

Abstract
It has been argued that the evolution of plant genome size is principally unidirectional and increasing owing to the varied action of whole-genome duplications (WGDs) and mobile element proliferation1. However, extreme genome size reductions have been reported in the angiosperm family tree. Here we report the sequence of the 82-megabase genome of the carnivorous bladderwort plant Utricularia gibba. Despite its tiny size, the U. gibba genome accommodates a typical number of genes for a plant, with the main difference from other plant genomes arising from a drastic reduction in non-genic DNA. Unexpectedly, we identified at least three rounds of WGD in U. gibba since common ancestry with tomato (Solanum) and grape (Vitis). The compressed architecture of the U. gibba genome indicates that a small fraction of intergenic DNA, with few or no active retrotransposons, is sufficient to regulate and integrate all the processes required for the development and reproduction of a complex organism.
Like other carnivorous plants, Utricularia (Lentibulariaceae) species derive nitrogen and phosphorus supplements by trapping and digesting prey organisms2,3. Lentibulariaceae are asterid angiosperms closely related to the model plants snapdragon (Antirrhinum) and monkey flower (Mimulus). Among Utricularia species, the intricate, water-filled suction bladders are variously arrayed on plant parts, and may even take the place of an embryonic leaf2,4. Whereas Utricularia vegetative structures are extremely diverse, its snapdragon-like flowers are stereotypical for plants of its asterid clade2 (Fig. 1a). Interestingly, these inhabitants of nutrient-poor environments do not bear true roots4.
Figure 1. Syntenic analysis of the Utricularia gibba genome.
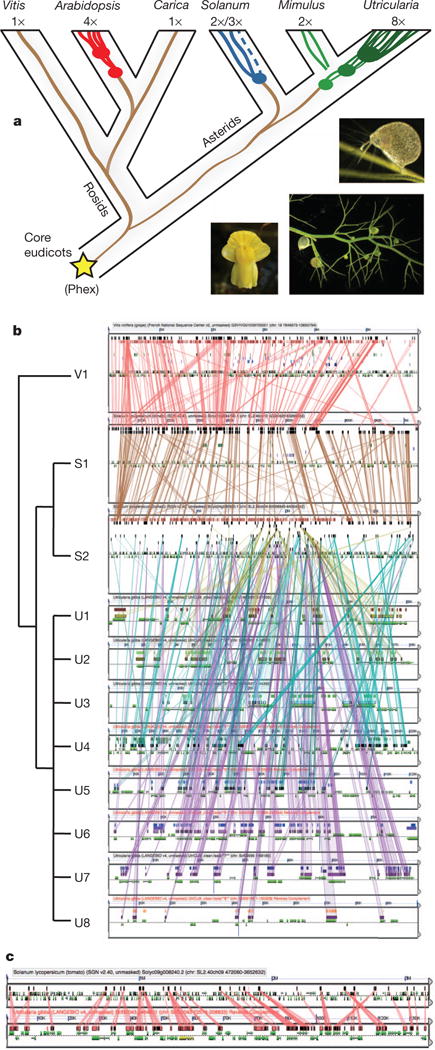
a, Whole-genome duplication (WGD) history highlighting the phylogenetic position of U. gibba. Vitis, Arabidopsis and Carica papaya are rosids; Arabidopsis has had two WGDs since the paleohexaploid (Phex) core eudicot ancestor. Tomato (Solanum), Mimulus and U. gibba are asterids; tomato has a mix of duplicated and triplicated regions; U. gibba has had three WGDs since common ancestry with tomato and the Phex ancestor. Mimulus has had a single WGD25 that may also be the most ancient WGD observed for U. gibba (see Supplementary Information section 7.1.3). U. gibba flowers are similar to those of Mimulus (that is, like snapdragons); tiny suction traps are borne on highly divided branching structures (insets, clockwise from left). b, A microsyntenic analysis shows that U. gibba (U) is 8:2:1 relative to homologous tomato (T) and Vitis (V) regions, respectively. As such, U. gibba is a 16-ploid with respect to Vitis, and the polyploidy of tomato is entirely independent (Supplementary Information section 7). Coloured lines connect high-scoring segment pairs (HSPs) on genome blocks masked for non-coding sequences. Gene models lie in the centres of each block, below the HSPs. This analysis may be regenerated by CoGe at http://genomeevolution.org/r/4wvh. c, Fractionation in a given U. gibba region can be massive with respect to tomato; the regions shown include an over 3 Mb block of the tomato genome (top), strongly syntenic and colinear to an approximately 130-kb block of U. gibba, representing an approximately 20:1 difference in total DNA. This analysis may be regenerated by CoGe at http://genomevolution.org/r/5cet.
Our U. gibba genome assembly, produced using a hybrid (454/Illumina/Sanger) sequencing strategy, closely matches the genome size estimated by flow cytometry (77 megabases (Mb)) (Supplementary Information section 1). Remarkably, despite its tiny size, the (G+C)-rich U. gibba genome accommodates about 28,500 genes, slightly more than Arabidopsis, papaya, grape or Mimulus, but less than tomato (Supplementary Information section 2). Indeed, the U. gibba genome has experienced a small, approximately 1.5% net gain across a conserved set of single-copy genes5 (Supplementary Information section 2.6). Synteny analysis reveals that U. gibba has undergone three sequential WGD events since last common ancestry with tomato and grape, with one of these duplications possibly shared by the closely related species Mimulus (Fig. 1a and Supplementary Information section 7). Consequently, the U. gibba genome seems to be 8× with respect to the palaeohexaploid (3×) core eudicot ancestor6 (Fig. 1b), whereas Arabidopsis is 4× with a genome 1.5-times larger7. Compared with independently polyploid tomato8, the U. gibba genome shows extremely fractionated gene loss (Fig. 1c), with almost two-thirds of syntenic genes shared with tomato having returned to single copy (Supplementary Information section 7.4 and Supplementary Table 39).
Intergenic sequence contraction in the U. gibba genome is particularly apparent in the paucity of repetitive DNA and mobile elements (Supplementary Table 8). Whereas repetitive DNA accounts for 10–60% of most plant genomes, in U. gibba it only amounts to 3%, including 569 mobile elements (Supplementary Information section 2). Notably, retrotransposable elements, which largely dominate angiosperm genomes, are rare in the U. gibba genome; we identified only 379, amounting to about 2.5% of the genome. Of these, only 95 seem complete and therefore potentially capable of further retrotransposition (Supplementary Information section 2.1 and Supplementary Tables 8 and 9). We found that all genes known to be involved in retrotransposon silencing have homologues in U. gibba (Supplementary Table 28), as well as a set of 75 microRNAs (miRNAs) belonging to 19 families (Supplementary Table 29 and Supplementary data 7). These results indicate that, despite its small genome, the general repertoire of miRNA-mediated gene regulation mechanisms in plants is conserved in U. gibba (Supplementary Table 29). Together, these data indicate that any influence of retrotransposon proliferation on U. gibba genome size must be countered by fractionation after WGDs and also by the silencing of these mobile elements.
The U. gibba genome contains a high percentage of small, putative promoters (Supplementary Fig. 11 and Supplementary Data 5) and tail-to-tail gene pairs with overlapping 3′ ends (Supplementary Tables 25 and 26). This configuration is similar to, but about 50% shorter than, that in Arabidopsis, which has led to denser packing in U. gibba gene islands (Fig. 2a). Using transient expression analysis, we confirmed that several short intergenic sequences function as transcriptional promoters, including a 400-base-pair region serving as a bidirectional promoter of a head-to-head gene pair (Fig. 2b and Supplementary Information section 3). These results indicate that the binding sites for transcription factors that direct the expression of U. gibba genes remain in their 5′ flanking regions, and that conserved cis-acting elements are compressed in at least a portion of the promoters of this carnivorous plant (Supplementary Fig. 11). Genome size contraction is also reflected at the level of introns, which showed smaller size and a slightly reduced number per gene (Supplementary Information section 5).
Figure 2. Architecture of the Utricularia gibba genome.
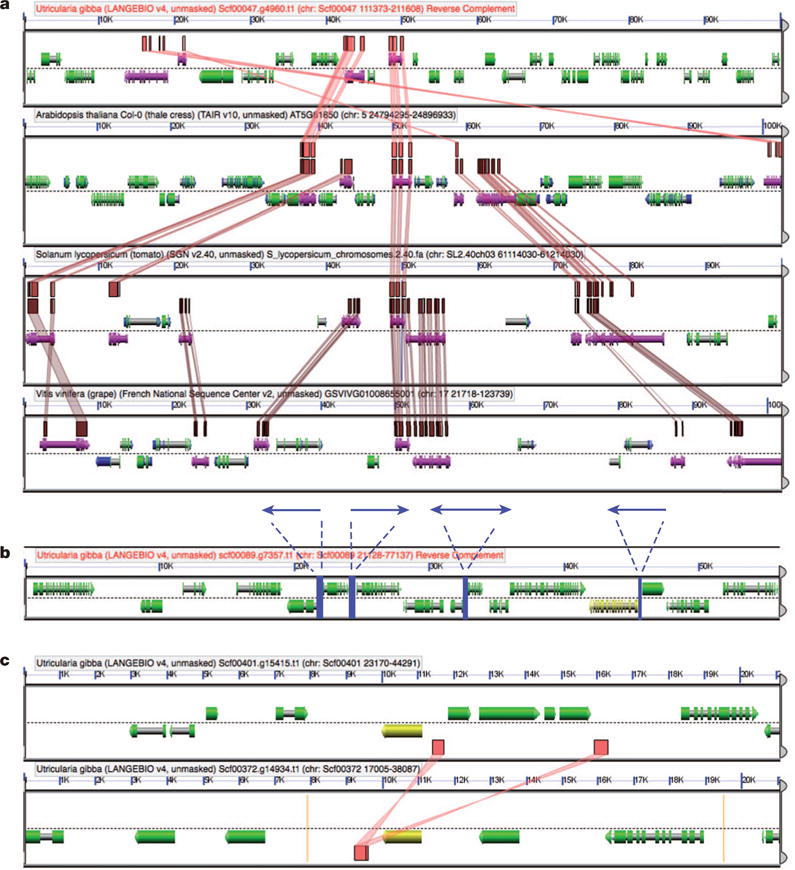
a, U. gibba gene islands are more compact than in Arabidopsis, and much higher in gene density than tomato or grape. For example, the Arabidopsis LEAFY gene lies directly in the middle of the second block from the top, which is an approximately 100-kb region from Arabidopsis chromosome 5. There are 28 genes in this view. In the corresponding U. gibba block (top), there are 34 genes within the same-sized region, which is therefore approximately 18% more densely packed. In tomato (3rd block) and grape (4th), there are many fewer genes (14 and 17, respectively) for a much lower density of gene space. b, Promoter spaces in U. gibba can be very short. Shown is part of a scaffold (scf00089), the sequence of which was verified by PCR walking. Four promoter regions (blue) showed reproducible activity in transient expression experiments (see Supplementary Information section 3). For example, the short bidirectional promoter between a divergent gene pair is approximately 400 bp. Other gene arrangements, tandem and convergent, can be seen in this example. c, Solo LTR remains of ectopically recombined mobile elements can be identified in the U. gibba genome. This example shows two blocks from U. gibba, the Solo LTR in the bottom block being homologous to the LTR pair present in the top block. In a, syntenic HSPs are shown as coloured lines connecting particular gene models (purple). Results from a and c can be regenerated at http://genomevolution.org/r/5kv5 and http://genomevolution.org/r/8lvv, respectively. See Supplementary Information for further discussion of b and c.
Compressed promoter spaces, fewer exons per gene than Arabidopsis (that is, net intron loss; Supplementary Table 12), and missing segments or whole genes in retroelements (Supplementary Fig. 4) support the notion that numerous microdeletions have occurred during U. gibba genome evolution, as previously observed in Arabidopsis9 and maize10. Furthermore, the presence of numerous solo long terminal repeat (LTR) elements (a single copy of an LTR that is the product of homologous recombination events between two identical or related LTR-retrotransposons) in the U. gibba genome (Fig. 2c and Supplementary Fig. 5) indicates that large-scale recombinational deletions have also occurred11. Unlike the contracted nuclear genome, the plastid and mitochondrial genomes of U. gibba are quite similar in structure to those of other angiosperms (Supplementary Information section 8 and Supplementary Figs 35–38) with no apparent shortening of intergenic regions (Supplementary Tables 41 and 43). Therefore, the evolutionary forces acting to reduce U. gibba genome size seem to have affected only the nucleus.
We investigated the coding DNA content of the U. gibba genome compared to the Arabidopsis, tomato, grape, Mimulus and papaya genomes in two complementary ways: (1) by predicted protein domains, and (2) by gene family classification. In the first approach, we compared protein domains and applied a likelihood ratio test to examine the significance of difference in numbers of Pfam domains (Supplementary Table 15). 97% of domain groups did not show significant differences among the plant species analysed, and of the remaining 3%, only 40% represented instances where U. gibba had fewer domain members than other plant species (Supplementary Table 16).
To gain insight into specific differences in the genic repertoire of U. gibba and their potential biological significance, in the second approach we classified gene families in the U. gibba, Arabidopsis, tomato, grape and papaya genomes using OrthoMCL12. Out of a total of 18,991 gene families, 1,275 have no U. gibba members (57% representing single-gene families, Supplementary Table 18), whereas 1,804 showed an increased number of genes in U. gibba (Supplementary Table 19). Several gene families specifically lost or conspicuously reduced in U. gibba may have functions related to its unusual embryogenesis (frequently involving asymmetrical production of shoot apical organs and absence of true cotyledons), its frequent shoot–leaf indistinction, and its lack of true roots (Supplementary Table 18; see references in Supplementary Information section 2.5). These include homologues of AT1G68170 (a nodulin MtN21-like transporter, differentially expressed in globular-stage embryos and cotyledons), PEI1 (an embryo-specific zinc finger transcription factor required for heart-stage embryo formation), and a paralogue of FD (involved in flowering but also expressed in embryos and cotyledons). In addition, compared to the two to three member gene family in all other species examined, U. gibba contains a single member of the CASPARIAN STRIP MEMBRANE DOMAIN PROTEIN family, which encodes proteins involved in Casparian strip formation in Arabidopsis roots. Other genes missing in U. gibba may also be involved in root development and physiology: homologues of WAK (a cell-wall-associated Ser/Thr kinase involved in cell elongation and lateral root development), NAXT1 (a nitrate efflux transporter mainly expressed in the cortex of adult roots), MYB48 and MYB59 (nitrogen-responsive genes involved in the regulation of cell cycle progression and root growth), and the MADS box genes ANR1 (ARABIDOPSIS NITRATE REGULATED 1 (AGL44)) and XAL1 (XAANTAL1 (AGL12)). ANR1 is a component of a signalling pathway that regulates lateral root growth in response to external NO3 supply, whereas XAL1 is involved in root-cell differentiation and flowering time. At least 50 MADS box genes are known to be expressed in Arabidopsis roots, of which the AGL17-like type II clade is noteworthy as all its members are expressed in roots, and four of them (AGL16, AGL17, AGL21 and AGL44) have been reported to be root-specific, as are the type I genes AGL26 and AGL56. Interestingly, contractions and losses in all of these root-expressed MADS box gene clades/subfamilies account for much of the global reduction of the MADS box gene family in U. gibba (Supplementary Fig. 7). In contrast, other MADS box gene subfamilies were found to be specifically expanded in U. gibba (see references in Supplementary Information section 2.5). One such example is SOC1, a gene expressed in shoots with a well-characterized role in regulating flowering time and a possible role in response to phosphorus and sulphur (but not nitrogen) availability. Because it has been reported in Utricularia vulgaris that trap formation is induced by low phosphorus availability but not by low nitrogen13, it is possible that the marked expansion of the U. gibba SOC1-like clade is related to the adaptive capacity for phosphorus scavenging from trapped prey. Three clusters representing members of different TCP (TEOSINTE BRANCHED1/CYCLOIDEA/PCF) transcription factor clades are also expanded in U. gibba. These genes regulate plant morphogenesis, including branching, and it is tempting to speculate that specific clade expansions may be related to the genus-wide diversity of branching patterns in Utricularia2.
Taken together, we infer from our analyses of U. gibba coding sequence that natural selection preserved a core set of gene functions, most of which have returned to single copy along with considerable genomic fractionation after three WGDs. Relaxed selection pressure for unnecessary functions probably led to gene losses, whereas in other cases, gene family expansions may have been promoted by selection. Evidence for localized selection on the U. gibba gene complement, however, does not provide support for the existence of genome-wide selective forces that might favour reduction of nonessential, non-coding DNA.
It has been argued that increased mutation pressure can enhance natural selection against non-essential DNA14. We proposed previously that enhanced molecular evolutionary rates caused by mutagens could have made the U. gibba genome more susceptible to natural selection3,15. This could now be evaluated, because information on the mutational diversity (θ) stored within a single genome is retrievable. θ, when small as in Arabidopsis16, closely approximates heterozygosity. We found that U. gibba does not have a θ value substantially different from that of Arabidopsis (Supplementary Information 6). As such, it is possible that the population genetic environment underlying U. gibba genome evolution did not engender special sensitivity to natural selection beyond that experienced by Arabidopsis with its larger proportion of non-coding DNA.
Collectively, our analyses highlighting total gene complement, sequential WGD and mutational diversity estimates for U. gibba raise quandaries regarding the evolution of its contracted genome. It is possible that inherent molecular mechanisms favouring deletion dominated nuclear genome size reduction in a population genomic background where selection was too weak to counteract such a burden. Some intrinsic molecular biases are known to correlate with genome size differences. For example, the net DNA deletion bias caused by double-strand break repair in Arabidopsis (120 Mb7) is greater than that of tobacco (5.1 gigabases (Gb)17), and deletions are larger as well18. A similar bias occurs in Arabidopsis thaliana compared to its larger-genome relative Arabidopsis lyrata9. Biased gene conversion, which is associated with (G+C)-rich sequences such as those found throughout the U. gibba genome19, leads to its own inherent deletion bias20, which has been argued to be an important neutral process behind other genome size reductions21. Of course, a molecular-mechanistic deletion bias does not preclude that selection still enhances fixation of such deletions.
Regarding a potential role of polyploidy in genome contraction, we propose that for small genomes facing a strong internal deletion bias, WGDs, by the creation of duplicates throughout the genome, might transiently buffer against loss of essential genes (Fig. 3). Interestingly, phylogenetic evidence indicates that genome evolution is highly dynamic in Lentibulariaceae, with nuclear DNA contents ranging from 60 Mb to 1.5 Gb22. Sequencing of additional Lentibulariaceae genomes is warranted to ascertain the basis for these differences. Moreover, because molecular dating analyses place the divergence of Utricularia from its carnivorous relative Pinguicula at approximately 40 million years before present (Myr BP)23, and that of U. gibba from other Utricularia species as recently as 5–15 Myr BP (Supplementary Information section 9), additional high-quality Lentibulariaceae genomes should permit phylogenetic dating of the sequential WGD events that occurred after common ancestry with tomato, approximately 87 Myr BP23.
Figure 3. A model of genome size reduction and the plant genome size evolutionary spectrum.
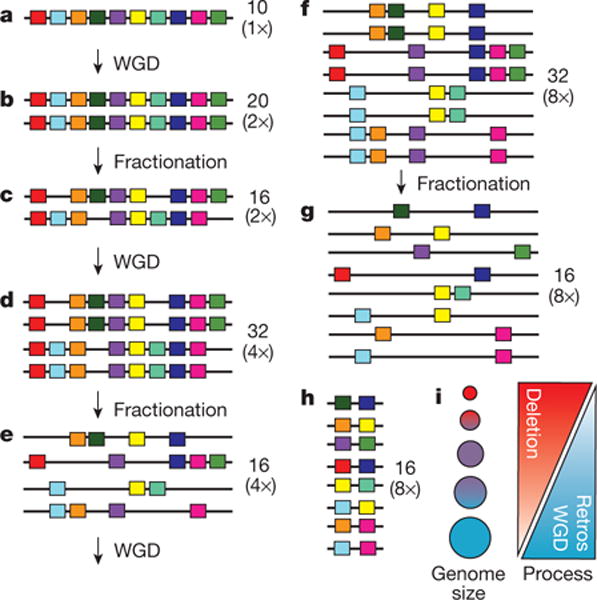
a, The initial diploid genome has 10 genes. b, c, After one WGD (b), there are 20 genes in the tetraploid, which fractionate into 16 genes (c). d–g, After another round of WGD (d), the octoploid genome (32 genes) fractionates again to yield 16 genes (e), which duplicate (to 32 genes) in yet another WGD (f), after which fractionation yields 16 genes in the 16-ploid (g). The resulting number of genes is the same as in the fractionated genome resulting from the first WGD (c), with only 6 more genes than the original diploid ancestor (a). h, The resulting genome after intergenic DNA contraction at any stage (a–g) has thus survived a high deletion rate via the net accrual of very few gene duplicates following sequential WGDs. U. gibba has in fact fractionated down to single copy two-thirds of its genes syntenic to tomato genes since its three WGDs. i, An interplay of deletion and retroelement proliferation rates relates to a continuum of plant genome size evolution, with WGDs providing short-term buffering against loss of crucial gene functions in small genomes affected by high endogenous deletion rates. Small genomes result when the recombinational deletion rate is high relative to retroelement proliferation and WGD, vice versa with large genomes.
In summary, U. gibba genome architecture demonstrates that angiosperms can evolve diverse gene landscapes while overall genome size contracts, not only during expansions. Furthermore, in contrast to recent publications that highlight a crucial functional role of non-coding DNA in complex organisms such as animals24, the necessary genomic context required to make a flowering plant may not require substantial hidden regulators in the non-coding ‘dark matter’ of the genome.
METHODS SUMMARY
Genomic DNA from U. gibba was subjected to a hybrid 454, Illumina and Sanger sequencing strategy. Approximately 5.2 Gb of sequence data were generated, consisting of 1.9 Gb of shotgun reads, 1.5 Gb of mate-pair reads, 1.5 Gb of paired-end reads and 119.5 Mb of Sanger reads; these were assembled using Newbler version 2.6. The assembly was filtered for organellar and environmental DNA, and validated by primer walking of representative scaffolds and random fosmid sequencing (Supplementary Information sections 1.4–1.6). A transcriptome from pooled plant parts served as a gene prediction and annotation aid (Supplementary Information section 2.3). Transposable elements were identified using the REPET package (Supplementary Information section 2.1). Non-coding RNAs were identified using tRNAscan-SE, RNAMMER, snoscan, and SRPscan (Supplementary Information sections 2.2 and 4). Gene models were predicted using AUGUSTUS with a transcriptome-derived training set (Supplementary Information section 2.3.2). Synteny to other plant genomes was analysed using CoGe (Supplementary Information section 7). Frequencies of Pfam domains among gene models, and their significant differences, were calculated for U. gibba and several other plant genomes (Supplementary Information section 2.4). Gene models from U. gibba and other plant species were clustered into orthogroups using OrthoMCL, annotated using Blast2GO, and studied for expansions and contractions of gene memberships (Supplementary Information section 2.5). Selected gene families from U. gibba, Arabidopsis and tomato were subjected to phylogenetic analysis (Supplementary Information section 2.5.2). The U. gibba genome was scanned for single-copy genes identified from other plant genomes (Supplementary Information section 2.6). Promoters and untranslated regions (UTRs) were studied in silico, selected UTRs were amplified by PCR and sequenced, and selected promoters were analysed in vivo using transient expression assays (Supplementary Information section 3). Genome compositional features were compared to Arabidopsis (Supplementary Information section 5). Population genomic parameters were calculated using the PSMC and mlRho applications (Supplementary Information section 6). Organelle genomes were assembled using Newbler version 2.6 and Megamerger, and annotated using DOGMA (Supplementary Information section 8). Molecular evolutionary rates and divergence times were estimated using BEAST and HyPhy (Supplementary Information section 9).
Full Methods and any associated references are available in the online version of the paper.
METHODS
Utricularia gibba was collected in the Umécuaro municipality, Michoacán, Mexico. For flow cytometry analysis, nuclei were isolated from shoot-like structures and flowers, stained with 1.5 ml 4′,6-diamidino-2-phenylindole, and their fluorescence measured after ultraviolet excitation. Arabidopsis thaliana was used as an internal standard to calculate U. gibba nuclear DNA content. The genome size estimated was 77.38 Mb.
Nuclear DNA was isolated from U. gibba shoot-like structures, then amplified and sheared to obtain DNA fragments ranked according to the sizes required for sequencing libraries (1 kb, 2 kb, 2–4 kb or 7–9 kb). For whole-genome shotgun sequencing, four distinct shotgun libraries (one 3 kb and three 8 kb mate-pair libraries) were constructed. Preparation, amplification and sequencing of these libraries were performed using Roche GS FLX Titanium Sequencing Kits and Genome Sequencer FLX Instruments following the manufacturer’s protocols. One additional shotgun library was constructed and sequenced using the GS FLX XL+ Sequencing kit and corresponding platform. Additionally, one paired-end library of ~450 bp was prepared using Illumina’s paired-end kit. The nuclear DNA was sheared with a Covaris S2 ultrasonicator and the library was sequenced (twice) as 2×250 bp on an Illumina MiSeq. Finally, conventional Sanger reads were generated with an ABI 3730xl sequencer using the Big Dye–terminator Cycle Sequencing kit. Recombinant clones (pJET1.2/blunt Cloning Vector) were used to transform DH10b cells to obtain two genomic libraries ((1) 43,968 clones, average insert size 1.2 kb, and (2) 55,680 clones, average insert size 4 kb), and clones were sequenced both uni- and bidirectionally. In total, ~5.2 Gb of sequence data was generated, consisting of 1.9 Gb of shotgun reads, 1.5 Gb of mate-pair reads, 1.5 Gb of paired-end reads and 119.5 Mb of Sanger reads (Supplementary Table 2).
The 454, Sanger and MiSeq reads were assembled using Newbler version 2.6 de novo genome assembler (with the -scaffold option). Vector and poor quality regions were masked in the Sanger reads using the LUCY2 software. Natural and artificial duplicates in pyrosequencing reads were eliminated using the CD-HIT pipeline. The MiSeq read pairs (2×250) were merged and adaptor-trimmed with SeqPrep using default settings. Paired-end reads that did not overlap with at least 10 bases were subjected to stringent read filtering and trimming before assembly. Reads were trimmed with a sliding window approach (window size 10 bases, shift 1 base). Illumina bases were kept until the average quality score Q of 10 adjacent bases was below Q = 25. Reads were removed if they were shorter than 30 bases after trimming, had at least one uncalled base, contained the adaptor sequence, or had less than two-thirds of the bases of the first half of the read with quality values of Q ≥ 30. Orphan reads were discarded to keep pairs only. Redundant read pairs that may originate from PCR artefacts were also removed by comparing the sequences of the read pairs. Out of 6,215,172 read pairs, 28% could be merged and 60% passed the stringent filtering. The average length of the merged reads was 459 bp. The filtered MiSeq pairs were exclusively used for scaffolding by trimming them to 49 bases. We generated a total of 4.7 billion high-quality base pairs from 20.3 million high-quality reads. After de novo assembly, contaminating sequences from organellar and environmental DNA were removed by a GC value and coverage-based filtering process. The U. gibba assembly spanned, with around 35-fold genome coverage, 81.87 Mb including embedded gaps (N50 = 80,839 bp, the weighted mean statistic such that 50% of the assembly is contained in contigs and scaffolds equal to or larger than this value). The total length of the assembled genome was about 5.73% greater than the genome size estimated by flow cytometry of isolated nuclei stained with DAPI (77.38 Mb).
Our assembly of the U. gibba genome was verified by single-pass primer walking resequencing of a ~100 kb window (total) from two randomly selected scaffolds. Additionally, using the pCC1FOS vector, a fosmid library with ~1,000 clones was generated. The complete sequences of 53 end-sequenced fosmids (with BLAST hits to the U. gibba genome) were obtained with an estimated coverage of ~250×. The complete alignments of fosmid sequences to the U. gibba whole genome sequence revealed that we were able to generate a shotgun assembly with only limited potential misassemblies.
Transposable elements in the U. gibba genome were identified both at the DNA and protein level. The REPET package was used to search for transposable elements within the U. gibba genome. To confirm the degree of completeness of U. gibba LTRretrotransposons, characteristic elements (both 5′-and 3′-long terminal repeats (LTRs), primer binding sites (PBSs), polypurine tracts (PPTs), and conserved protein domains and their positions) were identified using the LTR-Finder program. We took a computational approach to gain insight into the different RNA-mediated gene regulatory pathways present in U. gibba. Non-coding RNAs (ncRNAs), including miRNAs, small nuclear RNAs, tRNAs, ribosomal RNAs and H/ACA-box small nucleolar RNAs, were identified using INFERNAL software by searching against the Rfam database.
For transcriptome sequencing, total RNA was extracted from whole plants, shoot-like structures, inflorescences and traps using TRIzol according to the manufacturer’s instructions. To represent all U. gibba organs, 2 mg of RNA from each sample were pooled. cDNA synthesis was performed as described previously. The sequences were assembled with Newbler version 2.6.
The AUGUSTUS program was trained on the U. gibba genome using 37,799 Isotig sequences. First, using the AUGUSTUSbeta web server training tool and the U. gibba genome and transcriptome sequences (Isotigs), a data set with training gene structures was generated. Using this training set, parameters required by AUGUSTUS were calculated. Gene models in the U. gibba genome sequence were predicted ab initio as well as with hints, running AUGUSTUS locally with newly optimized parameters.
To analyse the distribution of gene families over different plant species, we identified the Pfam domains present from gene models predicted in the Arabidopsis, tomato, grape, Mimulus and papaya genomes. To compare the abundance of domains in proteins of different plant species we used a likelihood ratio test method (see Supplementary Information for more details). Clustering of homologous genes for the U. gibba, Arabidopsis, tomato, grape and papaya genomes was performed using OrthoMCL on the predicted protein sequences of all the five genomes. All U. gibba gene models were processed through the Blast2GO program to assign functions. We closely surveyed the first 100 OrthoMCL clusters showing U. gibba gene family member expansions, and then the first 100 showing contractions. We performed detailed phylogenetic classifications of five well-known transcription factor families (MADS, TCP, GRAS, ARF and AUX/IAA) using maximum likelihood and neighbour joining methods to provide highly focused views of gene family expansion and contraction in U. gibba relative to Arabidopsis and tomato. Using bidirectional best BLAST and synteny analysis (SynMap within CoGe), we calculated the proportions of previously reported single-copy genes (in Arabidopsis, Vitis, poplar and rice) that are also present as single copy in the U. gibba genome.
We estimated the average length of intergenic regions considering pairs of adjacent genes as either convergent (→ ←), divergent (← →), or tandem (→→ or ← ←). A total of 14 adjacent gene pairs (5 convergent, 4 divergent and 5 tandem) were selected to estimate UTR sizes in the U. gibba genome by random amplification of cDNA ends (RACE-PCR). For a rbcS gene promoter from U. gibba, we identified and studied the compaction of the I- and G-boxes and two other motifs almost always conserved in other species. The functionality of some promoters in U. gibba was tested by transient expression assay.
We applied the pairwise sequentially Markovian coalescent (PSMC) model, which was originally applied to human and other mammalian genomes, to study the mutational diversity of the U. gibba genome and effective population size (Ne) over time. The Arabidopsis thaliana genome (and reads from accession SRX158512) was treated similarly. In PSMC coalescent simulations, Ne is inferred from heterozygosity of the sequenced genome (θ 5 4Neμ). The mlRho application was similarly used to estimate genome-wide and window-based (100 kb, 75 kb, 50 kb and 25 kb) θ values.
For analyses of whole genome duplications, we focused on comparing the genomes of Solanum lycopersicum and U. gibba using the SynMap tool in the online CoGe portal (http://genomevolution.org/CoGe/). CoGe contains two major applications to help evaluate and estimate syntenic depth: SynMap and SynFind. We compared tomato to U. gibba using two parameter sets that differ in the window size of genes used to define a minimum number of colinear genes allowing two regions to be called syntenic. Fractionation depth refers to the number of syntenic genes that reduce to single-, double- or n-copy over the course of U. gibba’s three independent WGDs since common ancestry with tomato. Results were generated from SynMap via a master table of all genes in tomato along with their matching syntenic regions in U. gibba. GEvo microsyntenic analyses were performed on selected regions determined to be syntenic using SynMap and SynFind.
Scaffolds/contigs originating from the plastid and mitochondrial genomes of U. gibba were identified during the process of de novo assembly using Newbler version 2.6. These were further assembled and annotated using the Megamerger program and DOGMA web tool.
In order to investigate the divergence time of U. gibba from other Utricularia species, we obtained phylogenetic data sets for the family Lentibulariaceae from three regions of the chloroplast genome and one region of the mitochondrial genome. We applied the BEAST program to estimate divergence dates, and both this program and HyPhy to study molecular evolutionary rates.
Supplementary Material
Acknowledgments
We thank I. Bravo-Carmona for growing plants for this research. We also thank T. Mailund for discussion and critical comments on the manuscript, M. Stanke and K. Hoff for their assistance in the use of AUGUSTUS software, and P. Guzmán-Villate for providing the cell suspension used for transient expression assays. Special thanks goes to the members of the ‘laboratorio de servicios genómicos’ of LANGEBIO, CINVESTAV for sequencing services and their help. This research was supported by CONACyT (Mexico) via general support to LANGEBIO, HHMI grant 4367 (to L.H.-E.), the College of Arts and Sciences, University at Buffalo (to V.A.A.), and the NSF (0922742 to V.A.A.). E.I.-L. is indebted to CONACyT (Mexico) for a PhD fellowship. We acknowledge the US Department of Energy Joint Genome Institute for Mimulus data (available at http://www.phytozome.net/mimulus).
Footnotes
Author Contributions E.I.-L., V.A.A. and L.H.-E. conceived of and led the study. E.I.-L., V.A.A. and L.H.-E. wrote the paper with significant contributions by E.L., L.C.-P. and A.J.W.; E.I.-L., G.H.-G., C.A.P.-T., T.-H.C., T.L., M.J.A.J., S.C.S., A.O.-A., S.A.C.-P. and M.d.J.O.-E. collected data. E.I.-L., E.L., L.C.-P., T.-H.C., T.L., A.J.W., M.A.-V., E.G.-C., G.A.-H., H.H., A.E.M., S.X., M.L. and V.A.A. analysed data. J.S., T.P.M., T.M., D.B. and A.H.-E. provided materials. A.F.-C. and J.I.C.-L. provided bioinformatic support. All authors read and approved the final manuscript.
Supplementary Information is available in the online version of the paper.
Files containing raw sequence reads and quality scores were deposited in the Sequence Read Archive of the National Center for Biotechnology Information (NCBI). Primary accession numbers: SRS399135 (454 reads), SRS399163 (MiSeq reads), SRS399167 (fosmid Ion Torrent reads) and SRS399168 (RNAseq Ion Torrent reads). The U. gibba genome assembly and gene models are available on CoGe (http://genomevolution.org/CoGe/). Readers are welcome to comment on the online version of the paper.
The authors declare no competing financial interests.
References
- 1.Bennetzen JL, Kellogg EA. Do plants have a one-way ticket to genomic obesity? Plant Cell. 1997;9:1509–1514. doi: 10.1105/tpc.9.9.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taylor P. The Genus Utricularia: a Taxonomic Monograph. Kew Publishing: 1989. [Google Scholar]
- 3.Albert VA, Jobson RW, Michael TP, Taylor DJ. The carnivorous bladderwort (Utricularia, Lentibulariaceae): a system inflates. J Exp Bot. 2010;61:5–9. doi: 10.1093/jxb/erp349. [DOI] [PubMed] [Google Scholar]
- 4.Płachno BJ, Swiatek P. Unusual embryo structure in viviparous Utricularia nelumbifolia, with remarks on embryo evolution in genus Utricularia. Protoplasma. 2010;239:69–80. doi: 10.1007/s00709-009-0084-1. [DOI] [PubMed] [Google Scholar]
- 5.Duarte JM, et al. Identification of shared single copy nuclear genes in Arabidopsis, Populus, Vitis and Oryza and their phylogenetic utility across various taxonomic levels. BMC Evol Biol. 2010;10:61. doi: 10.1186/1471-2148-10-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jaillon O, et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449:463–467. doi: 10.1038/nature06148. [DOI] [PubMed] [Google Scholar]
- 7.The Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- 8.The Tomato Genome Consortium. The tomato genome sequence provides insights into fleshy fruit evolution. Nature. 2012;485:635–641. doi: 10.1038/nature11119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu TT, et al. The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nature Genet. 2011;43:476–481. doi: 10.1038/ng.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woodhouse MR, et al. Following tetraploidy in maize, a short deletion mechanism removed genes preferentially from one of the two homeologs. PLoS Biol. 2010;8:e1000409. doi: 10.1371/journal.pbio.1000409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Devos KM, Brown JK, Bennetzen JL. Genome size reduction through illegitimate recombination counteracts genome expansion in Arabidopsis. Genome Res. 2002;12:1075–1079. doi: 10.1101/gr.132102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li L, Stoeckert CJ, Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kibriya S, Jones JI. Nutrient availability and the carnivorous habit in Utricularia vulgaris. Freshw Biol. 2007;52:500–509. [Google Scholar]
- 14.Lynch M, Koskella B, Schaack S. Mutation pressure and the evolution of organelle genomic architecture. Science. 2006;311:1727–1730. doi: 10.1126/science.1118884. [DOI] [PubMed] [Google Scholar]
- 15.Ibarra-Laclette E, et al. Transcriptomics and molecular evolutionary rate analysis of the bladderwort (Utricularia), a carnivorous plant with a minimal genome. BMC Plant Biol. 2011;11:101. doi: 10.1186/1471-2229-11-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nordborg M, et al. The pattern of polymorphism in Arabidopsis thaliana. PLoS Biol. 2005;3:e196. doi: 10.1371/journal.pbio.0030196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leitch IJ, et al. The ups and downs of genome size evolution in polyploid species of Nicotiana (Solanaceae) Ann Bot. 2008;101:805–814. doi: 10.1093/aob/mcm326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kirik A, Salomon S, Puchta H. Species-specific double-strand break repair and genome evolution in plants. EMBO J. 2000;19:5562–5566. doi: 10.1093/emboj/19.20.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ibarra-Laclette E, Albert VA, Herrera-Estrella A, Herrera-Estrella L. Is GC bias in the nuclear genome of the carnivorous plant Utricularia driven by ROS-based mutation and biased gene conversion? Plant Signal Behav. 2011;6:1631–1634. doi: 10.4161/psb.6.11.17657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Assis R, Kondrashov AS. A strong deletion bias in nonallelic gene conversion. PLoS Genet. 2012;8:e1002508. doi: 10.1371/journal.pgen.1002508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nam K, Ellegren H. Recombination drives vertebrate genome contraction. PLoS Genet. 2012;8:e1002680. doi: 10.1371/journal.pgen.1002680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greilhuber J, et al. Smallest angiosperm genomes found in Lentibulariaceae, with chromosomes of bacterial size. Plant Biol. 2006;8:770–777. doi: 10.1055/s-2006-924101. [DOI] [PubMed] [Google Scholar]
- 23.Bell CD, Soltis DE, Soltis PS. The age and diversification of the angiosperms re-revisited. Am J Bot. 2010;97:1296–1303. doi: 10.3732/ajb.0900346. [DOI] [PubMed] [Google Scholar]
- 24.The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aagaard JE, Olmstead RG, Willis JH, Phillips PC. Duplication of floral regulatory genes in the Lamiales. Am J Bot. 2005;92:1284–1293. doi: 10.3732/ajb.92.8.1284. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.