incubated for 30 min at RT to bind surface‐tethered GlpGs having digoxigenin‐DNA handles (Fig. 1, bottom).
Magnetic tweezer instrumentation
The magnetic tweezer apparatus was custom‐built as previously described.21, 22, 40, 41 The tweezer setup was constructed on an inverted microscope (Olympus, IX73) with a motorized XY stage (ASI, MS‐2000 XY Automated Stage). Magnetic beads in a glass sample chamber were illuminated by 455 nm light‐emitting diode (Thorlabs, M455L3) and diffraction pattern images from the beads were captured at 60 Hz frame rate by a charge‐coupled device camera (JAI, CM‐040GE). We track the lateral (x,y) and vertical (z) motions of the beads by analyzing the diffraction images using customized software programs written in LabView (National Instruments).21, 22, 40, 41 The lateral movement was tracked by calculating the maximum self‐convolution for diffraction pattern intensity profiles (I(x), I(y)). The vertical movement (extension change) was tracked by calculating the minimum χ 2 estimate of a radial intensity profile (I(r)) with precalibration data, i.e., a stack of the radial intensity profiles as a function of bead height. The calibration data was measured by moving the focal plane in known increments with a nanopositioning piezoelectric stage (Mad City Labs, Nano‐F100S). Thermal drift of the microscope stage was corrected by simultaneously tracking the nonmagnetic polystyrene beads immobilized on the chamber surface.
To generate a magnetic field gradient, we employed a pair of two permanent neodymium magnets (10 × 10 × 12 mm) separated by 1 mm with antiparallel alignment of magnetic moments. Vertical and rotational motions of the magnet pair was manipulated with a translation stage (Physik Instrumente, M‐126.PD1) and a rotation stage (Physik Instrumente, DT‐50). We calibrated the mechanical tension applied to tethered molecules, as a function of the magnet height. At each magnet height, we measured the end‐to‐end distance (extension) of a tethered molecule (L) and lateral fluctuation of a magnetic bead (δx 2), and then calculated the applied tension from an equation F mag = k B TL/δx 2 where k B T is the thermal energy. The equation was derived by assuming an inverted pendulum for the trapped molecule in the magnetic potential.40, 42 The force range of our magnetic tweezers is approximately 0.01 pN to 70 pN. We controlled the pair of magnets with translational speeds of 0.1 mm/s during the gradual force ramp [average ∼0.3–0.4 pN/s, Fig. 3(a)]. The representative traces shown in Figure 3(a) were median‐filtered with a 50‐point window size.
Acknowledgments
Authors thank members of the lab for comments on the manuscript.
References
- 1. Bustamante C, Chemla YR, Forde NR, Izhaky D (2004) Mechanical processes in biochemistry. Annu Rev Biochem 73:705–748. [DOI] [PubMed] [Google Scholar]
- 2. Kedrov A, Janovjak H, Sapra KT, Muller DJ (2007) Deciphering molecular interactions of native membrane proteins by single‐molecule force spectroscopy. Annu Rev Biophys Biomol Struct 36:233–260. [DOI] [PubMed] [Google Scholar]
- 3. Borgia A, Williams PM, Clarke J (2008) Single‐molecule studies of protein folding. Annu Rev Biochem 77:101–125. [DOI] [PubMed] [Google Scholar]
- 4. Alegre‐Cebollada J, Perez‐Jimenez R, Kosuri P, Fernandez JM (2010) Single‐molecule force spectroscopy approach to enzyme catalysis. J Biol Chem 285:18961–18966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Puchner EM, Gaub HE (2012) Single‐molecule mechanoenzymatics. Annu Rev Biophys 41:497–518. [DOI] [PubMed] [Google Scholar]
- 6. Javadi Y, Fernandez JM, Perez‐Jimenez R (2013) Protein folding under mechanical forces: a physiological view. Physiology 28:9–17. [DOI] [PubMed] [Google Scholar]
- 7. Zoldak G, Rief M (2013) Force as a single molecule probe of multidimensional protein energy landscapes. Curr Opin Struct Biol 23:48–57. [DOI] [PubMed] [Google Scholar]
- 8. Woodside MT, Block SM (2014) Reconstructing folding energy landscapes by single‐molecule force spectroscopy. Annu Rev Biophys 43:19–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cordova JC, Das DK, Manning HW, Lang MJ (2014) Combining single‐molecule manipulation and single‐molecule detection. Curr Opin Struct Biol 28:142–148. [DOI] [PubMed] [Google Scholar]
- 10. Chen Y, Radford SE, Brockwell DJ (2015) Force‐induced remodelling of proteins and their complexes. Curr Opin Struct Biol 30:89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Olivares AO, Baker TA, Sauer RT (2016) Mechanistic insights into bacterial AAA plus proteases and protein‐remodelling machines. Nat Rev Microbiol 14:33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ryu JK, Jahn R, Yoon TY (2016) Progresses in understanding NSF‐mediated disassembly of SNARE complexes. Biopolymers 105:518–531. [DOI] [PubMed] [Google Scholar]
- 13. del Rio A, Perez‐Jimenez R, Liu R, Roca‐Cusachs P, Fernandez JM, Sheetz MP (2009) Stretching single talin rod molecules activates vinculin binding. Science 323:638–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Junker JP, Ziegler F, Rief M (2009) Ligand‐dependent equilibrium fluctuations of single calmodulin molecules. Science 323:633–637. [DOI] [PubMed] [Google Scholar]
- 15. He C, Genchev GZ, Lu H, Li H (2012) Mechanically untying a protein slipknot: multiple pathways revealed by force spectroscopy and steered molecular dynamics simulations. J Am Chem Soc 134:10428–10435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Popa I, Berkovich R, Alegre‐Cebollada J, Badilla CL, Rivas‐Pardo JA, Taniguchi Y, Kawakami M, Fernandez JM (2013) Nanomechanics of HaloTag tethers. J Am Chem Soc 135:12762–12771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cecconi C, Shank EA, Bustamante C, Marqusee S (2005) Direct observation of the three‐state folding of a single protein molecule. Science 309:2057–2060. [DOI] [PubMed] [Google Scholar]
- 18. Shank EA, Cecconi C, Dill JW, Marqusee S, Bustamante C (2010) The folding cooperativity of a protein is controlled by its chain topology. Nature 465:637–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim J, Zhang CZ, Zhang XH, Springer TA (2010) A mechanically stabilized receptor‐ligand flex‐bond important in the vasculature. Nature 466:992–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gao Y, Sirinakis G, Zhang YL (2011) Highly anisotropic stability and folding kinetics of a single coiled coil protein under mechanical tension. J Am Chem Soc 133:12749–12757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Min D, Kim K, Hyeon C, Cho YH, Shin YK, Yoon TY (2013) Mechanical unzipping and rezipping of a single SNARE complex reveals hysteresis as a force‐generating mechanism. Nat Commun 4:1705–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Min D, Jefferson RE, Bowie JU, Yoon TY (2015) Mapping the energy landscape for second‐stage folding of a single membrane protein. Nat Chem Biol 11:981–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ryu JK, Min D, Rah SH, Kim SJ, Park Y, Kim H, Hyeon C, Kim HM, Jahn R, Yoon TY (2015) Spring‐loaded unraveling of a single SNARE complex by NSF in one round of ATP turnover. Science 347:1485–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brenner MD, Zhou RB, Conway DE, Lanzano L, Gratton E, Schwartz MA, Ha T (2016) Spider silk peptide is a compact, linear nanospring ideal for intracellular tension sensing. Nano Lett 16:2096–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jahn M, Buchner J, Hugel T, Rief M (2016) Folding and assembly of the large molecular machine Hsp90 studied in single‐molecule experiments. Proc Natl Acad Sci USA 113:1232–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, Simpson D, Mendez J, Zimmerman K, Otto P, Vidugiris G, Zhu J, Darzins A, Klaubert DH, Bulleit RF, Wood KV (2008) HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol 3:373–382. [DOI] [PubMed] [Google Scholar]
- 27. Taniguchi Y, Kawakami M (2010) Application of HaloTag protein to covalent immobilization of recombinant proteins for single molecule force spectroscopy. Langmuir 26:10433–10436. [DOI] [PubMed] [Google Scholar]
- 28. Aubin‐Tam ME, Olivares AO, Sauer RT, Baker TA, Lang MJ (2011) Single‐molecule protein unfolding and translocation by an ATP‐fueled proteolytic machine. Cell 145:257–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Olivares AO, Nager AR, Iosefson O, Sauer RT, Baker TA (2014) Mechanochemical basis of protein degradation by a double‐ring AAA+ machine. Nat Struct Mol Biol 21:871–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yin J, Straight PD, McLoughlin SM, Zhou Z, Lin AJ, Golan DE, Kelleher NL, Kolter R, Walsh CT (2005) Genetically encoded short peptide tag for versatile protein labeling by Sfp phosphopantetheinyl transferase. Proc Natl Acad Sci USA 102:15815–15820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Maillard RA, Chistol G, Sen M, Righini M, Tan J, Kaiser CM, Hodges C, Martin A, Bustamante C (2011) ClpX(P) generates mechanical force to unfold and translocate its protein substrates. Cell 145:459–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kaiser CM, Goldman DH, Chodera JD, Tinoco I, Jr , Bustamante C (2011) The ribosome modulates nascent protein folding. Science 334:1723–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pippig DA, Baumann F, Strackharn M, Aschenbrenner D, Gaub HE (2014) Protein‐DNA chimeras for nano assembly. ACS Nano 8:6551–6555. [DOI] [PubMed] [Google Scholar]
- 34. Goldman DH, Kaiser CM, Milin A, Righini M, Tinoco I, Jr , Bustamante C (2015) Ribosome. Mechanical force releases nascent chain‐mediated ribosome arrest in vitro and in vivo. Science 348:457–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zakeri B, Fierer JO, Celik E, Chittock EC, Schwarz‐Linek U, Moy VT, Howarth M (2012) Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc Natl Acad Sci USA 109:E690–E697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li L, Fierer JO, Rapoport TA, Howarth M (2014) Structural analysis and optimization of the covalent association between SpyCatcher and a peptide Tag. J Mol Biol 426:309–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gibson DG (2011) Enzymatic assembly of overlapping DNA fragments. Methods Enzymol 498:349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ha T, Rasnik I, Cheng W, Babcock HP, Gauss GH, Lohman TM, Chu S (2002) Initiation and re‐initiation of DNA unwinding by the Escherichia coli Rep helicase. Nature 419:638–641. [DOI] [PubMed] [Google Scholar]
- 39. Joo C, Ha T, Single‐molecule FRET with total internal reflection microscopy In: Ha T, Selvin P, Ed. (2012) Single‐molecule techniques: A laboratory manual. New York: Cold Spring Harb Lab, pp 3–36. [DOI] [PubMed] [Google Scholar]
- 40. Gosse C, Croquette V (2002) Magnetic tweezers: micromanipulation and force measurement at the molecular level. Biophys J 82:3314–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ribeck N, Saleh OA (2008) Multiplexed single‐molecule measurements with magnetic tweezers. Rev Sci Instrum 79:094301. [DOI] [PubMed] [Google Scholar]
- 42. Strick TR, Allemand JF, Bensimon D, Bensimon A, Croquette V (1996) The elasticity of a single supercoiled DNA molecule. Science 271:1835–1837. [DOI] [PubMed] [Google Scholar]