

Abstract
ArnA from Escherichia coli is a key enzyme involved in the formation of 4‐amino‐4‐deoxy‐l‐arabinose. The addition of this sugar to the lipid A moiety of the lipopolysaccharide of pathogenic Gram‐negative bacteria allows these organisms to evade the cationic antimicrobial peptides of the host immune system. Indeed, it is thought that such modifications may be responsible for the repeated infections of cystic fibrosis patients with Pseudomonas aeruginosa. ArnA is a bifunctional enzyme with the N‐ and C‐terminal domains catalyzing formylation and oxidative decarboxylation reactions, respectively. The catalytically competent cofactor for the formylation reaction is N 10‐formyltetrahydrofolate. Here we describe the structure of the isolated N‐terminal domain of ArnA in complex with its UDP‐sugar substrate and N 5‐formyltetrahydrofolate. The model presented herein may prove valuable in the development of new antimicrobial therapeutics.
Keywords: N‐formyltransferase, 4‐amino‐4‐deoxy‐l‐arabinose, N10‐formyltetrahydrofolate, lipopolysaccharide, resistance to cationic antimicrobial peptides
Short abstract
PDB Code: 5J63
Abbreviations
- Ara4N
4‐amino‐4‐deoxy‐l‐arabinopyranose
- HEPPS
N‐2‐hydroxyethylpiperazine‐N'‐3‐propanesulfonic acid
- HPLC
high performance liquid chromatography
- NAD+
nicotinamide adenine dinucleotide
- Ni‐NTA
nickel nitrilotriacetic acid
- TEV
tobacco etch virus
- dTMP
thymidine monophosphate
- Tris
tris‐(hydroxymethyl)aminomethane
- UMP
uridine monophosphate.
Introduction
According to the Centers for Disease Control, the alarming rise of antibiotic resistance could bring about the “next pandemic.” Indeed, antibiotic resistant strains of bacteria are a significant threat to human existence. The main methods that bacteria utilize to confer resistance to antibiotics include modifying the drug, physically removing it from the cell, or altering the target site so it is no longer recognized by the antimicrobial agent.1
In addition to antibiotic resistance, some pathogenic bacteria, both Gram‐positive and Gram‐negative, are capable of evading the cationic antimicrobial peptides of the host immune system by modifications to their outermost envelopes.2 The cationic antimicrobial peptides or CAMPS are widely distributed in both animals and plants, and they represent some of the oldest host defense mechanisms. As the name implies, CAMPS are cationic in nature thereby allowing them to interact with the negatively charged outer components of the bacterium ultimately leading to cell death.3
Gram‐negative bacteria, in particular, contain a complex glycoconjugate referred to as the lipopolysaccharide or LPS that extends farthest away from the main body of the organism. The LPS is composed of a lipid A moiety, which anchors the macromolecule into the outer cell membrane. The lipid A is followed by the core polysaccharide and the O‐specific antigen.4 Importantly, there are phosphate groups attached to some of the sugars on the LPS that confer an overall negative charge, thus making these organisms susceptible to the action of CAMPS. Most Gram‐negative bacteria, however, have evolved mechanisms to avoid the bactericidal action of the CAMPs. One type of mechanism is the modification of the lipid A moiety by the addition of the positively charged 4‐amino‐4‐deoxy‐l‐arabinose group, hereafter referred to as Ara4N, which decreases the binding of CAMPS to the bacterial outer surface.5 The addition of Ara4N to the lipid A moiety not only results in a decrease of susceptibility to the CAMPS, but also to such therapeutics as polymyxin.
The first four steps in the pathway for the biosynthesis of Arn4N are highlighted in Scheme 1a6. Note that, in addition to the pmrE gene, the other genes encoding the enzymes required for the biosynthesis of Ara4N are located in the pmrHFIJKLM operon.7 As can be seen, UDP‐glucose is oxidized to UDP‐glucuronic acid by the action of PmrE. This is followed by the oxidative decarboxylation of UDP‐glucuronic acid to yield UDP‐4‐ketoarabinose, a reaction that is catalyzed by ArnA (also referred to as Pmrl). The keto group at the C‐4' carbon is aminated by the action of the pyridoxal 5'‐phosphate dependent enzyme ArnB (PmrH). The resulting amino group on C‐4' is transiently formylated by the action of ArnA (Pmrl), an N‐formyltransferase that requires N 10‐formyltetrahydrofolate for activity (Scheme 1b). It is thought that this formylation is an obligatory step in the production of l‐Ara4N‐modified lipid A, although its biological role is still not entirely clear.8
Scheme 1.
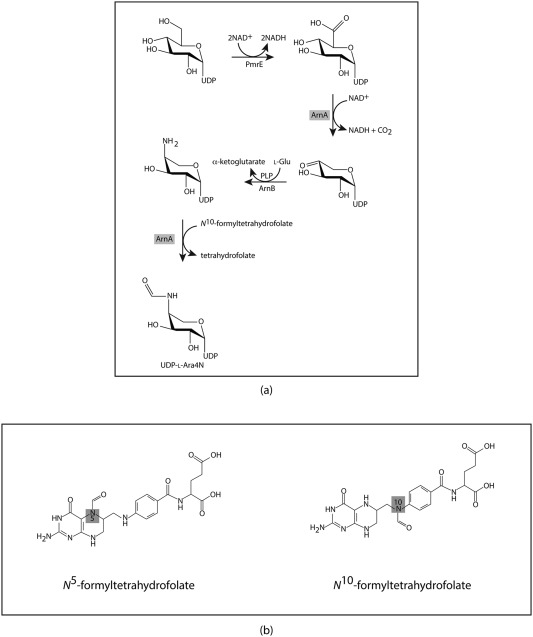
Biosynthetic pathway and relevant cofactors.
Strikingly, ArnA catalyzes both the second and fourth steps outlined in Scheme 1a. It is a bifunctional enzyme with the N‐ and C‐terminal domains catalyzing formylation and oxidative decarboxylation reactions, respectively. Given its role in the formation of Ara4N, it has garnered significant research attention as a possible target for structure‐based drug design. Not surprisingly, the structures of the individual domains as well as that of the full‐length protein have been determined by X‐ray crystallographic analyses.9, 10, 11, 12, 13 Whereas these investigations have yielded enormous insight into the structure of ArnA, none of the models deposited in the Protein Data Bank for the N‐formyltransferase domain contain a bound UDP‐sugar substrate. Given our interest in sugar N‐formyltransferases, we have now determined the structure of the Escherichia coli N‐terminal domain of ArnA in the presence of UDP‐Ara4N and N 5‐formyltetrahydrofolate. The model presented herein may prove valuable as a scaffold for the development of new antimicrobial therapeutics.
Results and Discussion
Overall structure
Crystals of the E. coli ArnA N‐formyltransferase domain/UDP‐Ara4N/N 5‐formyltetrahydrofolate complex utilized in this investigation belonged to the P1 space group with four subunits in the asymmetric unit. Note that the quaternary structure of the full length ArnA protein is hexameric.12 The model of the complex described here was refined to an overall R‐factor of 21.1% using X‐ray data from 2.5 to 20 Å. Relevant X‐ray data collection and model refinement statistics can be found in Table 1. The four subunits in the asymmetric unit are related by 222 symmetry, and their α‐carbons superimpose with a root‐mean‐square deviation of between 0.24 and 0.51 Å. For the sake of simplicity the following discussion refers only to subunit A in the X‐ray coordinate file.
Table 1.
X‐ray Data Collection Statistics and Model Refinement Statistics
| ArnA complex | |
|---|---|
| Resolution limits (Å) | 20–2.5 (2.55–2.5)a |
| Number of independent reflections | 45,712 (3035) |
| Completeness (%) | 91.3 (88.7) |
| Redundancy | 2.5 (2.2) |
| Avg I/avg σ(I) | 28.5 (11.8) |
| R sym (%)b | 5.6 (14.4) |
| R‐factor (overall)%/no. reflections | 21.1/45712 |
| R‐factor (working)%/no. reflections | 20.8/43367 |
| R‐factor (free)%/no. reflections | 27.3/2345 |
| Number of protein atoms | 9227 |
| Number of heteroatoms | 609 |
| Average B values | |
| Protein atoms (Å2) | 34.8 |
| Ligand (Å2) | 28.7 |
| Solvent (Å2) | 25.2 |
| Weighted RMS deviations from ideality | |
| Bond lengths (Å) | 0.014 |
| Bond angles (º) | 1.80 |
| Planar groups (Å) | 0.008 |
| Ramachandran regions (%)c | |
| Most favored | 89.1 |
| Additionally allowed | 10.5 |
| Generously allowed | 0.4 |
Statistics for the highest resolution bin.
.
R‐factor = (Σ|F o ‐ F c|/Σ|F o|) × 100 where F o is the observed structure‐factor amplitude and F c. is the calculated structure‐factor amplitude.
Distribution of Ramachandran angles according to PROCHECK.29
Shown in Figure 1(A) is a ribbon representation of the ArnA N‐formyltransferase domain. The polypeptide chain extends from Met 1 to Asn 305 with one break between Pro 249 and Lys 253. The protein adopts a bilobal type appearance with the N‐terminal motif containing a seven‐stranded mixed β‐sheet flanked on one side by three α‐helices and on the other by two. The N‐terminal domain harbors the active site, which is quite shallow and solvent exposed. The C‐terminal domain contains six β‐strands and two α‐helices.
Figure 1.
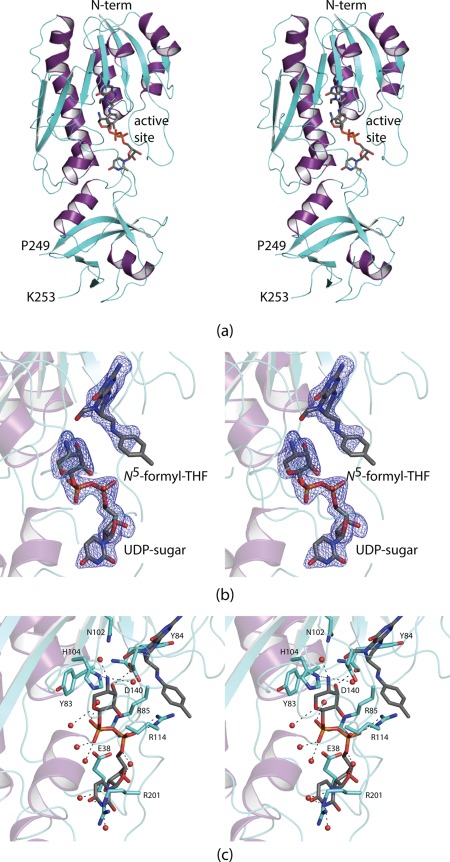
Structure of the ArnA N‐formyltransferase domain. A ribbon representation of the subunit is shown in (a) with the β‐strands and α‐helices colored in aquamarine and purple, respectively. There is one break in the polypeptide chain backbone between Pro 249 and Lys 253. The bound ligands are displayed in stick representations. Electron density corresponding to the bound ligands is shown in (b). The map, contoured at 3σ, was calculated with coefficients of the form F o−F c, where F o was the native structure factor amplitude and F c was the calculated structure factor amplitude. Although the map was calculated to a nominal resolution of 1.9 Å, because of radiation damage it was only possible to refine the model to an acceptable R‐factor using X‐ray data to 2.5 Å resolution. Note that the ligands were not included in the coordinate file used to calculate the initial map. A close‐up view of the active site is presented in (c). Ordered water molecules are represented by the red spheres. The bound ligands are highlighted in gray bonds. Potential hydrogen bonding interactions are indicated by the dashed lines. All figures were prepared using PyMOL.30
Active site architecture
Electron densities corresponding to the bound UDP‐Ara4N and N 5‐formyltetrahydrofolate ligands are displayed in Figure 1(B). The electron density was well defined for the UDP‐sugar and for the 2‐amino‐4‐oxo‐6‐methylpteridine ring of the cofactor. It was weak for the p‐aminobenzoic and glutamate portions of the cofactor (glutamate moiety was removed from the model). A close‐up view of the active site is presented in Figure 1(C). The uracil ring of the UDP‐sugar is anchored into the active site by the guanidinium group of Arg 201. There is an electrostatic interaction between the pyrophosphoryl moiety of the substrate and Arg 114. A salt bridge between Arg 85 and Glu 38 abuts one side of the substrate pyrophosphoryl group. As often observed in enzymes that bind sugars, one side of the pyranosyl ring of the UDP‐Ara4N substrate is surrounded by an aromatic group, in this case Tyr 83. Strikingly, there are few interactions between the pyranosyl ring and the protein. The C‐2' hydroxyl group does not participate in hydrogen bonding interactions with the protein, and the C‐3' hydroxyl group lies only within ∼3 Å of the backbone carbonyl group of Tyr 84. The C‐4' amino nitrogen that is ultimately formylated interacts with His 104, which is the presumed catalytic base. His 104, along with Asn 102 and Asp 140, are absolutely conserved amongst the N‐formyltransferases including E. coli glycinamide ribonucleotide transformylase, E. coli l‐methionyl‐tRNA N‐formyltransferase, and the sugar N‐formyltransferases solved in this laboratory (WlaRD, WbtJ, VioF, and QdtF).14, 15, 16, 17, 18, 19 In the case of WlaRD, for example, the structurally equivalent residues, His 96, Asn 94, and Asp 132, when changed to N94A, H96N, and D132N by site‐directed mutagenesis, resulted in the loss of measurable enzymatic activity.16
Comparison with previously reported ArnA models
Whereas the N‐formyltransferase domain of ArnA that is described here has been previously investigated by two other groups, in neither case was a model determined in the presence of UDP‐AraN4.10, 11 One of the structures was that of the apo form of the enzyme.11 In that model, there were breaks between Asn 35 and Ala 40 and His 250 and Ser 252. When UDP‐Ara4N is bound, the polypeptide chain between Asn 35 and Ala 40 becomes ordered allowing Glu 38 to project into the active site where it participates in a salt bridge with Arg 85 [Fig. 2(A)]. Other than the ordering of this loop, there are little changes in the protein structure upon binding UDP‐Ara4N. Water molecules that fill the cavity in the absence of substrate are simply expelled from the active site upon UDP‐sugar binding.
Figure 2.
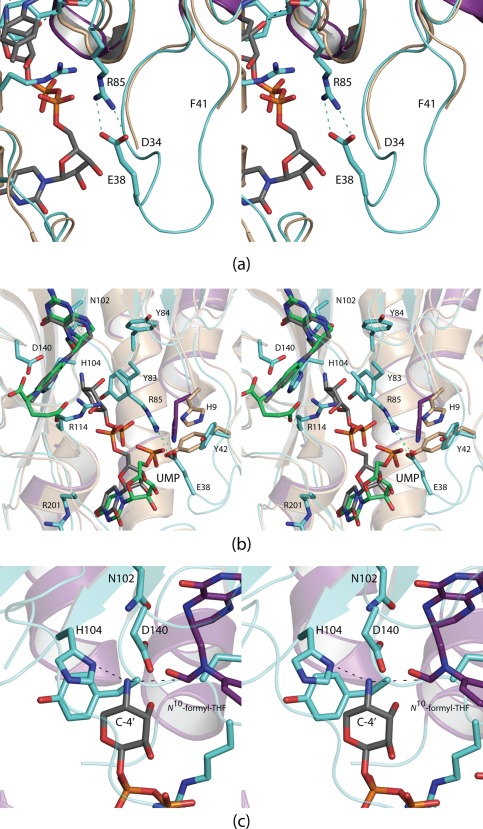
Comparison of the ArnA N‐formyltransferase complexes. The polypeptide chain of ArnA, in the absence of ligands, is disordered in two regions: Asn 35 to Ala 40 and His 250 to Ser 252. When UDP‐Ara4N binds, the loop defined by Asn 35 to Ala 40 becomes ordered as Glu 38 projects into the active site to form a salt bridge with Arg 85 as shown in (a). The apo enzyme structure is colored in wheat. Shown in (b) is a superposition of the active sites of ArnA when either UMP or UDP‐Ara4N is bound. The polypeptide chain for the protein/UMP model is highlighted in wheat, and the UMP ligand is displayed in green bonds. Besides the ordering of the Asn 35 to Ala 40 loop, the other significant conformational changes that occur upon substrate binding take place at His 9 and Tyr 42. A model of the Michaelis complex, constructed on the basis of the WlaRD/N 10‐formyltetrahydrofolate/dTMP complex, is depicted in (c).
The other reported structure of the ArnA N‐formyltransferase domain contained N 5‐formyltetrahydrofolate and UMP.10 Shown in Figure 2(B) is a superposition of the active site of this model onto that of the ternary complex described here. In the ArnA/UMP complex, the Asn 35 to Ala 40 loop is still disordered suggesting that it is the pyrophosphoryl group of the UDP‐sugar that triggers loop closure. As can be seen, when UMP binds to ArnA, its phosphoryl group projects away from the active site and towards the salt bridge that forms when UDP‐Ara4N is bound. It would thus be difficult to construct a model for UDP‐Ara4N binding based simply on the observed position for UMP.
Interest in the sugar N‐formyltransferases has risen in the past few years since the discovery of N‐formylated sugars on the lipopolysaccharides of such Gram‐negative pathogenic organisms as Brucella abortus,20 Salmonella enterica O60,21 Providencia alcalifaciens O40,22 Francisella tularensis,17 and Campylobacter jejuni.16 The three‐dimensional structures of several have been determined including WlaRD, WbtJ, VioF, and QdtF.16, 17, 18, 19 One in particular, WlaRD, was solved in the presence of the catalytically competent cofactor N 10‐formyltetrahydrofolate. The α‐carbons for WlaRD and ArnA superimpose with a root‐mean‐square deviation of 2.3 Å. Using the WlaRD/N 10‐formyltetrahydrofolate complex as a guide, a model of the Michaelis complex for ArnA was constructed as shown in Figure 2(C). The C‐4' amino group that is ultimately formylated is sandwiched between His 104 and the carbonyl group of the N 10‐formyltetrahydrofolate cofactor. A catalytic mechanism can be envisioned whereby His 104 serves as a general base to promote attack of the sugar amino group on the formyl carbonyl carbon of the cofactor leading to a tetrahedral intermediate. The source of the proton required for the collapse of the tetrahedral intermediate is presently unknown. This is in sharp contrast to that previously proposed whereby it was suggested that His 104, in its protonated form, interacts with the formyl group of the cofactor.11 In the model presented in Figure 2(C), the distance between His 104 and the formyl oxygen of the cofactor is ∼5 Å.
In conclusion, the structure of the N‐formyltransferase domain of ArnA has now been determined in the presence of its substrate, UDP‐Ara4N. Given the fact that modification of the lipopolysaccharide by Ara4N reduces sensitivity of bacteria to both CAMPS and polymyxin type antibiotics, ArnA is clearly a target for drug design. The model presented here adds new biochemical insight into this enzyme, and the X‐ray coordinates may eventually prove useful in the design of inhibitors with potential therapeutic value.
Materials and Methods
Cloning of the arnA gene
The N‐terminal portion of the bifunctional ArnA gene (residues 1–306) was cloned from E. coli W3110 (ATCC 39936) using Platinum Pfx DNA polymerase (Invitrogen). Primers were designed that incorporated NdeI and XhoI restriction sites. The PCR product was digested with NdeI and XhoI and ligated into pET28T, a laboratory pET28b(+) vector that had been previously modified to incorporate a TEV protease cleavage recognition site after the N‐terminal polyhistidine tag.23
Protein expression and purification
The pET28T‐arnA plasmid was utilized to transform Rosetta2(DE3) E. coli cells (Novagen). The cultures were grown in lysogeny broth supplemented with kanamycin and chloramphenicol at 37°C with shaking until an optical density of 0.8 was reached at 600 nm. The flasks were cooled in an ice bath, and protein expression was initiated with the addition of 1 mM isopropyl β‐d‐1‐thiogalactopyranoside. The cells were then allowed to grow at 22°C for 18 h.
The cells were harvested by centrifugation and disrupted by sonication on ice. The lysate was cleared by centrifugation, and the N‐terminal region of ArnA was purified with Ni‐NTA resin (Qiagen) according to the manufacturer's instructions. The polyhistidine tag was removed by digestion with TEV protease. The protein was dialyzed against 10 mM Tris‐HCl (pH 8.0) and 200 mM NaCl and concentrated to 17 mg/mL based on an extinction coefficient of 0.80 (mg/mL)−1cm−1.
The UDP‐Ara4N substrate utilized in this investigation was prepared in vitro. Specifically the reaction mixtures contained 5 mM UDP‐glucuronic acid, 12 mM NAD+, 40 mM monosodium glutamate, 50 mM HEPPS (pH 8), 0.5 mM pyridoxal 5'‐phosphate, 0.75 mg/mL of the C‐terminal domain of ArnA, and 1.5 mg/mL ArnB (Scheme 1). The reaction was incubated overnight at 22°C. Enzymes were removed by passage through an Amicon Ultra 10 kDa cutoff membrane. The reaction mixture was diluted and loaded onto a HiLoad 26/10 Q Sepharose column and purified by HPLC at a buffered pH of 4.0 with an ammonium acetate gradient of 0‐450 mM over six column volumes. The UDP‐Ara4N product was eluted at a concentration of 250 mM ammonium acetate.
Crystallization and structural analysis
Crystallization conditions were surveyed by the hanging drop method of vapor diffusion using a laboratory based sparse matrix screen at both room temperature and 4°C. X‐ray diffraction quality crystals of the enzyme were subsequently grown from precipitant solutions containing 20–22% poly(ethylene glycol) 5000, 50 mM MgCl2, 5 mM N 5‐formyltetrahydrofolate, 10 mM UDP‐Ara4N, and 100 mM HEPPS (pH 8.0) at 21°C. The crystals belonged to the space group P1 with unit cell dimensions of a = 76.0 Å, b = 76.2 Å, c = 84.9 Å, α = 89.9°, β = 63.8°, and γ = 61.5° The asymmetric unit contained four subunits.
For X‐ray data collection, the crystals were transferred to a cryoprotectant solution containing 30% poly(ethylene glycol) 5000, 250 mM NaCl, 50 mM MgCl2, 15% ethylene glycol, 5 mM N 5‐formyltetrahydrofolate, 10 mM UDP‐Ara4N, and 100 mM HEPPS (pH 8.0). The X‐ray data were collected at the Advanced Photon Source, Structural Biology Center Beamline 19‐BM. These X‐ray data were processed with HKL3000.24 Relevant X‐ray data collection statistics are listed in Table 1. The structure was determined via molecular replacement with the software package PHASER25 and using as a search model the X‐ray coordinates 2BLN10 from the Protein Data Bank. Iterative cycles of model building with COOT and refinement with REFMAC reduced the R work and R free to 21.1% and 27.3%, respectively, from 20 to 2.5 Å resolution.26, 27, 28
Acknowledgments
We thank Professor Peter Tipton for helpful comments. A portion of the research described in this paper was performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source (U. S. Department of Energy, Office of Biological and Environmental Research, under Contract DE‐AC02‐06CH11357).
The authors have no competing financial interests.
X‐ray coordinates have been deposited in the Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, N. J. (accession no. 5J63).
Broader statement: ArnA from Escherichia coli is a key enzyme involved in the formation of 4‐amino‐4‐deoxy‐l‐arabinose. The addition of this sugar to the lipid A moiety of the lipopolysaccharide of pathogenic Gram‐negative bacteria allows these organisms to evade the cationic antimicrobial peptides of the host immune system. Here we present a model for the enzyme in the presence of its UDP‐sugar substrate. This model provides a new scaffold for structure‐based drug design.
References
- 1. Blair JM, Webber MA, Baylay AJ, Ogbolu DO, Piddock LJ (2015) Molecular mechanisms of antibiotic resistance. Nat Rev Microbiol 13:42–51. [DOI] [PubMed] [Google Scholar]
- 2. Peschel A (2002) How do bacteria resist human antimicrobial peptides? Trends Microbiol 10:179–186. [DOI] [PubMed] [Google Scholar]
- 3. Scott MG, Hancock RE (2000) Cationic antimicrobial peptides and their multifunctional role in the immune system. Crit Rev Immunol 20:407–431. [PubMed] [Google Scholar]
- 4. Raetz CR, Whitfield C (2002) Lipopolysaccharide endotoxins. Ann Rev Biochem 71:635–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gunn JS, Ryan SS, Van Velkinburgh JC, Ernst RK, Miller SI (2000) Genetic and functional analysis of a PmrA‐PmrB‐regulated locus necessary for lipopolysaccharide modification, antimicrobial peptide resistance, and oral virulence of Salmonella enterica serovar typhimurium. Infect Immun 68:6139–6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Breazeale SD, Ribeiro AA, Raetz CR (2002) Oxidative decarboxylation of UDP‐glucuronic acid in extracts of polymyxin‐resistant Escherichia coli. Origin of lipid a species modified with 4‐amino‐4‐deoxy‐l‐arabinose. J Biol Chem 277:2886–2896. [DOI] [PubMed] [Google Scholar]
- 7. Gunn JS, Lim KB, Krueger J, Kim K, Guo L, Hackett M, Miller SI (1998) PmrA‐PmrB‐regulated genes necessary for 4‐aminoarabinose lipid A modification and polymyxin resistance. Mol Microbiol 27:1171–1182. [DOI] [PubMed] [Google Scholar]
- 8. Breazeale SD, Ribeiro AA, McClerren AL, Raetz CR (2005) A formyltransferase required for polymyxin resistance in Escherichia coli and the modification of lipid A with 4‐amino‐4‐deoxy‐l‐arabinose. Identification and function of UDP‐4‐deoxy‐4‐formamido‐l‐arabinose. J Biol Chem 280:14154–14167. [DOI] [PubMed] [Google Scholar]
- 9. Gatzeva‐Topalova PZ, May AP, Sousa MC (2004) Crystal structure of Escherichia coli ArnA (PmrI) decarboxylase domain. A key enzyme for lipid A modification with 4‐amino‐4‐deoxy‐l‐arabinose and polymyxin resistance. Biochemistry 43:13370–13379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Williams GJ, Breazeale SD, Raetz CR, Naismith JH (2005) Structure and function of both domains of ArnA, a dual function decarboxylase and a formyltransferase, involved in 4‐amino‐4‐deoxy‐l‐arabinose biosynthesis. J Biol Chem 280:23000–23008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gatzeva‐Topalova PZ, May AP, Sousa MC (2005) Crystal structure and mechanism of the Escherichia coli ArnA (PmrI) transformylase domain. An enzyme for lipid A modification with 4‐amino‐4‐deoxy‐l‐arabinose and polymyxin resistance. Biochemistry 44:5328–5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gatzeva‐Topalova PZ, May AP, Sousa MC (2005) Structure and mechanism of ArnA: conformational change implies ordered dehydrogenase mechanism in key enzyme for polymyxin resistance. Structure 13:929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fischer U, Hertlein S, Grimm C (2015) The structure of apo ArnA features an unexpected central binding pocket and provides an explanation for enzymatic cooperativity. Acta Crystallogr D71:687–696. [DOI] [PubMed] [Google Scholar]
- 14. Almassy RJ, Janson CA, Kan CC, Hostomska Z (1992) Structures of apo and complexed Escherichia coli glycinamide ribonucleotide transformylase. Proc Natl Acad Sci USA 89:6114–6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schmitt E, Blanquet S, Mechulam Y (1996) Structure of crystalline Escherichia coli methionyl‐tRNA(f)Met formyltransferase: comparison with glycinamide ribonucleotide formyltransferase. EMBO J 15:4749–4758. [PMC free article] [PubMed] [Google Scholar]
- 16. Thoden JB, Goneau MF, Gilbert M, Holden HM (2013) Structure of a sugar N‐formyltransferase from Campylobacter jejuni . Biochemistry 52:6114–6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zimmer AL, Thoden JB, Holden HM (2014) Three‐dimensional structure of a sugar N‐formyltransferase from Francisella tularensis . Protein Sci 23:273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Genthe NA, Thoden JB, Benning MM, Holden HM (2015) Molecular structure of an N‐formyltransferase from Providencia alcalifaciens O30. Protein Sci 24:976–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Woodford CR, Thoden JB, Holden HM (2015) New role for the ankyrin repeat revealed by a study of the N‐formyltransferase from Providencia alcalifaciens . Biochemistry 54:631–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu AM, Mackenzie NE (1987) Structural and immunochemical characterization of the O‐haptens of Brucella abortus lipopolysaccharides from strains 19 and 2308. Mol Cell Biochem 75:103–111. [DOI] [PubMed] [Google Scholar]
- 21. Perepelov AV, Liu B, Senchenkova SN, Shashkov AS, Feng L, Knirel YA, Wang L (2010) Structure and gene cluster of the O‐antigen of Salmonella enterica O60 containing 3‐formamido‐3,6‐dideoxy‐d‐galactose. Carbohydr Res 345:1632–1634. [DOI] [PubMed] [Google Scholar]
- 22. Ovchinnikova OG, Liu B, Guo D, Kocharova NA, Bialczak‐Kokot M, Shashkov AS, Feng L, Rozalski A, Wang L, Knirel YA (2012) Structural, serological, and genetic characterization of the O‐antigen of Providencia alcalifaciens O40. FEMS Immunol Med Microbiol 66:382–392. [DOI] [PubMed] [Google Scholar]
- 23. Thoden JB, Holden HM (2005) The molecular architecture of human N‐acetylgalactosamine kinase. J Biol Chem 280:32784–32791. [DOI] [PubMed] [Google Scholar]
- 24. Minor W, Cymborowski M, Otwinowski Z, Chruszcz M (2006) HKL‐3000: the integration of data reduction and structure solution‐from diffraction images to an initial model in minutes. Acta Crystallogr D62:859–866. [DOI] [PubMed] [Google Scholar]
- 25. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Crystallogr D60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 27. Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Crystallogr D66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum‐likelihood method. Acta Crystallogr D53:240–255. [DOI] [PubMed] [Google Scholar]
- 29. Laskowski RA, Moss DS, Thornton JM (1993) Main‐chain bond lengths and bond angles in protein structures. J Mol Biol 231:1049–1067. [DOI] [PubMed] [Google Scholar]
- 30. DeLano WL (2002) The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA, USA, The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA, USA. [Google Scholar]