

Abstract
Here we present the generation and function of two sets of bacterial plasmids that harbor fluorescent genes encoding either blue, cyan, yellow or red fluorescent proteins. In the first set, protein expression is controlled by the strong and constitutive nptII promoter whereas in the second set, the strong tac promoter was chosen that underlies LacIq regulation. Furthermore, the plasmids are mobilizable, contain Tn7 transposons and a temperature-sensitive origin of replication. Using Escherichia coli S17-1 as donor strain, the plasmids allow fast and convenient Tn7-transposon delivery into many enterobacterial hosts, such as the here-used E. coli O157:H7. This procedure omits the need of preparing competent recipient cells and antibiotic resistances are only transiently conferred to the recipients. As the fluorescence proteins show little to no overlap in fluorescence emission, the constructs are well suited for the study of multicolored synthetic bacterial communities during biofilm production or in host colonization studies, e.g. of plant surfaces. Furthermore, tac promoter-reporter constructs allow the generation of so-called reproductive success reporters, which allow to estimate past doublings of bacterial individuals after introduction into environments, emphasizing the role of individual cells during colonization.
Keywords: fluorescent proteins, Escherichia coli O157:H7, reproductive success, CUSPER
We present the generation and function of two sets of bacterial plasmids encoding different fluorescent proteins to generate synthetic communities and reproductive success bioreporters in Enterobacteriaceae.
INTRODUCTION
To study bacteria at the micrometer scale or single-cell resolution (Kreft et al.2013), it is imperative to visualize them. This can be achieved by microscopy in conjunction with fluorescent markers, e.g. dyes such as propidium iodide and SYTO9 allowing to determine the membrane integrity of bacterial cells (Berney et al.2007), fluorescence in situ hybridization (Remus-Emsermann et al.2014), or by labeling bacteria using fluorescent proteins (FPs) (Ledermann et al.2015). Often, FPs were used under the control of inducible promoters as bioreporters, which allow the assessment of the availability of inducing agents, such as fructose (Leveau and Lindow 2001a), or under the control of constitutive promoters to identify bacteria in situ (Bloemberg et al.2000, Ledermann et al.2015).
The use of miniTn7-transposon delivery plasmids is widespread in molecular and environmental microbiology, e.g. to chromosomally integrate promoter-reporter gene constructs (Choi and Schweizer 2006, McKenzie and Craig 2006, Lagendijk et al.2010) or for the complementation of knockout mutants (Crepin, Harel and Dozois 2012). The bacterial Tn7 transposon provides an excellent non-destructive tool for several reasons: (i) it integrates with high affinity at the attTn7 site within a wide range of Gram-negative bacteria; (ii) it does not disrupt genes, omitting potential pleiotropic effect; (iii) and antibiotics are not required to maintain the inserted transposons (Choi and Schweizer 2006, Choi and Kim 2009). Most Tn7-transposon delivery systems have in common that they employ a suicide plasmid-based approach using vectors carrying a pUC origin of replication (Lambertsen, Sternberg and Molin 2004, Choi and Schweizer 2006, Lagendijk et al.2010). Plasmids featuring this origin of replication do not replicate in bacterial recipients and classically, the transposon machinery is provided on a helper plasmid. In Enterobacteriaceae, however, the used pUC origin of replication works efficiently, thereby excluding Escherichia coli and Salmonella spp. as possible recipients.
Using a temperature-sensitive Tn7-delivery system constructed by McKenzie and Craig (2006), we provide a ready-to-use alternative for fluorescent labeling of Enterobacteriaceae. We provide two different promoters, the synthetic, constitutive, LacIq-repressible tac promoter (Ptac) (de Boer, Comstock and Vasser 1983) and the constitutive nptII promoter (PnptII) (Ledermann et al.2015) in combination with fluorescent marker genes, encoding blue, cyan, yellow or red FPs. In combination with two-parental mating employing E. coli S17-1, which provides the tra operon necessary for the mobilization of plasmids as a chromosomal insertion, we overcome the often tedious low efficiency of transposition and preparation of competent recipient cells. The here-proposed sets of plasmids offer a convenient alternative to generate multicolored sets of stably fluorescently labeled bacteria. The FPs allow for multichannel fluorescent microscopy analysis with little to no overlap of FP emissions to study multistrain synthetic communities (Fig. S2, Supporting Information).
Employing the repressability of the Ptac by LacIq, e.g. by coexpressing LacIq, it is possible to construct so-called reproductive success, or CUSPER, bioreporter strains (reproductive success backwards = CUSPER) (Remus-Emsermann and Leveau 2010, Remus-Emsermann et al.2012, Remus-Emsermann, Kowalchuk and Leveau 2013). CUSPER bioreporters are equipped with a Ptac-controlled FP and express LacIq, thereby the expression of the FP is repressed. When derepressing the expression, i.e. by adding lactose or isopropyl β-D-1-thiogalactopyranoside to growing cells, cells can be loaded with FP (Leveau and Lindow 2001b). If the derepressor is subsequently removed, no de novo production of FP occurs and the preformed FP is then diluted from growing cells (Remus-Emsermann and Leveau 2010). The fluorescence intensity of growing cells relative to their ancestors thereby becomes a proxy for the number of divisions individual cells underwent (Remus-Emsermann and Leveau 2010). CUSPER bioreporters were used to study the probability of successful colonization of bacterial cells on plant leaves, the heterogeneity of microhabitats on plant leaf surface, the probability of secondary bacterial colonization on precolonized leaves (Remus-Emsermann and Leveau 2010, Remus-Emsermann et al.2012, Remus-Emsermann, Kowalchuk and Leveau 2013), and to estimate reproductive success in a spatial context in mice spleens and plant leaves (Helaine et al.2010, Tecon and Leveau 2012).
Using the model strain E. coli O157:H7 Δstx, we demonstrate the transposon delivery of the promoter fluorescence reporter constructs and resulting fluorescence at the population and single-cell resolution.
MATERIAL AND METHODS
Bacteria and growth conditions
Escherichia coli NEB 5-alpha (New England Biolabs, Ipswich, MA, USA), E. coli S17-1 and E. coli O157:H7 Δstx (NCTC 12900) were routinely grown on Lysogeny broth (LB) or LB agar at 37°C. To prevent plasmids loss, all pGRG36 plasmid-carrying cells were grown at 32°C. For counterselection of auxotroph E. coli S17-1 after mating, MM2 medium agar (4 g L−1 L-asparagine, 2 g L 1 K2HPO4, 0.2 g L−1 MgSO4, 3 g L−1 NaCl, 10 g L−1 sorbitol, 15 g L−1 agar) was used. Where necessary, media were supplemented with 100 μg mL−1 ampicillin, 50 μg mL−1 kanamycin or 20 μg mL−1 tetracycline. For microtiter plate reader experiments, cells were grown in M9 minimal medium (20 mL L−1 20% (w/v) casamino acids (Amresco), 40 mL L−1 10% (w/v) carbon source, 2 mL L−1 1 M MgSO4, 1 mL L−1 0.1 M CaCl2, 100 mL L−1 10 × M9 salts (85.1 g L−1 Na2HPO4 × 2 H2O, 30 g L−1 KH2PO4, 5 g L−1 NaCl, 10 g L−1 NH4Cl, pH 7)) containing either glucose or lactose as sole carbon source.
Plasmid construction
All plasmids used in this study are given in Table 1, a generic map of all produced plasmids is given in Fig. 1. All PCRs were conducted using Phusion polymerase (New England Biolabs, Ipswich, MA, USA) following the manufacturer's recommendations and annealing temperatures were chosen based on the respective melting temperature of the primers (Table 2). All plasmids that were used for PCR amplification were isolated using the NucleoSpin plasmid extraction kit (Macherey-Nagel, Oensingen, Switzerland) following the manufacturer's recommendations. The designated plasmid backbone of the herein constructed plasmids, pGRG36 (a gift from Nancy Craig (Addgene plasmid # 16666)), was isolated using the NucleoBond Xtra Plus Midiprep kit (Macherey-Nagel, Oensingen, Switzerland) following the manufacturer's recommendations. All restriction enzymes were acquired at New England Biolabs.
Table 1.
Plasmids and bacterial strains used in this study.
| Name | Promoter and fluorescent gene | Antibiotic resistance | Reference |
|---|---|---|---|
| pGRG36 | Not applicable | Ampicillin | McKenzie and Craig (2006) |
| pUC18T-mini-Tn7T-Zeo-ecfp | tac promoter, eCFP | Ampicillin, Zeocin | Choi and Schweizer (2006) |
| pUC18T-mini-Tn7T-Zeo-eyfp | tac promoter, eYFP | Ampicillin, Zeocin | Choi and Schweizer (2006) |
| pMP7607 | tac promoter, mCherry | Kanamycin | Lagendijk et al. (2010) |
| pFru97 | nptII promoter, dsRed | Kanamycin | Tecon and Leveau (2012) |
| pRJaph_eBFP2 | nptII promoter, eBFP2 | Tetracycline | Ledermann et al. (2015) |
| pRJaph_mtq2 | nptII promoter, mTurquoise2 | Tetracycline | Ledermann et al. (2015) |
| pRJaph_mChe | nptII promoter, mCherry | Tetracycline | Ledermann et al. (2015) |
| pCPP39 | Not applicable | Tetracycline | Leveau and Lindow (2001b) |
| pMRE100 | tac promoter, mCherry | Ampicillin | This study |
| pMRE101 | tac promoter, eCFP | Ampicillin | This study |
| pMRE102 | tac promoter, eYFP | Ampicillin | This study |
| pMRE103 | nptII promoter, mCherry | Ampicillin | This study |
| pMRE104 | nptII promoter, eCFP | Ampicillin | This study |
| pMRE105 | nptII promoter, eYFP | Ampicillin | This study |
| pMRE106 | nptII promoter, eBFP2 | Ampicillin | This study |
| pMRE107 | nptII promoter, mTurquoise2 | Ampicillin | This study |
| E. coli O157:H7::mre100 | Chromosomally inserted, inducible mCherry gene | This study | |
| E. coli O157:H7::mre101 | Chromosomally inserted, inducible eCFP gene | This study | |
| E. coli O157:H7::mre102 | Chromosomally inserted, inducible eYFP gene | This study | |
| E. coli O157:H7::mre103 | Chromosomally inserted, constitutive mCherry gene | This study | |
| E. coli O157:H7::mre104 | Chromosomally inserted, constitutive eCFP gene | This study | |
| E. coli O157:H7::mre105 | Chromosomally inserted, constitutive eYFP gene | This study | |
| E. coli O157:H7::mre106 | Chromosomally inserted, constitutive eBFP2 gene | This study | |
| E. coli O157:H7::mre107 | Chromosomally inserted, constitutive mTurquoise2 gene | This study |
Figure 1.
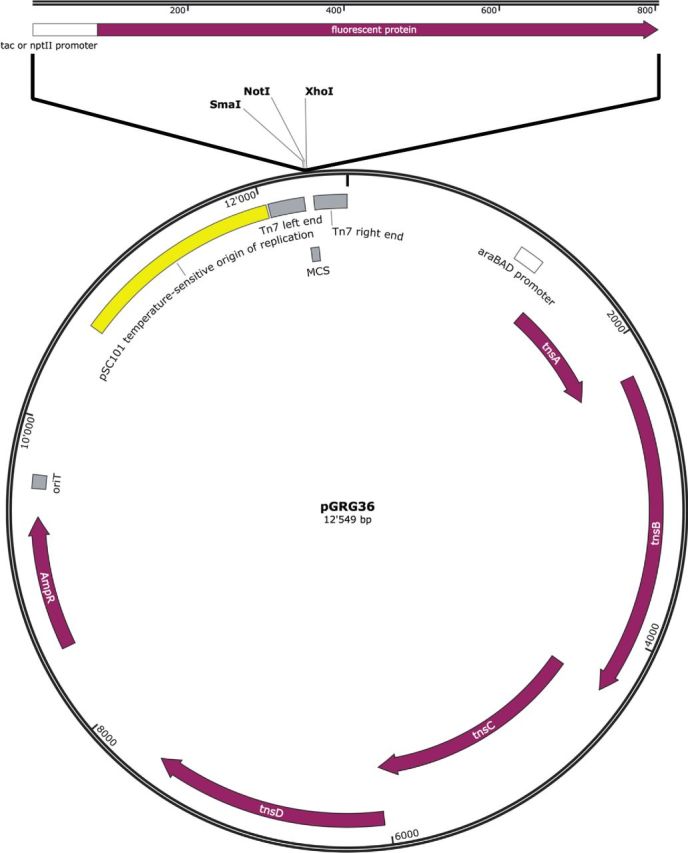
Schematic representation of pMRE100-pMRE107 plasmids. The map shows pGRG36 including the insertion site of the different promoter FP constructs and other relevant features. araBAD promoter = arabinose-inducible promoter, tnsA-D = transposase genes, AmpR = ampicillin resistance conferring beta-lactamase, oriT = origin of transfer, pSC101 = origin of replication, Tn7 left end = left border of Tn7 transposon, MCS = multiple cloning site, Tn7 right end = right border of Tn7 transposon.
Table 2.
Primers used in this study.
| Primer name | Sequence 5′- 3′ | Tm* (°C) | Target and notes |
|---|---|---|---|
| Xho_Ptac_mChe.for | AAA ACT CGA GGG GGA ATT CTT GAC AAT TAA TCA TC | 60 | tac promoter; includes a 5′ XhoI restriction site |
| NotI_mChe.rev | AAA GCG GCC GCA AAA ACC GCC CTG CAA GG | 69 | mCherry; includes a 3′ NotI restriction site |
| Ptac_C/YFP.for | AAA ACT CGA GGG GGA ATT CTT GAC AAT TAA TCA TCG GCT CGT ATA ATG TGT GGA ATT GTG AGC GGA TAA CAA TTT TCA CAC AGG AAA CAG CTA AAT GCT GAG CAA GGG CG | 56 | CFP and YFP; the tac promoter is included in the primer |
| Ptac_C/YFP.rev | AAA AGC GGC CGC TAT TAC TTG TAC AGC TCG TCC ATG | 55 | eYFP and eCFP |
| Gib_Ptac+C/YFP.fwd | GTT TTA ATT AAT CAG ATC CCG GGA ATT CTT GAC AAT TAA TC | 48 | tac promoter; contains overlap to SmaI digested pGRG36 |
| Gib_Ptac+C/YFP.rev | GTG GCG GCC GCT ATT GAC CCT TAC TTG TAC AGC TCG TCC | 52 | eCFP and eYFP; contains overlap to SmaI digested pGRG36 |
| Gib_PnptII.fwd | CAC GTT TTA ATT AAT CAG ATC CCA ATT GGG GAT CGG AAG CTT GAC | 58 | nptII promoter from pFru97; contains overlap to SmaI digested pGRG36 |
| Gib_PnptII.rev | CCT TTG CTC ATA TGT TTT TCC TCC TTA TAA AGT TAA TCT TTA GTT AGT TAG GG | 59 | nptII promoter from pFru97; contains overlap to mCherry |
| Gib_mChe.fwd | GGA AAA ACA TAT GAG CAA AGG AGA AGA AGA TAA CAT GG | 58 | mCherry; contains overlap to the nptII promoter |
| Gib_mChe.rev | GGC CGC TAT TGA CCC TTA TTT GTA AAG CTC ATC CA | 47 | mCherry; contains overlap to SmaI digested pGRG36 |
| Gib_PnptII_C/YFP.for | GAA AAA CAT ATG CTG AGC AAG GGC GAG | 62 | eCFP and eYFP; contains overlap to the nptII promoter |
| Gib_PnptII_C/YFP.rev | CTG TAC AAG TAA GGG TCA ATA GCG GCC | 62 | eCFP and eYFP; contains overlap to SmaI digested pGRG36 |
| Gib_C/YFP_PnptII.rev | TGC TCA GCA TAT GTT TTT CCT CCT TAT AAA GTT AAT CTT TAG TTA GTT AGG G | 59 | nptII promoter from pFru97; contains overlap to eCFP and eYFP |
| Gib_PnptII_ngFPs.for | GGC CGC TAT TGA CCC TTA CTT GTA CAG CTC GTC CAT G | 55 | nptII promoter from pRJaph plasmids; contains overlap to SmaI digested pGRG36 |
| Gib_ngFPs.rev | CAC GTT TTA ATT AAT CAG ATC CCC ACG CTG CCG CAA GC | 55 | mTurquoise and eBFP; contains overlap to SmaI digested pGRG36 |
*Tm specific to the overlap to the specified targets, overlaps for isothermal assembly were designed to anneal at 50°C; ABR = antibiotic resistance: Amp = Ampicillin, Kan = Kanamycin, Zeo = Zeocin, Tet = Tetracycline.
As source of Ptac mCherry gene fragment, plasmid pMP7607 was used. The fragment was amplified and equipped with restriction sites XhoI and NotI using primers Xho_Ptac_mChe_for and NotI_mChe_rev. The resulting amplicon was NotI/XhoI digested and ligated into equally digested pGRG36 to generate pMRE100-Ptac-mChe. The ligation reaction was transformed into E. coli NEB 5-alpha chemically competent cells following the manufacturer's recommendations and selected on LB agar containing ampicillin. To construct pMRE101-Ptac-eCFP and pMRE102-Ptac-eYFP, eCFP and eYFP were amplified from pUC18T-miniTn7-Zeo-eCFP and pUC18T-miniTn7-Zeo-eYFP, respectively, using primers Ptac_C/YFP.for, which contains Ptac, and Ptac_C/YFP.rev. The resulting amplicons were amplified using primers Gib_Ptac+C/YFP.fwd and Gib_Ptac+C/YFP.rev to generate amplicons with overlaps to SmaI-digested pGRG36. Each amplicon was fused into pGRG36 using isothermal assembly as described by Gibson et al. (2009). Briefly, amplicons were mixed in equimolar ratios with 100 ng SmaI-linearized pGRG36 each in a total of 5 μL before 15 μL isothermal assembly reaction mix was added (for 1.2 mL isothermal assembly mix, combine 320 μL isothermal assembly buffer (25% PEG-8000 (Amresco, Cleveland, OH, USA), 500 mM Tris-HCl pH 7.5 (Sigma, Saint Louise, MO, USA), 50 mM MgCl2 (Rockland, Limerick, PA, USA), 50 mM DTT (Amresco, Cleveland, OH, USA), 1 mM each of the four dNTPs (Amresco, Cleveland, OH, USA) and 5 mM NAD) with 1.2 μL T5 Exonuclease (New England Biolabs, Ipswich, MA, USA), 20 μL Phusion polymerase (New England Biolabs, Ipswich, MA, USA), 160 μL Taq DNA ligase (New England Biolabs, Ipswich, MA, USA) and 700 μL ddH2O). After incubation at 50°C for 15 min, 10 μL of the reactions were used to transform E. coli NEB 5-alpha chemically competent cells.
To construct pMRE103-PnptII-mChe, PnptII was amplified from pFru97 using primers Gib_PnptII.fwd and Gib_PnptII.rev, yielding an amplicon with 5′ overlap to SmaI-digested pGRG36 and 3′ overlap to the 5′ end of Gib_mChe.fwd and Gib_mChe.rev amplified mCherry. The two amplicons and SmaI-digested pGRG36 were isothermal assembled as described above, yielding pMRE103-PnptII-mChe. pMRE104-PnptII-eCFP and pMRE105-PnptII-eYFP were constructed by amplifying PnptII from pFru97 using primers Gib_PnptII.for and Gib_C/YFP_PnptII.rev and amplifying eCFP and eYFP from pMRE101-Ptac-eCFP and pMRE102-Ptac-eYFP, respectively, using primers Gib_PnptII_C/YFP.for and Gib_PnptII_C/YFP.rev. The resulting amplicons were assembled into SmaI-digested pGRG36 as described above. Plasmids pMRE106-PnptII-eBFP2 and pMRE107-PnptII-mTq2 were constructed by amplifying the respective fluorophore genes and PnptII using primers Gib_PnptII_ngFPs.for and Gib_ngFPs.rev from pRJaph_eBFP2 or pRJaph_mTq2. The resulting amplicons were isothermal assembled with SmaI-digested pGRG36 as described above.
All generated plasmids were purified from E. coli NEB 5-alpha, verified by PCR sequencing and transformed into E. coli S17-1 to allow two-parental mating of the mobilizable pMRE-series plasmids. Plasmid sequences can be found in the supplementary material. Plasmid nucleotide sequences were deposited under GenBank accession numbers: KU973693–KU973700.
Two-parental mating and Tn7 transposition
For two-parental mating, donor E. coli S17-1 containing miniTn7-delivery plasmids were grown overnight as a lawn on LB agar containing ampicillin at 32°C and recipient E. coli O157:H7 were grown overnight as a lawn on LB agar at 37°C. Freshly grown lawns of donor and recipient were harvested using inoculation loops, resuspended in 1×PBS (8 g L−1 NaCl, 0.24 g L−1 KCl, 1.42 g L−1 Na2HPO4, 0.24 g L−1 KH2PO4), washed twice by centrifugation at 3500 × g and resuspension in 1×PBS. Donor and recipient were mixed to reach the same number of cells before they were harvested by centrifugation at 3500 × g. The donor/recipient mixture was resuspended in 1×PBS to reach a final OD600 nm of ∼20. 100 μL of this suspension was spotted onto LB containing no antibiotics and left to dry in a laminar flow. Afterwards, spotted cell mixes were incubated at 32°C overnight before they were harvested using an inoculation loop. Harvested cells were resuspended in 1 × PBS and plated on MM2 containing ampicillin where they were incubated at 32°C, preventing auxotrophic E. coli S17-1 to grow. Transconjugants were picked from MM2 and propagated on LB agar containing ampicillin. Tn7 integration was then performed as described by McKenzie and Craig (2006). Briefly, E. coli were cultivated in LB containing ampicillin at 220 rpm and 32°C overnight to promote transposition (in E. coli leaky expression of the arabinose promoter-driven TnsABCD, transposase genes were sufficient for transposition into attTn7 locus, for other species and strains it might be advantageous to add 0.1% arabinose to the medium to induce the transposons’ promoter). Overnight cultures were plated onto LB agar containing no ampicillin and were cultivated at 42°C to block replication of the heat-sensitive plasmid. Colonies growing on non-selective agar were screened for their ability to fluoresce on a fluorescence microscope. To verify that the ampicillin resistance conferring plasmid was lost, 10 individual colonies were streaked onto LB agar containing the antibiotic as well as onto LB agar plates without. Colonies that were not able to grow on ampicillin but grew on LB were screened once more for their ability to fluoresce.
FP production from chromosomally inserted genes followed in microtiter plates
Bacterial strains containing chromosomally inserted FPs under the control of Ptac were cultivated in M9 containing lactose and casamino acids or in M9 containing glucose and casamino acids. Strains containing chromosomally inserted FPs under the control of PnptII were cultivated in M9 containing glucose and casamino acids. Strains were cultivated in triplets of 300 μL at 28°C and 3 s of orbital shaking every 10 min in a polystyrene, 96-well, flat bottom microtiter plate (Greiner Bio-One, Frickenhausen, Germany) in an Infinite M200 microtiter plate reader (Tecan, Männedorf, Switzerland). The initial density of the cultures was between 0.001 and 0.005 OD600 as measured by the plate reader. Growth was determined by monitoring absorbance at 600nm. Fluorescence was determined by exciting the culture at 383 ± 9 nm, 420 ± 9 nm, 497 ± 9 or 560 ± 9 nm and measuring emission at 448 ± 20 nm, 481 ± 20 nm, 533 ± 20 nm or 610 ± 20 nm for eBFP2, cyan FPs (eCFP and mTurquoise2), eYFP or mCherry, respectively. Fluorescence and absorbance were determined every 10 min for a total of 48 h or 288 cycles, respectively. The determined fluorescence intensity was background subtracted using the respective T0 fluorescence intensity.
Microscopy
Microscopy was performed on a Zeiss AxioImager.Z2 microscope equipped with a Zeiss Axiocam MRm for image acquisition. Images were acquired using the Zeiss Zen 2012 blue software. eBFP2 was visualized using Zeiss filter set 49 (G 365 nm/FT 395 nm/BP 445/50 nm), eCFP and mTurquoise2 were visualized using Zeiss filter set 47 HE (BP 436/25 nm/FT 455 nm/BP 480/40 nm), eYFP was visualized using Zeiss filter set 46 HE (BP 500/25 nm/FT 515 nm/BP 535/30 nm) and mCherry was visualized using Zeiss filter set 43 HE (BP 550/25 nm/FT 570 nm/BP 605/70 nm). To visualize fluorescence of individual bacterial cells, an EC Plan-Neofluar 100× objective (1.30 NA, Oil, Ph3) or an EC Plan-Neofluar 40× objective (0.75 NA, air, Ph2) was used. Individual colonies were investigated using a Plan-Apochromat 5× objective (0.16 NA).
To measure fluorescence intensity of individual cells, freshly grown colonies were harvested using an inoculation loop and resuspended in 1 × PBS. Serial dilutions of the suspensions were spotted onto gelatin coated 10-well microscope slides (Thermo Scientific, Dreieich, Germany) and dried for 15 min to bind cells to the surface of the slide. Dilutions of appropriated densities, e.g. containing several well-separated individual cells per field of view at 100× magnification, were further investigated. Single-cell fluorescence intensity was determined of at least 100 cells per strain by acquiring multichannel images of respective fluorescence signals and phase contrast signals. Multichannel images were imported to the program Fiji (Schindelin et al.2012). Cells were separated from the background based on their phase contrast using the Fiji thresholding command and standard settings. Cells were added to the Fiji region of interest manager using the analyze particles command. The ‘multimeasure’ command of the region of interest manager was then used to determine the average fluorescence of individual cells.
RESULTS AND DISCUSSION
When grown in media lacking lactose, Escherichia coli O157:H7 cells harboring ::MRE100, ::MRE101 or ::MRE102 exhibited weak fluorescence (Fig. 2) due to the presence of the Ptac repressor gene lacIq in E. coli O157:H7. Addition of lactose to minimal medium led to the induction of FPs in E. coli O157:H7 harboring ::MRE-Ptac constructs causing an accumulation of FP in cells compared to their repressed controls as shown by microscopy and microtiter plate reader (Fig. 2). Chromosomally inserted mCherry in E. coli O157:H7::MRE100 exhibited a significantly stronger signal after induction and the strongest observed signal of the here-introduced reporter set (Fig. 2B). Escherichia coli O157:H7::MRE101 expressing eCFP exhibited a slightly stronger, however not significant, fluorescence signal after induction, which could be shown only using microscopy. Possibly, eCFPs short excitation wavelength led to a high autofluorescent signal of the medium and/or microtiter plate plastic in the plate reader which decreased the signal-to-noise ratio. Lastly, E. coli O157:H7::MRE102 expressing eYFP exhibited the second strongest fluorescence, however not significantly increased, signal after induction.
Figure 2.

Analysis of chromosomally inserted Ptac fluorescence protein gene labels using microscopy and microtiter plate reader. (A) Phase contrast and fluorescence micrographs of E. coli O157:H7 carrying chromosomally integrated Tn7-transposons containing mCherry, eCFP or eYFP encoding genes under the control of the lactose derepressible tac promoter. The left two columns show phase contrast and fluorescence images of non-induced cells, the right two columns show lactose-induced cells. For fair comparisons of fluorescence intensities and background, a linear contrast was applied to the images. (Exposure times for the respectively measured fluorophores are given in fluorescence micrographs, scale bar = 5 μm.) (B) Average single-cell fluorescence intensity per millisecond exposure after background subtraction. Non-induced cells are represented as white bars, and induced cells as black bars. Statistical differences in fluorescence intensity between treatments were assessed by performing a one-way ANOVA. ** = P < 0.01; **** = P < 0.0001. (C) Background-subtracted fluorescence of E. coli O157:H7 carrying the Tn7 insertions or wild-type cells cultivated under non-induced or induced conditions. Lines reflect floating averages of three replicates; stippled lines reflect the standard deviation of the mean.
These results show that the repression of the Ptac by the in E. coli endogenously present LacIq was not sufficient to tightly control the activity of Ptac as also non-induced bacterial cells exhibited fluorescence, albeit at lower average intensities, which were, however, only significantly lower in the case of mCherry expressing E. coli O157:H7::MRE100 (Fig. 2B). To show that the activity of Ptac can be controlled more tightly, the Ptac repressor lacIq was expressed from the multicopy plasmid pCPP39 (Leveau and Lindow 2001b, Remus-Emsermann and Leveau 2010) and transformed into E. coli O157:H7::MRE100. In E. coli O157:H7::MRE100 (pCPP39), absolutely no fluorescence was detected in cells grown on minimal medium containing glucose as sole carbon, while cells grown on minimal medium containing lactose as sole carbon were fluorescent (Fig. S1, Supporting Information). This tightly controlled gene expression system can be employed to generate CUSPER bioreporters (Remus-Emsermann and Leveau 2010, Remus-Emsermann et al.2012, Remus-Emsermann, Kowalchuk and Leveau 2013). Cells can then be loaded with a fluorophore by adding the derepressor and after its removal, the constitutive repression abolishes the de novo production of the FP. The now FP-loaded cells can be used as CUSPER bioreporters, i.e. while growing, they will evenly dilute the FP to daughter cells, which will contain less FP than the mother cell.
All FPs that were placed under the control of PnptII were functionally expressed in E. coli O157:H7 while present on plasmids (data not shown) or after chromosomal insertion (Fig. 3). However, the different protein varieties resulted in fluorescent cells yielding different fluorescent intensities. The strongest discernable fluorescence, as assessed by microscopy and microtiter plate reader, was exhibited by mCherry expressing E. coli O157:H7::MRE103, followed by cells E. coli O157:H7::MRE107 expressing mTurquoise2, E. coli O157:H7::MRE105 expressing eYFP, E. coli O157:H7::MRE104 expressing eCFP and finally E. coli O157:H7::MRE106 expressing eBFP2. Notably, the fluorescence of chromosomally inserted eBFP2 was very low using the available fluorescence microscope and plate reader, with signals barely above the limit of detection. Furthermore, the fluorescence of cells expressing eCFP was far inferior to cells expressing mTurquoise2.
Figure 3.
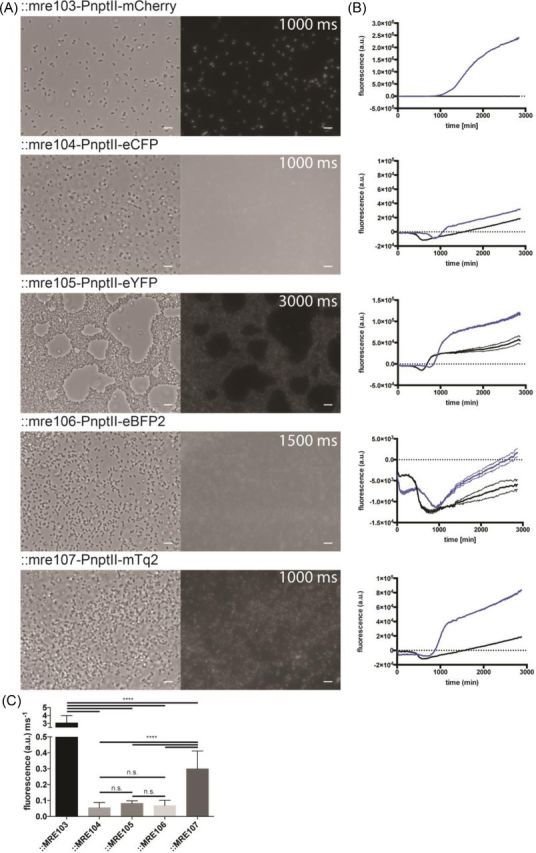
(A) Phase contrast and fluorescence micrographs of E. coli O157:H7 carrying chromosomally integrated Tn7 transposons containing mCherry, eCFP, eYFP, eBFP2 or mTurquoise2 encoding genes under the control of the constitutive nptII promoter. The first column shows phase contrast images, the second corresponding fluorescence images (exposure times are given in the fluorescent images, scale bars = 5 μm). For fair comparisons of fluorescence intensities and background, a linear contrast was applied to the images. (B) Fluorescence intensity of E. coli O157:H7 carrying the Tn7 insertions or wild-type cells. Lines reflect the mean of three replicates; stippled lines reflect the standard deviation of the mean. (C) Average single-cell fluorescence intensity per millisecond exposure after background subtraction. Statistical differences in fluorescence intensity between treatments were assessed by performing a one-way ANOVA, and significant differences are indicated in the graph. **** = P < 0.0001.
The here-presented plasmids feature a narrow-host range origin of replication and should be functional in many relevant Enterobacteriaceae including E. coli, Salmonella spp. and Shigella spp (McKenzie and Craig 2006). The Tn7-transposon delivery machinery and promoters are considered to be functional in a broad host range including all Enterobacteriaceae (Miller, Leveau and Lindow 2000, Peters and Craig 2001). Thereby the plasmids are of value for many different studies and allow to rapidly setup and perform experiments at single-cell resolution, for example, the investigation of multispecies biofilms and interactions within (Sternberg et al.1999, Stewart and Franklin 2008, Burmølle et al.2014) and bacterial behavior in microbe–microbe or microbe–host interactions (Bloemberg et al.2000, Remus-Emsermann, Kowalchuk and Leveau 2013, Janissen et al.2015, Ledermann et al.2015). As the promoter-reporter constructs are inserted chromosomally, there is no need for antibiotic selection to maintain the labels. Furthermore, by avoiding the integration of antibiotic resistances it is possible to add additional genetic elements that require antibiotics pressure, such as plasmids or transposons, in the host bacteria.
With non-optimized wide-field microscopy systems, it is possible to combine the following three FPs to allow for overlap-free observations of subpopulations of bacteria: eCFP/mTurquoise2, eYFP and mCherry (Fig. 4; Fig. S2, Supporting Information). To add a fourth population, i.e. an eBFP2-labeled strain, it is necessary to optimize the detection system by using optimized fluorescence emission and excitation filters or a filter-free confocal microscopy system. The observation of several subpopulations at the same time allows for the application of spatial statistics (Daims, Lücker and Wagner 2006, Remus-Emsermann et al.2014), which will give deep insights into the behavior of bacterial strains towards each other in a given environment, e.g. if they aggregate or segregate.
Figure 4.

Artificial mixtures of E. coli O157:H7 containing different Tn7-transposon inserted promoter fluorescent-protein labels. (A) A mix of induced E. coli O157:H7::MRE100, E. coli O157:H7::MRE101 and E. coli O157:H7::MRE102. Left image, phase contrast image, right image, false color overlay of mCherry (red), eYFP (green) and eCFP (blue). The contrast of each channel was adjusted. (B) A mix of E. coli O157:H7::MRE103, E. coli O157:H7::MRE105 and E. coli O157:H7::MRE107. Left image, phase contrast image; right image, false color overlay of mCherry (red), eYFP (green) and mTurquoise2 (blue). The contrast of each channel was adjusted. The scale bars represent 10 μm.
CONCLUSION
With this novel set of FP labels harbored on miniTn7-transposon delivery plasmids in combination with the conjugation strain E. coli S17-1, we provide a convenient ready-to-go tool to generate fluorescently labeled enterobacterial strains for microbe–microbe and microbe–host interaction studies as well as reproductive success bioreporters.
AUTHOR CONTRIBUTIONS
MRE and DD conceived the study, MRE planned the experiments, MRE and PG performed the experiments, MRE analyzed the data and MRE wrote the manuscript with critical input from DD.
Supplementary Material
Acknowledgments
Daniela Remus is acknowledged for critically reading and commenting on the manuscript. The authors thank Raphael Ledermann and Hans-Martin Fischer for providing fluorescent spectra data of eBFP2 and mTurquoise2. The authors also thank Johan Leveau for pFru97 and pCPP39, Ellen Lagendjik for pMP7607, Herbert Schweizer for pUC18T-miniTn7-Zeo-eCFP and pUC18T-miniTn7-Zeo-eYFP and Nancy Craig for access to pGRG36.
SUPPLEMENTARY DATA
FUNDING
This work was financially supported by the and persistent Microorganisms along food chains (REDYMO).
Conflict of interest. None declared.
REFERENCES
- Berney M, Hammes F, Bosshard F, et al. Assessment and interpretation of bacterial viability by using the LIVE/DEAD BacLight kit in combination with flow cytometry. Appl Environ Microb. 2007;73:3283–90. doi: 10.1128/AEM.02750-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloemberg GV, Wijfjes AHM, Lamers GEM, et al. Simultaneous imaging of Pseudomonas fluorescens WCS365 populations expressing three different autofluorescent proteins in the rhizosphere: new perspectives for studying microbial communities. Mol Plant Microbe In. 2000;13:1170–6. doi: 10.1094/MPMI.2000.13.11.1170. [DOI] [PubMed] [Google Scholar]
- Burmølle M, Ren D, Bjarnsholt T, et al. Interactions in multispecies biofilms: do they actually matter? Trends Microbiol. 2014;22:84–91. doi: 10.1016/j.tim.2013.12.004. [DOI] [PubMed] [Google Scholar]
- Choi KH, Kim KJ. Applications of transposon-based gene delivery system in bacteria. J Microbiol Biotechn. 2009;19:217–28. doi: 10.4014/jmb.0811.669. [DOI] [PubMed] [Google Scholar]
- Choi KH, Schweizer HP. mini-Tn7 insertion in bacteria with single attTn7 sites: example Pseudomonas aeruginosa. Nat Protoc. 2006;1:153–61. doi: 10.1038/nprot.2006.24. [DOI] [PubMed] [Google Scholar]
- Crepin S, Harel J, Dozois CM. Chromosomal complementation using Tn7 transposon vectors in Enterobacteriaceae. Appl Environ Microb. 2012;78:6001–8. doi: 10.1128/AEM.00986-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daims H, Lücker S, Wagner M. daime, a novel image analysis program for microbial ecology and biofilm research. Environ Microbiol. 2006;8:200–13. doi: 10.1111/j.1462-2920.2005.00880.x. [DOI] [PubMed] [Google Scholar]
- de Boer HA, Comstock LJ, Vasser M. The tac promoter: a functional hybrid derived from the trp and lac promoters. P Natl Acad Sci USA. 1983;80:21–5. doi: 10.1073/pnas.80.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang R-Y, et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–5. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- Helaine S, Thompson JA, Watson KG, et al. Dynamics of intracellular bacterial replication at the single cell level. P Natl Acad Sci USA. 2010;107:3746–51. doi: 10.1073/pnas.1000041107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janissen R, Murillo DM, Niza B, et al. Spatiotemporal distribution of different extracellular polymeric substances and filamentation mediate Xylella fastidiosa adhesion and biofilm formation. Sci Rep. 2015;5:9856. doi: 10.1038/srep09856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreft J-U, Plugge CM, Grimm V, et al. Mighty small: Observing and modeling individual microbes becomes big science. P Natl Acad Sci USA. 2013;110:18027–8. doi: 10.1073/pnas.1317472110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagendijk EL, Validov S, Lamers GE, et al. Genetic tools for tagging Gram-negative bacteria with mCherry for visualization in vitro and in natural habitats, biofilm and pathogenicity studies. FEMS Microbiol Lett. 2010;305:81–90. doi: 10.1111/j.1574-6968.2010.01916.x. [DOI] [PubMed] [Google Scholar]
- Lambertsen L, Sternberg C, Molin S. Mini-Tn7 transposons for site-specific tagging of bacteria with fluorescent proteins. Environ Microbiol. 2004;6:726–32. doi: 10.1111/j.1462-2920.2004.00605.x. [DOI] [PubMed] [Google Scholar]
- Ledermann R, Bartsch I, Remus-Emsermann MNP, et al. Stable fluorescent and enzymatic tagging of Bradyrhizobium diazoefficiens to analyze host-plant infection and colonization. Mol Plant Microbe In. 2015;28:959–67. doi: 10.1094/MPMI-03-15-0054-TA. [DOI] [PubMed] [Google Scholar]
- Leveau JHJ, Lindow SE. Appetite of an epiphyte: Quantitative monitoring of bacterial sugar consumption in the phyllosphere. P Natl Acad Sci USA. 2001a;98:3446–53. doi: 10.1073/pnas.061629598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leveau JHJ, Lindow SE. Predictive and interpretive simulation of green fluorescent protein expression in reporter bacteria. J Bacteriol. 2001b;183:6752–62. doi: 10.1128/JB.183.23.6752-6762.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie G, Craig N. Fast, easy and efficient: site-specific insertion of transgenes into Enterobacterial chromosomes using Tn7 without need for selection of the insertion event. BMC Microbiol. 2006;6:39. doi: 10.1186/1471-2180-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller WG, Leveau JHJ, Lindow SE. Improved gfp and inaZ broad-host-range promoter-probe vectors. Mol Plant Microbe In. 2000;13:1243–50. doi: 10.1094/MPMI.2000.13.11.1243. [DOI] [PubMed] [Google Scholar]
- Peters JE, Craig NL. Tn7: smarter than we thought. Nat Rev Mol Cell Bio. 2001;2:806–14. doi: 10.1038/35099006. [DOI] [PubMed] [Google Scholar]
- Remus-Emsermann MN, Lücker S, Müller DB, et al. Spatial distribution analyses of natural phyllosphere-colonizing bacteria on Arabidopsis thaliana revealed by fluorescence in situ hybridization. Environ Microbiol. 2014;16:2329–40. doi: 10.1111/1462-2920.12482. [DOI] [PubMed] [Google Scholar]
- Remus-Emsermann MNP, Kowalchuk GA, Leveau JH. Single-cell versus population-level reproductive success of bacterial immigrants to pre-colonized leaf surfaces. Environ Microbiol Rep. 2013;5:387–92. doi: 10.1111/1758-2229.12040. [DOI] [PubMed] [Google Scholar]
- Remus-Emsermann MNP, Leveau JHJ. Linking environmental heterogeneity and reproductive success at single-cell resolution. ISME J. 2010;4:215–22. doi: 10.1038/ismej.2009.110. [DOI] [PubMed] [Google Scholar]
- Remus-Emsermann MNP, Tecon R, Kowalchuk GA, et al. Variation in local carrying capacity and the individual fate of bacterial colonizers in the phyllosphere. ISME J. 2012;6:756–65. doi: 10.1038/ismej.2011.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–82. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg C, Christensen BB, Johansen T, et al. Distribution of bacterial growth activity in flow-chamber biofilms. Appl Environ Microb. 1999;65:4108–17. doi: 10.1128/aem.65.9.4108-4117.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart PS, Franklin MJ. Physiological heterogeneity in biofilms. Nat Rev Microbiol. 2008;6:199–210. doi: 10.1038/nrmicro1838. [DOI] [PubMed] [Google Scholar]
- Tecon R, Leveau JHJ. The mechanics of bacterial cluster formation on plant leaf surfaces as revealed by bioreporter technology. Environ Microbiol. 2012;14:1325–32. doi: 10.1111/j.1462-2920.2012.02715.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.