

Facial shaping results from oriented divisions and crowd movements of ectomesenchymal cells during morphogenetic events.
Keywords: Early face development, embryonic development, neural crest cells, migration, clonal envelopes, morphogenesis
Abstract
Cranial neural crest cells populate the future facial region and produce ectomesenchyme-derived tissues, such as cartilage, bone, dermis, smooth muscle, adipocytes, and many others. However, the contribution of individual neural crest cells to certain facial locations and the general spatial clonal organization of the ectomesenchyme have not been determined. We investigated how neural crest cells give rise to clonally organized ectomesenchyme and how this early ectomesenchyme behaves during the developmental processes that shape the face. Using a combination of mouse and zebrafish models, we analyzed individual migration, cell crowd movement, oriented cell division, clonal spatial overlapping, and multilineage differentiation. The early face appears to be built from multiple spatially defined overlapping ectomesenchymal clones. During early face development, these clones remain oligopotent and generate various tissues in a given location. By combining clonal analysis, computer simulations, mouse mutants, and live imaging, we show that facial shaping results from an array of local cellular activities in the ectomesenchyme. These activities mostly involve oriented divisions and crowd movements of cells during morphogenetic events. Cellular behavior that can be recognized as individual cell migration is very limited and short-ranged and likely results from cellular mixing due to the proliferation activity of the tissue. These cellular mechanisms resemble the strategy behind limb bud morphogenesis, suggesting the possibility of common principles and deep homology between facial and limb outgrowth.
INTRODUCTION
The facial compartment is arguably the most sophisticated and heterogeneous part of our body. Despite the keen attention to the biology of facial development, the functional and adaptive placement and spatial integration of sensory organs, skeletal structures, nervous system, and feeding apparatus are still not well understood. Numerous congenital craniofacial abnormalities affect the form and function of the face. These may result in, for instance, deformities, distractions, elongations, shortenings, asymmetries, and generally aberrant structures. Explanations to these malformations still await the fundamental understanding of the underlying failure of morphogenesis (1). Cells of the facial compartment arise from four main sources: neural crest stem cells, paraxial mesoderm, epidermis, and endoderm. The nonepithelial tissues in the facial region originate from migratory neural crest stem cells and the paraxial mesoderm (2). Embryonic epidermis and endoderm generate epithelialized structures, including covering tissues, various glands, epithelial compartments of the hair follicles, and teeth (3, 4). The paraxial mesoderm produces progenitors of striated muscles and endothelial cells and forms the vascular tree in the face and the head in general (5, 6). The neural crest, the largest contributor to the developing face, gives rise to cartilage, bone, dentin and pulp of the teeth, dermal papillae of hair follicles, smooth muscles of the vessel walls, ligaments, fascia, adipose tissue, dermis, pericytes in the forebrain, epithelial cells in the ear, pigment cells, peripheral glial cells, subpopulation of sensory neurons in trigeminal ganglia, sympathetic and parasympathetic neurons, and some other cell types found in the head (7). Neural crest cells have long been considered to be multipotent within the neural tube and later during their active migration (8–11). However, current evidence has suggested that early cell fate restrictions in the neural crest may already exist at the level of the neural tube (12, 13). Still, a very recently performed clonal analysis of neural crest populations in the trunk has supported the concept of multipotency of the premigratory and migratory neural crest (14). Despite this, questions concerning neural crest heterogeneity, multipotency, and progressive specification are far from resolved. This is especially evident when considering the intrinsic differences between neural crest populations that delaminate from different parts along the anterioposterior axis and also along the time scale. For example, only cranial neural crest cells give rise to the ectomesenchyme as an intermediate embryonic cell type that, in turn, will produce most of the structures in a developing face (7, 15). These neural crest–derived early ectomesenchymal cells are the main focus of this study. Little is known about their behavior before the formation of bones, cartilages, and other tissues in the face.
In the limb bud, similar mesenchymal cells of a different origin also play a major role in tissue morphogenesis and shape development (16). Previous studies have demonstrated that elongation and shape formation of the embryonic limb are achieved mostly as a result of directional cell activities that include oriented cell divisions, cell allocation, and migration. The shaping largely results from orientation signals and their interpretation in the tissue, rather than only from local differences in proliferation speed (17, 18). The directional activities are regulated by Wnt5a/JNK (c-Jun N-terminal kinase), whereas continuous rearrangements of mesenchymal cells are controlled by the FGF (fibroblast growth factor)/MAPK (mitogen-activated protein kinase) pathway (17). Planar cell polarity (PCP) pathway members (including Wnt5a) are well known to control orientation of cells (19) and are heavily involved in coordinated outgrowth and shape development of multiple embryonic structures (20). As an example, knockout and overexpression of Wnt5a result in the shortening and widening of both the limbs and the face (21–23).
After the arrival of neural crest cells to the face, the embryo is still very small, and significant growth and expansion of the cranial structures will follow. Very little is known about the role of clonal dynamics and the coordinated and directional cell behavior in the ectomesenchyme that eventually shape the face. Here, we attempted to address questions concerning the process of face morphogenesis after the stage of migratory neural crest: How do the newly arrived individual neural crest cells occupy and build different regions of the face? Are there any defined regions occupied by unique clones? Is there a somatotopical mapping at the level of the dorsal neural tube, and what is the degree of cell and tissue polarization, clonal mixing, and migratory behavior of ectomesenchymal cells? Using a variety of methods, we demonstrated that the early outgrowth and shaping of the face are driven by oriented cell divisions and allocations of daughter cells, as well as organized relocations of large cellular groups with minimal individual migration. These are features shown to be of utmost importance in limb shaping, and our findings might support the concept of conserved programs in limb and facial outgrowth.
RESULTS
The early face is organized by well-defined overlapping ectomesenchymal clones
To understand clonal arrangements in relation to facial shape and outgrowth, we took advantage of genetic tracing with the help of Sox10-CreERT2 and PLP-CreERT2 mouse strains coupled to an R26Confetti reporter (24). Both Sox10-CreERT2 (25) and PLP-CreERT2 (26, 27) recombine in the cranial neural crest when the pregnant females are injected with tamoxifen at embryonic day 8.5 (E8.5), whereas the R26Confetti reporter enables efficient color coding of individual cells by 10 individual color combinations suitable for clonal analysis. There are unequal chances of activating different color combinations (14). For example, clones with activated yellow fluorescent protein (YFP) + red fluorescent protein (RFP), RFP + cyan fluorescent protein (CFP), RFP + YFP, and green fluorescent protein (GFP) are rare compared to clones expressing only YFP, RFP, or CFP and are always significantly underrepresented as compared to these single-color clones. Clones carrying GFP together with another fluorescent protein were never detected. For details about proportions of recombined color combinations, see Baggiolini et al. (14). Sox10-CreERT2 and PLP-CreERT2 demonstrate different recombination efficiencies and can be selectively used to achieve the desired tracing outcomes and to confirm the specificity of neural crest recombination in cross-comparisons.
With the help of the Sox10-CreERT2 line, we focused on single-color solitary clones in the whole head, which we successfully achieved by titrating the amount of the injected tamoxifen (28). First, we induced genetic recombination in the neural crest at E8.5 and analyzed the progeny 24 to 36 hours later, using three-dimensional (3D) imaging and analysis. The results showed that genetically traced neural crest cells give rise to spatially defined clonal patches of early ectomesenchyme (“clonal envelopes”) after arrival to the facial region (Figs. 1, A and B, and 2A). A clonal envelope can be strictly defined as a region of 3D space demarcated by a graph connecting all cells belonging to one neural crest–derived clone. Such a clonal envelope occupies only a local portion of the face. Together with the identification of clonal envelopes, we could assess the amount of mixing or overlapping of several clones in a particular location that was analyzed. In cases of rare recombination events, we observed a single compact group of cells in one solitary facial location. We termed this an ectomesenchymal neural crest–derived clone (Fig. 1A). Such single clones, from multiple embryos, were analyzed in detail for cell number, occupied volume, clonal density, and variation of distances between individual cells comprising the clone (Figs. 1 and 2). Observations of such single compact clones in the whole embryo head allowed us to rule out the possibility of a long-range migration of individual early ectomesenchymal cells.
Fig. 1. Size, shape, and distribution of neural crest–derived ectomesenchymal clones.
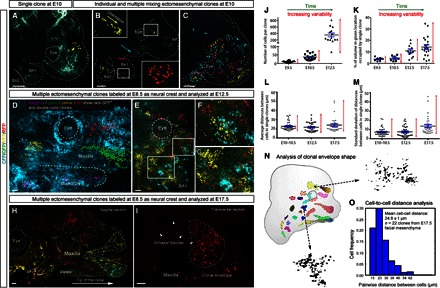
(A to C) Genetic tracing of neural crest cells and their progenies induced at E8.5 in Sox10-CreERT2/R26Confetti embryos and analyzed at E9.5 to E10. (A) Head of the E10 embryo with single YFP+ ectomesenchymal clone. Note the compact structure of the clone. (B) Multiple separated clones in different regions of embryo face. Yellow and blue arrowheads show the orientation of cellular groups. (C) Example of multiple overlapping clones in the early developing face. Note the intense local clonal mixing. (D to I) Genetic tracing of neural crest cells and their progenies induced at E8.5 in PLP-CreERT2/R26Confetti embryos and analyzed at E12.5. (D) Reconstruction of rare (RFP+CFP, YFP+CFP, RFP+YFP, and GFP-expressing) individual clones in the facial region of an E12.5 embryo. Note that some clones are markedly stretched in the anterior facial region. (E to G) Distribution of ectomesenchymal single-color–labeled clones in the periocular posterior maxillary region. Note the irregular geometry of clonal envelopes and their well-defined borders. (F and G) Magnified regions outlined in (E). (H) Sagittal section through the head of a genetically traced embryo starting from E8.5 and analyzed at E17.5. Area of the maxilla and frontonasal prominence with individual traced clones acquiring conical shape (dotted line) in the anterioposterior direction. (I) Transversal section through the upper jaw of the genetically traced E17.5 embryo. Note the compact shape and defined borders of the RFP+ clone (outlined by the dotted line). Arrowheads point at whisker follicles. (J) Graph showing the increasing size and variability of individual ectomesenchymal clones during facial development. (K) Graph showing the proportional occupied clonal volume and related variability of individual ectomesenchymal clones at different developmental stages. (L and M) Graphs visualizing developmental dynamics of clonal density (L) and its heterogeneity (M) measured as an average distance between cells of one clone (closest-neighbor approach) and SD of this parameter per clone, respectively. Bars show mean (black) and SEM (blue). (N) Examples of ectomesenchymal clonal envelopes from an E12.5 embryo with traced neural crest–derived progenies. Note the isotropic structure of clones and well-defined borders with irregular curvature. (O) Histogram showing spatial isotropicity based on pairwise distances between cells sharing a common clonal origin in experimentally obtained ectomesenchymal clones at E17.5. All images are maximum-intensity projections of confocal stacks, except (C) and (D) with volume rendering of isosurfaces. BA1 and BA2, the first and the second branchial arches; NP, nasal prominence. The eye is outlined by the dashed line. Scale bars, 200 μm (A to C and E to O) and 30 μm (D).
Fig. 2. Analysis of clonal structure in the faces of E10 control and cyclopamine-treated embryos.
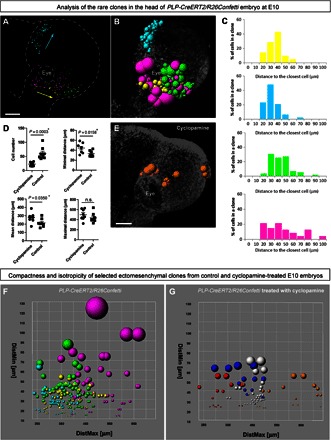
(A) Distribution of analyzed clones from PLP-CreERT2/R26Confetti head. Purple, yellow, and green colors mark different rare dual-color clones, whereas cyan represents the rare GFP+ clone. (B) Spatial structure of clones: Radiuses of colored spheres (representing single cells) correspond to minimal distances to the closest neighbors. When all cells in a clone are analyzed, this parameter reflects the compactness of the clone. (C) Histograms showing distances to the closest neighbor for every cell in the clone. (D) Graphs showing differences between wild-type and cyclopamine-treated ectomesenchymal clones in the faces of E10 embryos. Each point represents the average of the following parameters determined for each cell within a single clone: minimal distance, distance to the closest cell; mean distance, mean distance between one cell and all other cells in the clone; and maximal distance, distance to the furthest cell in the same clone. (E) Spatial structure of a clone from a cyclopamine-treated embryo. Radiuses of colored spheres (representing single cells) correspond to minimal distances to the closest neighbors. (F and G) Plots reflecting the compactness and clonal structure of analyzed clones from control (left) and cyclopamine-treated (right) E10 embryos. Colored spheres on these plots correspond to individual cells. Color defines a clone. The radius of spheres reflects the distance to the most proximal neighbor cell (DistMin) in a clone. DistMax is the maximal distance between a given cell and the most distal cell from the same clone. When the entire clone is analyzed, this parameter describes the spatial dimension of the clone. Scale bars, 200 μm (A and E). n.s., not significant.
For clonal analysis, with the help of the PLP-CreERT2 line, we focused on the rare double color combination–labeled clones in the front part of the head to clearly distinguish individual regions occupied by single clones (fig. S1). Results obtained from rare double-colored and GFP+ clones in PLP-CreERT2/R26Confetti embryos and from clonally titrated recombination events in Sox10-CreERT2/R26Confetti were in agreement with each other.
The cells inside the defined clonal envelopes appeared sparse at all investigated developmental stages and, as a result, we observed a high degree of clonal overlapping between the progenies of individual neural crest cells in any given location (Fig. 1, C and E to H). Despite such an extensive overlapping of the clones, all clones appeared to be spatially distinct with recognizable borders (Fig. 1, A to I).
Next, we looked into the cellular structure of ectomesenchyme clones at E10 to address their compactness, spatial heterogeneity, and size. The results showed that the cellular density in a typical clone drops from the center to the periphery of the clone. This was analyzed through measuring minimum and maximum distances between all neighboring cells of the same clone and plotting such distances in 3D space as attributes of every cell (Fig. 2). In terms of compactness, we observed a natural variation within clones. Still, they were rather similar to each other in wild-type embryos (Fig. 2, A to C and F). However, we managed to challenge the clonal arrangements, compactness, and heterogeneity by treating the embryos with cyclopamine 1 day before analysis. Facial development is known to be affected by cyclopamine, and the effects have been extensively studied in the past (29). In treated embryos, the clones were smaller and often dissimilar from each other, especially when compared to the control (Fig. 2, D to E and G). We often observed spatially distinct or even connected rare clonal patches of the same color code located in close proximity to each other in E9.5 to E10 embryonic heads (see Fig. 1B, next to the arrows). This suggests the presence of a dividing migratory neural crest cell producing progenies in a few neighboring locations within a restricted facial region.
In contrast to the arrangements in the face, migrating neural crest in the trunk did not show a similar clonal compact clustering with resolvable clonal borders (fig. S2, A to C). This suggests extensive mixing due to intense migration of individual cells.
Regardless of the significant increase in the number of cells in each clone in the growing face (Fig. 1J), the borders of the clonal envelopes remained visually defined (Fig. 1, A, I, and N), and the average distances between cells in the clone remained largely stable (Fig. 1L). However, 4 days after tamoxifen-induced genetic tracing of neural crest progenies, at E12.5 and onward, we observed an increasing variability in the size of the analyzed clones and in the proportional volume occupied by the cells of the clone within the clonal envelope (showing proportional contribution of this clone to the location) (Fig. 1, J and K). We also observed an increase in dispersion of the individual intercellular distances within a clone over time (Fig. 1M). Furthermore, the clones tended to attain a stretched asymmetric shape in regions with extensive growth in the head, which suggests a mechanism of directional clonal expansion in such locations (Fig. 1, D, H, and N). Surprisingly, the clonal progenies of labeled neural crest cells retained a compact structure even at later developmental stages, such as E17.5, as visualized on sagittal and transversal sections (Fig. 1, H, I, and O).
Clonal arrangements in the branchial arches did not differ from the nasal prominence and other facial locations in terms of defined clonal envelopes and clonal overlapping (fig. S2, D to F). Analysis of heart neural crest also showed a high degree of clonal overlapping that simultaneously occurs with clonal clustering of individual neural crest progenies (fig. S2, E and F, arrowheads).
Coexistence of neural crest–derived ectomesenchyme and mesodermal derivatives in the early face
The existence of well-defined clonal envelopes is supported by the manifestation of the border between neural crest– and mesoderm-derived mesenchymal derivatives that persists to postnatal stages. To visualize this border at postnatal stages, we used conditional knockout of Sox2 (30) in neural crest cells, using Wnt1-Cre (31).
The results of this experiment showed that only facial follicles corresponding to Sox2-deficient areas demonstrated a change in hair color shade—instead of black, it became brownish, which highlighted the border between neural crest– and mesoderm-derived dermis in young postnatal animals (fig. S3, A to Q). This border between neural crest– and mesoderm-derived mesenchyme was also observed during embryonic stages (fig. S3, R to T). Expression of dopachrome tautomerase (DCT) showed that melanocytes survived well in the targeted tissue and that the pigment-producing pathway was in place in both wild-type and Sox2-deficient melanocytes. The melanocytes did not express Sox2, as opposed to dermal papilla cells (fig. S3, B to Q). This is in line with the fact that expression of Sox2 is generally incompatible with melanocyte fate (32). Therefore, the outcome of Sox2 deficiency in skin must result from an interplay between melanocytes and dermal papilla cells. Indeed, melanocytes communicate with dermal papilla cells to adjust the amounts and the ratio of different pigments (33), which involves the Agouti pathway and explains how animals dynamically change pigmentation patterns (34). Because dermal papilla cells in the head are produced from neural crest cells and the mesoderm, and loss of Sox2 affected only the neural crest–derived dermal papillae, we were able to detect the border between numerous ectomesenchyme (NCC)– and mesoderm-derived dermal papillae in head skin. We suggest that the presence of such visual border is an interesting and important observation, especially because the dermis is organized by fibroblast-like cells that presumably are capable of long-range migration during late embryonic development and afterward.
Both neural crest and paraxial head mesoderm contribute to a range of mesenchyme-derived structures, including skeletal elements that fuse without signs of different cellular origin. This indicates that clonal behavior and morphogenetic cell dynamics in paraxial mesoderm derivatives are similar to the clonal dynamics in neural crest–derived tissues. We used Mesp1-Cre/R26Confetti animals in 2D and 3D imaging to trace the progeny of paraxial mesoderm in the face (35). Mesoderm-derived mesenchyme showed a high degree of clonal overlapping not only in branchial arches but also in the dorsolateral aspects of maxillary and occipital regions (fig. S4, A to I). Additionally, local angiogenic progenitors in multiple places demonstrated an impressive diversity of clonal origins based on their color code (fig. S4, J and K). Numerous mesoderm-derived mesenchymal domains sharing the same color code appeared compact and local, similar to ectomesenchymal clones in the face (fig. S4, E to I; see arrowheads). These data point toward the possibility that similar morphogenetic mechanisms operate in neural crest– and mesoderm-derived mesenchymal tissues.
Next, we wanted to investigate the volume proportions occupied by the mesoderm and the neural crest, respectively, in different localities in the developing face. We also wanted to calculate the number of neural crest–derived clones that contribute to a given location within selected regions of interest (ROIs). For this, we made 3D analyses of the traced mesodermal (high efficiency of recombination) and neural crest (low efficiency) progenies in various facial regions, including the nasal prominence and branchial arches. The analyses showed that mesodermal derivatives occupied 21.01 ± 3.46% of ROI volume (mean and SEM; n = 9) in the face at E10.5 (fig. S4, L to P). Furthermore, we calculated that single neural crest–derived clones contributed to 4.465 ± 0.8844% of ROI volume (mean and SEM; n = 7) at the same stage (see Fig. 1K). Thus, we concluded that up to 17 neural crest clones may contribute to one ROI location.
Modeling ectomesenchymal cell dynamics in 2D and 3D confirms the minimal role of individual cell migration in clonal overlapping
As seen from above, most of the observed ectomesenchyme-derived clones in the face show complex clonal envelope shapes (Fig. 1N), with the borders representing sharp drops of cellular density at the periphery of the clone (Figs. 1O and 2C). Highly variable and complex shapes of the clonal envelopes cannot be explained without the assumption that complex morphogenetic processes operate in the tissue.
Given that the ectomesenchyme is a potentially migratory tissue, we asked whether migratory behavior contributes to the development of facial shape and, if this is the case, how the selectivity and directionality of cell migration can be achieved. On the basis of our previous results, we wanted to understand how the experimentally observed sharp borders of clonal envelopes can be maintained in the case of migratory behavior of facial neural crest–derived cells. To address this issue, we devised a mathematical model that operates virtual cells in 3D space plus time. We tested a group of variables, such as cell division speed, migration, and allocation of daughter cells in random or defined directions, as well as pushing of newborn mitotic products during proliferation. We compared the readouts from the series of in silico simulations to the results of our experimental clonal analyses. We looked for parameter combinations in the model that gave rise to dynamics with degrees of overlapping and clonal shapes that were compatible with those observed in the experiments. Unexpectedly, the 2D version of the model showed that proliferation-driven cell pushing/place-exchanging and related short-distance movements are sufficient to achieve efficient clonal overlapping over time (Fig. 3, A and B). In this model, we assumed (based on the biological data including live imaging experiments described below) that products of cell divisions would push neighboring cells to obtain space. Alternatively, they could intermingle with close neighboring cells by exchanging positions with them, as a way of accommodating the pushing forces in the growing tissue. Such short-ranged spatial rearrangements require some dynamics of cell shape. This can be rendered as a very short range migratory behavior. Indeed, simulations in 3D space confirmed that clonal overlapping is compatible with zero or minimal individual migratory behavior, whereas proliferation appeared to be the main driving force for the mixing of the cell division products. Moreover, well-defined clonal envelopes were maintained only when individual migration (in relation to neighboring cells) was minimal in the simulated system. An in silico model, while showing defined clonal patches, also demonstrated a drop in clonal compactness from the center of the clone to the periphery in agreement with experimental data (Fig. 2, B, C, and F). Together, this strongly suggests that clonal overlap in authentic facial development may occur owing to the spatial allocations and mingling of daughter cells that result from cell divisions exclusively. In such a case, the products of mitoses will push and blend in the directions of surrounding domains, and through this, the clonal progenies will intermix.
Fig. 3. Mathematical modeling of clonal expansion and overlapping in 3D space.
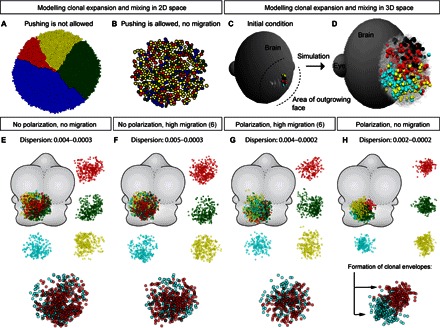
(A and B) 2D modeling of cell dynamics; initially traced clones are labeled by different colors. Note that in the condition where cell pushing is allowed (B), clonal overlapping is efficiently achieved without any migratory behavior. (C and D) Visualization of initial conditions and example of in silico cell dynamics simulation in 3D space. (E to H) Results of in silico cell dynamics simulation in 3D space performed with different settings. Note the formation of defined clonal envelopes under the condition where polarization probabilistically directs oriented cell divisions with minimal individual cell migration (H).
Oriented divisions and crowd movements of ectomesenchymal cells participate in facial shaping
The modeling of fine borders of clonal envelopes remained imperfect as compared to the experimental data, unless the concept of polarizing morphogen gradients was considered. Therefore, we introduced a local radial gradient that affects the directionality of cell division and allocation of daughter cells in 3D space into the in silico model. The results of these simulations demonstrated that the oriented cell divisions (or controlled allocations of the progeny after mitosis) together with minimal individual migratory behavior are the keys to achieving the distribution of virtual clonal progenies that resemble the actual clonal patterns in facial development (Figs. 3, C to H, and 4). Experimental data on the stretched geometry of clonal envelopes in the most outgrowing parts of the face additionally point toward oriented cell divisions during progressive shape development (consistent with Fig. 1, D and H). At the same time, the distribution of cells within the analyzed clonal envelope appeared to be relatively uniform as defined by variation in individual cell-to-cell distances (measured as a distance to the closest neighbor) (Figs. 1O and 2C).
Fig. 4. Mathematical model predicts widening versus elongation strategies of in silico facial development depending on the conditions.
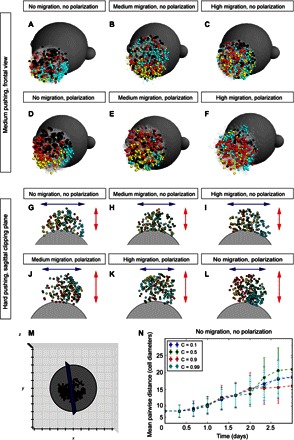
(A to F) Simulations of cell dynamics in a 3D environment with medium pushing power (technical value in a model c = 0.5). (G to L) Clipping planes, view from the top on simulations of facial development. Note how different conditions lead to widening or elongation of the cellular cluster with clonal envelopes. The technical value for high pushing power in a model was c = 0.9. (M) Example of the clipping plane, frontal view. (N) Graph showing the changes in the spatial homogeneity (isotropicity) of simulated clones during rounds of simulation.
This was similar to the in silico simulations, where we found that polarization (and no migration) was characterized by the lowest dispersion of distances between the cells (Fig. 3H). In silico simulations demonstrated a clear difference in elongation versus widening of the modeled facial structure, depending on the presence of an anterioposterior polarizing gradient (Fig. 4, G to L).
To conclude, the mathematical model predictions pointed toward the likelihood of locally controlled allocations of daughter cells after mitosis. Such allocations can result from oriented cell divisions and controlled spindle orientation following anterioposterior polarization cues in the tissue.
In such a case, most of the allocations of newborn cells occur predominantly in one direction. This reduces the “lateral” clonal mixing because the pushing power of clonal mitotic products that invade lateral clonal domains during proliferation is eased. This should lead to reduced clonal blending and to the efficient maintenance of defined clonal envelope borders while having an impact on the resulting shape in terms of widening versus elongation (Fig. 4, especially G to L).
To directly test the predictions from the model, we analyzed Wnt5a knockout mouse embryos (36). Wnt5a is a noncanonical Wnt and a member of the PCP pathway that is involved in polarizing epithelial and mesenchymal tissues. Indeed, the knockout of Wnt5a leads to massive shortening and widening of the face, starting from preskeletogenic time points (Fig. 5, A to J). The lower jaw appeared much shorter and wider. At the same time, our data showed that the volume of the lower jaw was not reduced as compared to the control (Fig. 5, F and H). This indicates that there was no deficit in proliferation and in the number of cells that formed the face. This is similar to the modeling predictions shown in Fig. 4 (G to L). Moreover, this phenotype does not influence the induction of all necessary facial structures, including cartilages, bones, sensory compartments, muscles, glands, and developing teeth, as monitored by x-ray micro–computed tomography (μ-CT) scans and visualizations (Fig. 5, E to P). Additionally, we did not observe any changes in the general position of major anisotropic proliferative zones, as analyzed with ethynyl deoxyuridine (EdU) incorporation assay at E12.5 (Fig. 6, Q and R). Strikingly, when we examined the orientation of cell divisions using an antibody against phospho-histone 3 (PH3; commonly used to visualize metaphase plate) in the anterior face, we found that the predominant anterioposterior orientations of mitotic products were no longer prevalent in Wnt5a mutants at E12.5 (Fig. 5, S to V). This result is in full accordance with the in silico simulations (Figs. 3 and 4, G to L).
Fig. 5. Disruption of Wnt/PCP-driven cell polarization and subsequent changes in facial shape.
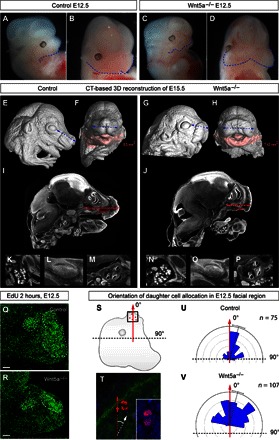
(A to D) Representative images of E12.5 embryos: littermate controls (A and B) and Wnt5a full knockout embryos (C and D). (E to H) μ-CT–based 3D reconstructions of the E15.5 embryonic heads: littermate controls (E and F) and Wnt5a full knockout (G and H). Note the short snout and wider face of the mutant embryos as compared to the control littermates. Pink color code shows the mandible and its volume calculated for control and mutant embryos. (I to P) μ-CT–based sagittal section through the head of the control (I and K to M) and mutant (J and N to P) embryos with the measurements of the length between the posterior part of the olfactory system and the anterior tip of the snout. (K to P) Tomographic slices through the salivary gland, developing the molar and the inner ear from control (K to M) and Wnt5a mutant (N to P) embryos. (Q and R) Distribution of high proliferation zones in control (Q) and Wnt5a knockout mutant (R) E12.5 embryonic mandibles that were treated with EdU for 2 hours before analysis. Dotted lines outline stereotypic, highly proliferative zones. (S) Scheme of the ROIs where the oriented cell divisions were quantified in the head of E12.5 embryos. (T) Example of chromosome PH3–based staining on the section of an E12.5 embryonic head; the white arrow points toward the outgrowing part of nasal prominence, whereas the red arrows show the orientation of cell division and the allocation of daughter cells. (U and V) Rose diagram of quantified orientations of cell divisions in the anterior face of E12.5 control (U) and Wnt5a knockout mutant (V) embryos. Note the disruption of the directionality of cell divisions in the mutant embryo as compared to the control littermate. Scale bars, 200 μm (Q and R).
Fig. 6. Live imaging of early zebrafish craniofacial development.
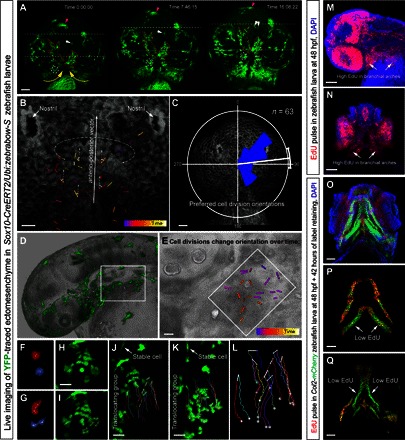
(A to L) Live imaging of genetically traced neural crest–derived progenies in Sox10-CreERT2/Ubi:zebrabow-S zebrafish larvae between 30 and 56 high-power field (hpf) (A to C) and 30 and 42 hpf (D to L). (A) Ventral view on zebrafish larva head with genetically labeled groups of YFP+ cells. Note the spatial stability of translocating labeled groups during organized cell movements in the regions of expanding brachial arches and around the stomodeum. The dotted circle and white arrows show small defined trackable groups of cells. Purple arrows point at the melanocyte. The red dotted line shows how the borders of YFP+ groups change over time. Yellow arrows demonstrate the major direction of crowd movement. (B and C) Analysis of oriented cell divisions during live imaging of developing zebrafish head, ventral view. (B) Bars show orientations of individual cell divisions and color code corresponds to the timing of cell division. (C) Rose diagram of orientations of cell divisions. (D to L) Cell divisions in the branchial arch of zebrafish at 30 to 42 hpf, side view. (E) Magnified region from (D). Note that cell divisions change the predominant orientation over time. (F to L) Analysis of the group of cells from the region outlined by the white rectangle in (D). (F and G) Frames from time lapse with two dividing cells from the branchial arch. (H and I) Frame before (H) and immediately after (I) mitosis of several ectomesenchymal cells in the region, side view on the branchial arch. (J to L) Tracking of dividing ectomesenchymal cells and their progeny in the forming branchial arch. (J) First frame of tracking. (K) Final frame of tracking (12 hours). Arrows in (J) and (K) point at the stable YFP+ cell that does not change the position in the embryo and serves as a stable orientation anchor for measuring translocation/crowd movement of the labeled group of ectomesenchymal cells. Note that during the displacement of the entire group, most of the cell division products stay proximally close to each other with some rare exceptions [dark brown track in (L)]. Despite this, high intensity of local cellular mixing is achieved owing to proliferation in accordance with modeling results presented in Fig. 2 (A to C). (M and N) EdU incorporation shows proliferation rates in different parts of the developing zebrafish head. Note that a 5-min EdU pulse at 48 hpf immediately followed by the analysis showed high proliferation rates in branchial arches (arrows). (O to Q) Transgenic Col2a1aBAC:mCherry zebrafish larva’s entire head (O) and skeletal elements (P and Q) at 4 days postfertilization (dpf) with incorporated EdU, administered at 48 hpf for 5 min. Note that this EdU label–retaining experiment highlights uneven proliferation in ectomesenchymal chondrogenic progenitors at 48 hpf (5-min pulse). Arrows point at low EdU-retaining regions in the facial cartilages. Scale bars, 50 μm (A, C, M to O, and Q) and 20 μm (B, E, and H to K).
Live imaging in zebrafish reveals oriented crowd movements of large cellular groups
To directly observe the dynamics of early shape development in the facial region, and to connect it with individual cellular behavior, we used live imaging of genetically traced zebrafish larvae. For this, we took advantage of the Sox10-CreERT2/Ubi:zebrabow fish line, which is suitable for clonal color coding and inducible lineage tracing in neural crest cells. The nonrecombined cells express RFP, whereas cells after recombination can acquire stable YFP or CFP expression in the lineage. Because of imaging constraints, we followed mostly YFP+ neural crest–derived clones during early craniofacial development.
First, the results of this experiment demonstrated that neural crest cells labeled at clonal density give rise to spatially separate and compact groups of cells. These groups are analogous to the ectomesenchymal clones that we observed in mouse embryos (fig. S5, A to D, arrows). Second, live imaging clearly demonstrated that individual ectomesenchymal cells do not migrate significant distances but rather rearrange their shape and accommodate their position within their local microdomain (Fig. 6 and movies S1 to S4). This complies with the theoretical prediction from the mathematical model (see Fig. 4). As a result, the labeled cellular clusters change their shape over time (Fig. 6A) but do not alter their basic cellular structure. This structure includes cell density and coordinated orientation of cell divisions that may change over time (Fig. 6, B to L, and especially E).
The progenies of dividing ectomesenchymal cells remain in close proximity to each other in most of the cases (see tracks of the progeny in Fig. 6, J to L). In agreement with the inference from the mathematical model, these ectomesenchymal cells mostly divide in certain orientations within spatial microdomains, as observed in live imaging (Fig. 6, B to E). This, too, is consistent with the results obtained in the mouse model (see Fig. 5, S to V).
At the same time, ectomesenchymal cells relocate to new distant positions in the embryonic head in synchronized crowd movements (Fig. 6, J to L, and fig. S5, E to P). These movements stem from pushing activities in major proliferative centers, located in, for example, branchial arches, as evident from EdU analysis (Fig. 6, M to Q, and movie S5) and direct live observations (movies S1 to S4). Anisotropic proliferation is apparent in larval ectomesenchyme. This is reflected in results from an acute EdU incorporation experiment (Fig. 6, M and N) as well as in EdU label–retaining analysis in ectomesenchyme-derived cranial cartilage (Fig. 6, O to Q). Application of EdU for 15 min at 48 hours and analysis at 4 dpf showed an uneven, symmetrical, and specific pattern of EdU incorporation and retention in skeletal structures that form after 48 hours of development from ectomesenchymal cells. Note that the translocating groups of ectomesenchymal cells behave similarly to viscous liquid (fig. S5, E to P, and movies S1 to S4). Such morphogenetic movements may account for specific distortions and complex geometries of clonal envelopes, as shown in Fig. 1N.
To sum up, these results agree with computer simulations and are consistent with observations of clonal patterns in mouse embryos. Together, they suggest that crowd movements, anisotropic proliferation, and cell divisions with natural-like variation of the cell cycle length and phase play a significant role in facial morphogenesis, whereas long-range individual migration is limited in the face after neural crest cells transform into neuroglial and ectomesenchymal components.
Early ectomesenchymal cells are oligopotent within a given locality
The establishment of facial structures by ectomesenchymal clones raises the question whether each clone displays restricted and defined competence to generate only specific cell types. Hypothetically, overlapping clones may have committed to distinct fates and later give rise to different derivatives within a single locality. We tested this assumption using lineage tracing and fate analysis of rare double-colored and single-colored clones (Fig. 7). The results instead demonstrated that multiple fates are often generated within one compact clone. Thus, an individual clone may contribute to, for example, local mesenchyme and to dermal papillae of whisker follicles (Fig. 7, A and B). In other situations, single clones contributed to the dental mesenchymal compartment and the surrounding osteogenic tissue (Fig. 7, C to E). We often observed that perichondrial cells, chondrocytes, and neighboring mesenchymal patches shared the same clonal origin (Fig. 7, F to K). Osteogenic cells of membranous bones and neighboring mesenchyme also showed clonal relationships (Fig. 7, L and M).
Fig. 7. Cell fate potential of individual neural crest–derived ectomesenchymal clones.
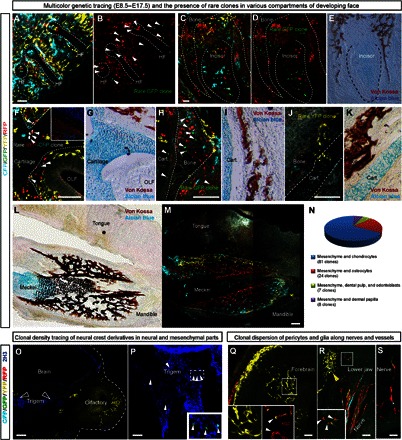
(A to I) Genetic tracing of neural crest–derived progeny in PLP-CreERT2/R26Confetti embryos. Tamoxifen was injected at E8.5, and the analysis of cell types was performed at E17.5. (A and B) Close-up of the region in the anterior face with outlined whisker follicles. Note that cells of the GFP+ rare clone (arrowheads) contribute to both the surrounding mesenchyme and dermal papillae. (C to E) A developing incisor is shown with the dotted line. Note the contribution of the rare GFP+ clone to the peridental mesenchyme, osteoblasts, and the dental mesenchymal compartment. (F and G) Cells of the rare CFP+/RFP+ clone, pointed out by arrowheads, give rise to perichondrial flat cells lining the cartilage, chondrocytes in the olfactory cartilage, and mesenchyme surrounding the olfactory neuroepithelium. (H and I) Arrowheads show cells of the rare GFP+ clone that contribute to cartilage (chondrocytes), trabecular bone (osteocytes), and the surrounding mesenchyme. (J and K) Rare YFP+ clone contributes to the mesenchyme and chondrocytes of the nasal capsule. (L and M) Localized YFP+ and CFP+ clones occupy only a part of a lower jaw and contribute to mandibular bone, Meckel’s cartilage (in the case of the CFP+ clone), and the surrounding loose mesenchyme. (E, G, I, K, and L) Von Kossa staining highlights trabeculae of the facial bones, whereas Alcian blue staining shows cartilage. (N) Diagram representing the occurrence of different cell fate combinations observed within individual clones. (O) Clonal density genetic tracing induced at E8.5 and analyzed at E12.5 in Sox10-CreERT2/R26Confetti embryos. Note the absence of genetically traced cells in the neuroglial compartment despite numerous ectomesenchymal clones present in the location. (P) Example of a GFP+ rare clone (arrowheads) in the neuroglial compartment that does not include ectomesenchymal derivatives. (Q) Part of the network of pericytes in the forebrain is organized by the single yellow clone. NG2 is a pericyte marker, shown in the inset. (R and S) Several traced neural crest–derived glial clones, including a rare YFP+/RFP+ double-colored clone (magnified in the inset), follow the nerve fibers in the lower jaw. For comparison, note the compact ectomesenchymal YFP+ clone in the upper left corner of the image. Scale bars, 200 μm. HF, hair follicle.
None of the analyzed unique ectomesenchymal clones showed any progeny in the neuroglial compartment (Fig. 7O). Consistently, several rare analyzed clones in the neuroglial compartment did not appear to share the same color code with cells in the ectomesenchymal domain (Fig. 7P).
Embryonic ectomesenchyme-derived pericytes (marked by NG2) were found to spread clonally by dividing along the vessels. This is similar to how glial cells clonally spread along nerve fibers (Fig. 7, Q to S). To conclude, labeled neural crest cells give rise to oligopotent clonal local patches of ectomesenchyme that generate different fates that are necessary in a specific location.
DISCUSSION
The facial region is largely composed of neural crest–derived progenies. The purpose of the present study was to investigate how those progenies dynamically build the face. Previously, an elegant clonal analysis has been performed in the trunk neural crest derivatives that include neurons, glial cells, and melanocytes. Many important questions related to both trunk crest multipotency and techniques concerning Confetti multicolor tracing were resolved in that study (14). However, trunk neural crest and cranial neural crest are different in their ability to give rise to mesenchymal structures. Consequently, knowledge of the clonal structure and cell behavior in relation to early morphogenesis of the face has remained obscure.
We addressed this question with clonal analysis using a multicolor Confetti reporter and found that neural crest cells give rise to well-defined clonal patches (clonal envelopes) of ectomesenchymal tissue in the prospective facial compartment. They demonstrate conspicuous borders and consist of clonal progeny formed from a single labeled neural crest cell. Once formed, these clonal patches persist until at least late embryonic development and most likely postnatally: They occupy certain locations in the face and retain recognizable borders. The patches do not dissolve with developmental time despite some growing heterogeneity inside the clone in terms of intercellular distances. The cells within the clonal envelope are loosely packed and mix locally with the progenies of other neural crest cells. Developmental robustness of the facial compartment might be rooted in a high degree of clonal overlapping in any given locality that is populated by the progenies of at least several individual neural crest cells. Hypothetically, negative somatic mutations occurring in a single migrating neural crest cell or at the level of the neural tube can be compensated by other unaffected neural crest–derived local clonal progenies. Moreover, the intense clonal overlapping among neural crest progenies argues against any somatotopic mapping (37) at the level of the neural tube. The increasing variability in geometrical shape and size of the clones might reflect directional cell behavior unique for a given locality, with different proliferation rates in various compartments of the outgrowing face.
We took advantage of mathematical modeling to sort out these potential mechanisms of clonal behavior. The 3D in silico simulations of cell dynamics showed that highly efficient clonal overlapping can be achieved without migration of individual cells, solely due to the pushing and mingling force of cells as a result of multiple cell divisions in the area. Moreover, the model predicted the existence of a chemical gradient that controls the orientation of cell divisions, to achieve defined clonal envelopes. We set out to test the suggestions from the model regarding both the minor migratory behavior and the directional cell dynamics related to cell divisions with subsequent allocations of daughter cells. Live imaging experiments in zebrafish confirmed our computer model predictions that ectomesenchymal cells do not perform extensive individual migrations and predominantly divide in few orientation planes within a local microdomain. Moreover, similar to the situation in the developing limb (17), we discovered that ectomesenchymal cells execute large-scale, collective, coordinated morphogenetic movements, where the cellular arrangements of microdomains remain well preserved. Such crowd movements, reminiscent of the behavior of viscous fluid, have been previously described (17, 38). The viscous fluid approach was previously used for analyses of biological systems on cellular and organismal levels (39). According to our results, the ectomesenchymal cells in the zebrafish lower jaw translocate or perform crowd movement in relation to the eyes and the brain. This takes place while the stomodeum is displaced anteriorly, presumably as a result of lower jaw mesenchyme outgrowth. The massive relocation of ectomesenchymal cells occurs because of proliferation in the lateral regions of the branchial arches and does not involve independent migration of each cell. This is supported by the fact that differentiating chondrocytes are still moving forward together with the surrounding tissue during lower jaw extension (see movie S3). Thus, it seems that individual ectomesenchymal clones mix and overlap as cells are added because of cell divisions. This causes subsequent mingling with the neighbors without obvious middle- or long-range individual cell migration. However, translocations of large groups of cells (that is, crowd movements) might be responsible for the changes in clonal envelope 3D geometry during critical morphogenetic rearrangements.
The individual shape of clonal envelopes reflects the anisotropic growth of the structure (40, 41) following local orientation cues. Our results show that the cues that orient the plane of cell divisions in the face, at least in part, are represented by the gradient of Wnt5a, which influences the allocation of daughter cells after mitosis and, through this, the general shape. The idea of a Wnt5a gradient is strongly supported by the similarity of the phenotypes resulting from full Wnt5a knockout and overexpression of Wnt5a (21–23). In both cases, the gradient is erased from the tissue, which is phenotypically manifested by the shortening and widening of the face and limbs. Thus, the molecular mechanisms controlling the emission, detection, and interpretation of polarity signals (including noncanonical Wnts) can be partly responsible for the early body plans as well as for the developmental origins of facial diversity (42), especially during early preskeletogenic stages. Despite the strong phenotype in Wnt5a and in other PCP mutants, many parameters related to the shape and placement of different structures are not seriously affected. Therefore, there must be other mechanisms unrelated to directional cell divisions that control the facial shape. Anisotropic proliferation rates in different facial compartments and resulting crowd movements/translocations of cells can provide an alternative way of governing the shape.
Progressively, ectomesenchymal cells that belong to the same clone adopt different fates following odontogenic, chondrogenic, osteogenic, adipogenic, and other directions of differentiation within a spatial microdomain. Therefore, single neural crest–derived ectomesenchymal early progenitors are oligopotent in the face. Our data do not support the fact that different pools of neural crest cells contribute to prespecified ectomesenchymal populations restricted in their fate potential within the mesenchymal spectrum of fates. However, the results may partly support previously described early fate restrictions in the cranial neural crest and the existence of an early choice between neuroglial and mesenchymal directions of differentiation (15). This renders cranial neural crest cells different in early fate restrictions as compared to cells of the trunk neural crest (14).
Various early embryonic mesenchymal populations, similar to neural crest–derived ectomesenchyme, give rise to multiple differentiated cell types that organize muscle, connective, and skeletal tissues in the head and other compartments, including limbs. Our results show that cellular mechanisms of early facial shaping seem to be very similar to those operating in the limb. Furthermore, recent discoveries of epithelial-to-mesenchymal transition (EMT), a classical property of neural crest (43), in limb bud initiation (16) suggest that similarities between limb and face development are stronger than previously thought. Moreover, a recent study demonstrated that cranial neural crest cells that give rise to mesenchymal derivatives in the head may undergo EMT from a neural fold domain that might not express neural markers (15). If true, it can be defined as a non-neural ectoderm and is possibly similar to analogous sites in regions of future limb buds. For instance, it is widely accepted that facial growth and patterning are regulated by the frontonasal ectodermal zone (FEZ), which includes SHH and FGF8 expression domains (44, 45). The roles of BMPs, endothelins, and other soluble factors in facial development and outgrowth have been thoroughly investigated (44, 46). Variation of signals that affect cartilage and bone development may also influence shaping programs at later stages. This is suggested, for example, by studies on BMP3 mutations associated with the size and varying geometry of the vertebrate skull (47). Apparently, these key signals, including SHH, FGFs, and BMPs, play critical instructive roles in both facial and limb development. It could be speculated that the apical ectodermal ridge secreting FGF8 and the zone of polarizing activity emitting SHH in limb buds might be considered to be deeply homologous to the FEZ in the face. Thus, a blueprint of the cellular and molecular logics that operate in the mesenchyme of the anterior head could become a starting point for the induction of appendages in the more posterior parts of the ancestral body. Some evidence suggest that the origin of paired appendages involved redeployment of genetic programs from the paraxial to the lateral mesoderm (48, 49). Our data highlight a great degree of similarity in clonal dynamics between neural crest– and paraxial mesoderm–derived mesenchyme in the face and branchial arches. Together, our results support a profound similarity between vertebrate face and limb development and, in turn, raise questions concerning a deep homology (50) between these seemingly unrelated structures.
MATERIALS AND METHODS
Experimental design
This study heavily relied on clonal density genetic tracing and on the investigation of clonal envelopes in facial ectomesenchyme both as a series of static pictures and as a dynamic live imaging of mouse and zebrafish embryos, respectively. In silico modeling of clonal envelopes in developing ectomesenchyme helped to visualize and define principles of clonal overlapping in 3D envelope border formation and other important aspects leading to the prediction of a gradient that orients cell divisions. Investigation of a Wnt5a mouse mutant with deficient polarizing signals confirmed the importance of oriented cell dynamics in facial development.
Mouse strains and animal information
All animal work was approved and permitted by the Ethical Committee on Animal Experiments (Norra Djurförsöksetiska Nämnd; www.djurforsok.info/lagar-regler/) and conducted according to the Swedish Animal Agency’s Provisions and Guidelines for Animal Experimentation, and international guidelines and regulations were followed (Institutional Review Board/Institutional Animal Care and Use Committee). Glia-specific genetic tracing mouse strains PLP-CreERT2 and Sox10-CreERT2 were previously described (25, 26). Both PLP-creERT2 and Sox10-creERT2 strains were coupled to R26Confetti mice that were received from the laboratory of H. Clevers (24). To induce genetic recombination of adequate efficiency, pregnant females were injected intraperitoneally with tamoxifen (Sigma, T5648) dissolved in corn oil (Sigma, C8267). Tamoxifen concentration ranged from 1 to 5 mg per animal to obtain a range of recombination efficiency. Wnt5a full knockout embryos were obtained from Wnt5a+/− mice (36) at the expected Mendelian proportions. Mesodermal tracing was obtained using Mesp1-Cre mouse strain (35) coupled to R26Confetti reporter strain. Sox2fl/fl mice have been previously described (30) and were coupled to Wnt1-Cre (31) that were ordered from the Jackson Laboratory (stock number 003829). For embryo analyses, heterozygous mice of the relevant genotype were mated overnight, and by noon, the plug was considered to be E0.5. Mice were sacrificed by isoflurane (Baxter, KDG9623) overdose, and embryos were dissected and collected into ice-cold phosphate-buffered saline. Subsequently, the samples were placed on freshly prepared 4% paraformaldehyde and, depending on the developmental stage, were fixed for 3 to 6 hours at 4°C on a roller. Afterward, the embryos were cleared in Scale-A2 reagent [4 M urea, 0.1% (w/v) Triton X-100, 10% (w/w) glycerol, distilled water] for 6 hours and imaged in whole-mount mode or, alternatively, the embryos, after fixation, were cryopreserved in 30% sucrose (VWR, C27480) overnight at 4°C, embedded in optimal cutting temperature medium (Histolab, 45830), and cut into 14- to 150-μm sections on a cryostat (Microm), depending on the subsequent application.
For cyclopamine treatments, three time-mated PLP-CreERT2/R26Confetti females were injected with cyclopamine (LC Laboratories) solution that was administered in corn oil via double intraperitoneal injections (30 mg/kg in each injection; days 8 and 9 of gestation). On gestation day 10, we took out and analyzed more than 16 individual embryos.
μ-CT analysis
We used a GE Phoenix v|tome|x L 240 equipped with a 180-kV/15-W maximum-power nanofocus x-ray tube and a high-contrast flat panel detector DXR 250 with 2048 × 2048 pixels and a 200 × 200 μm pixel size. The exposure time was 900 ms for every 2000 positions. The μ-CT scan was carried out at an acceleration voltage of 60 kV and at an x-ray tube current of 200 μA. The voxel size of the obtained volumes was in the range of 4 to 6 μm, depending on the size of the embryo head. The tomographic reconstruction was realized using GE Phoenix datos|x 2.0 3D computed tomography software. The 3D and 2D cross section visualizations were performed with VGStudio MAX 2.2 software. The histogram of the images was adjusted to reach a better contrast of soft tissues.
Fish in vivo experiments
For genetic tracing in the zebrafish model, we used heterozygous hybrids obtained from crossings between Sox10:ERT2 (51) and ubi:Zebrabow-S (52) transgenic fish strains. The recombination was induced by application of 1 μM 4-hydroxytamoxifen (Sigma-Aldrich) into E3 medium at 16 hpf, and embryos were incubated for 12 hours at 28.5°C. All other manipulations with embryos were performed according to the standard methods described by Westerfield (53). Live imaging was performed using a Zeiss LSM 780 (Carl Zeiss) confocal microscope through existing protocols with minor modifications (54). For further image processing and calculations, we used ZEN 2012 (Carl Zeiss) and Imaris software (Bitplane). For EdU analysis in developing zebrafish, we applied EdU to E3 medium at a concentration of 1.5 mg/ml for 5 min. We used Col2a1aBAC:mCherry transgenic fishes to visualize a cartilage in EdU-stained individuals.
Computational model
Individual cells were represented by their location on an underlying unstructured lattice, such as those typically used in finite element computations. The lattice resolution was chosen such that the average cell size is close to the desired biological cell size. Cell division and migration events were executed by inserting a new cell at a lattice site adjacent to the mother cell (division) or by swapping location with an adjacent cell (migration). Unlike many similar implementations of multicellular on-lattice models, we accounted for cell pushing; that is, if a dividing cell attempts to place its progeny at an occupied site, there is a probability that the occupying cell gets pushed to make room for the daughter cell. This means that cells can continue to divide even if all neighboring sites become occupied, rather than becoming quiescent. Because we are interested in capturing naturally occurring variations in the process, our model is stochastic. Stochasticity enters by letting the cell division time and the migration time become normally distributed random variables where the mean and variances are parameters that can be tuned to vary the degree of noise in those processes. The division direction and the migration direction are, in the absence of a polarizing field, uniformly distributed random variables. The degree of randomness in the cell division directions can be modulated by introducing a polarizing field and by tuning via a parameter how strongly the cells respond to this field, allowing us to vary the cell division direction from being completely random to becoming highly directionalized. The model is simulated as an event-driven system by a kinetic Monte Carlo algorithm, where the event with the shortened sampled waiting time is executed in each iteration. Detailed information about the mathematical model is provided in the Supplemental Materials.
Microscopy
Frozen samples were sectioned at 14 to 50 μm, and the sections were stored at −20°C after drying for 1 hour at room temperature. Confocal microscopy was performed with Zeiss LSM 710 CLSM and Zeiss LSM 780 CLSM instruments. The settings for the imaging of Confetti fluorescent proteins were as previously described (24). The imaging of the confocal stacks was performed using a Zeiss LSM 780 CLSM, Plan-Apochromat 10×/0.45 M27 Zeiss air objective, with 23 to 79 optical slices of 12 μm each with a z-axis shift of 9 μm for every step. Before whole-mount imaging, the embryos (from E9.5 to E12.5) were cleared in Scale-A2 reagent for 6 hours. For basic image processing and analysis, we used ZEN 2012 software.
Immunohistochemistry, tissue stains, and EdU analysis
The following primary antibodies were used: goat anti-GFP (fluorescein isothiocyanate) (Abcam; 1:500), goat anti-Sox10 (Santa Cruz Biotechnology; 1:500), anti-neurofilament 2H3 (generated by Developmental Studies Hybridoma Bank; 1:100), Tuj1 (Promega, G712A; 1:1000), NG2 (Millipore, AB5320; 1:200), anti-PH3 (Millipore, clone MC463; 1:1000), 4′,6-diamidino-2-phenylindole (DAPI; with Vectashield mounting medium for fluorescence; H-1200, Vector Laboratories Inc.), DCT (gift from V. Hearing; 1:1000), and rabbit anti-Sox2 (Abcam, AB97959; 1:1000). For the detection of the abovementioned primary antibodies, we used secondary antibodies produced in donkey conjugated with Alexa Fluor 405, 488, 555, or 647 (Invitrogen; 1:1000). Slices were mounted with 87% glycerol mounting medium (Merck). EdU (Life Technologies) was injected 2 hours before the embryos were harvested at a concentration of 65 μg/g. Cells with incorporated EdU were visualized using a Click-iT EdU Alexa Fluor 647 Imaging Kit (Life Technologies). For von Kossa staining of the bone, we submerged the cryosections into silver solution (50 g/liter of silver nitrate in distilled water). Then, we exposed sections to white light from the laboratory lamp for 20 to 50 min. Next, silver nitrate was extensively removed with distilled water during three 7-min washes. After the washes, we placed cryosections into thiosulfate solution (50 g/liter in distilled water) for 5 min. Finally, we performed three sequential 5-min washes in distilled water and mounted microscopic slides with glycerol for imaging. For Alcian blue staining of the cartilage, we used Alcian blue solution (pH 2.5; 1 g of Alcian blue 8GX in 100 ml of 3% glacial acetic acid). Cryoslides were hydrated in distilled water and then kept in 3% acetic acid for 3 min. Then, the slides were transferred to Alcian blue solution and microwaved for 30 s. Afterward, depending on the strength of the signal, the slides were left with Alcian blue solution or were immediately washed in water twice for 5 min and mounted with glycerol.
Image analysis and statistics
All statistical data in the figures are represented as means ± SEM. Every dot on the graphs in Fig. 1 corresponds to one analyzed clone. The unpaired version of Student’s t test was used to calculate the statistics (P value). All results were replicated in at least three different animals. We used Bitplane Imaris software for volume rendering and digital quantifications of occupied clonal volumes, measuring intercellular distances, automated cell counting, and producing maximum intensity projection images. For example, in Fig. 1 and fig. S1, rare double-colored clones in E9.5 and E10.5 and rare green clones in E17.5 were identified, segmented, and visualized in Imaris. For every clone on the graphs in Fig. 1 (M and N), we calculated 6 to 22 intercellular distances depending on the clonal size in a particular location from several embryos. For analysis of the percentage of occupied clonal volume in Fig. 1L and fig. S4P, we used a 150 × 150 × 200 μm ROI volume (except for the locations where mesenchymal cells represented a narrow layer between the developing brain and the ectoderm; in such cases, one dimension of the ROI volume was reduced accordingly), where we segmented the clonal surfaces using Imaris, calculated total volume encapsulated in cells of the clone, and then subtracted such volume from the total volume of the ROI for every analyzed clone or in different anatomical positions for all color-coded mesodermal derivatives. In Fig. 2, the distances between cells in single clones were measured using Spots to Spots Closest Distance Xtension in Imaris.
For the analysis of oriented cell divisions in Wnt5a mutants with corresponding controls, we performed immunohistochemistry with PH3 antibody on frozen sagittal sections of embryonic heads. Angle of the cell division was calculated as an angle between the cell division axis and the vector drawn through the basisphenoid toward developing nostrils. At least three individual Wnt5a mutant embryos were assessed together with higher numbers of littermate wild-type controls. In zebrafish live imaging experiments, dividing cells were tracked manually through the time series. Coordinates of each cell center after cell division were determined. Angle of the cell division in 3D was calculated as an angle between the cell division axis and the vector drawn through the fish body midline from the mouth to the midpoint between nostrils. We performed the actual calculations using vector coordinates derived from 3D space in Imaris. We counted orientations of cell divisions in three fishes between 30 and 52 hpf.
Supplementary Material
Acknowledgments
We thank A. Schier, A. Pan, and C. Nuesslein-Volhard for the fish strains; S. Warner for excellent technical support; M. Karlen and O. Kharchenko for the illustrations; and J. Jaros for technical support with μ-CT. We also thank V. Dyachuk for essential help with mouse experiments. Funding: M.K. was supported by the European Molecular Biology Organization Long-Term Fellowship. The work of E.I. was supported by the Russian Fund for Basic Research (grants 14-04-32027, 15-04-07573, and 15-29-02650). K.F., A.S.C., and I.A. were supported by grants from the Swedish Research Council and Karolinska Institutet. I.A. is supported by the Hållsten Research Foundation and the Åke Wiberg Foundation. T.Z. and J.K. were supported by the Ministry of Education, Youth and Sports of the Czech Republic under the project Central European Institute of Technology 2020 (LQ1601). The laboratory of P.S. was supported by grants from the Medical Research Council and the National Institute of Dental and Craniofacial Research. The laboratory of U.S. was supported by the Swiss National Science Foundation. A.H. was supported by the Swedish strategic research program eSSENCE, Vetenskapsrådet (VR), and the NIH under award number R01-EB014877-01. This work is solely the responsibility of the authors and does not necessarily reflect the views of the NIH. H. Brismar and H. Blom acknowledge funding support from VR and the Science for Life Laboratory. Author contributions: M.K., A.H., and I.A. designed the study and wrote the paper. M.K., D.G., T.Z., M.T., E.I., M.X., A.H., T.Y., J.P., and I.A performed the experiments and analyzed the data. K.F., J.K., V.P., S.K.N., P.S., E.A., H. Brismar, H. Blom, H.C., U.S., and A.S.C. contributed to the experimental work and study design. All authors read and approved the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/8/e1600060/DC1
fig. S1. Identification of rare double-color and GFP+ clones in neural crest ectomesenchyme in E9.5 to E10.5 embryonic faces.
fig. S2. Clonal mixing and distribution of NCC-derived clones in the embryonic trunk and head through the development.
fig. S3. Defined borders between mesoderm- and neural crest–derived progenies at postnatal and embryonic stages.
fig. S4. Genetic tracing of mesoderm-derived mesenchymal progenies reveals similarities with the neural crest–derived ectomesenchyme.
fig. S5. Live imaging of ectomesenchymal clones and progenitors in the eye shows difference between organized crowd movements and individual migrations.
movie S1. Live imaging of genetically traced neural crest–derived progenies in Sox10-CreERT2/Ubi:zebrabow-S zebrafish larvae between 30 and 56 hpf, ventral view.
movie S2. Live imaging of translocating ectomesenchymal clusters in Sox10-CreERT2/Ubi:zebrabow-S zebrafish larvae between 39 and 52 hpf, ventral view.
movie S3. Live imaging of genetically traced neural crest–derived progenies in Sox10-CreERT2/Ubi:zebrabow-S zebrafish larvae between 30 and 88hpf, ventral view.
movie S4. Live imaging of translocating ectomesenchymal clusters in Sox10-CreERT2/Ubi:zebrabow-S zebrafish larvae between 39 and 52 hpf, ventral view.
movie S5. 3D EdU analysis of Col2a1aBAC:mCherry zebrafish larva’s entire head at 4 dpf corresponding to Fig. 5 (O to Q).
Supplementary Materials and Methods
REFERENCES AND NOTES
- 1.Buchanan E. P., Xue A. S., Hollier L. H. Jr, Craniofacial syndromes. Plast. Reconstr. Surg. 134, 128e–153e (2014). [DOI] [PubMed] [Google Scholar]
- 2.Trainor P. A., Tam P. P., Cranial paraxial mesoderm and neural crest cells of the mouse embryo: Co-distribution in the craniofacial mesenchyme but distinct segregation in branchial arches. Development 121, 2569–2582 (1995). [DOI] [PubMed] [Google Scholar]
- 3.Franklin V., Khoo P. L., Bildsoe H., Wong N., Lewis S., Tam P. P. L., Regionalisation of the endoderm progenitors and morphogenesis of the gut portals of the mouse embryo. Mech. Dev. 125, 587–600 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Pispa J., Thesleff I., Mechanisms of ectodermal organogenesis. Dev. Biol. 262, 195–205 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Mootoosamy R. C., Dietrich S., Distinct regulatory cascades for head and trunk myogenesis. Development 129, 573–583 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Couly G., Coltey P., Eichmann A., Le Douarin N. M., The angiogenic potentials of the cephalic mesoderm and the origin of brain and head blood vessels. Mech. Dev. 53, 97–112 (1995). [DOI] [PubMed] [Google Scholar]
- 7.Achilleos A., Trainor P. A., Neural crest stem cells: Discovery, properties and potential for therapy. Cell Res. 22, 288–304 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKinney M. C., Fukatsu K., Morrison J., McLennan R., Bronner M. E., Kulesa P. M., Evidence for dynamic rearrangements but lack of fate or position restrictions in premigratory avian trunk neural crest. Development 140, 820–830 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bronner-Fraser M., Fraser S. E., Cell lineage analysis reveals multipotency of some avian neural crest cells. Nature 335, 161–164 (1988). [DOI] [PubMed] [Google Scholar]
- 10.Baroffio A., Dupin E., Le Douarin N. M., Clone-forming ability and differentiation potential of migratory neural crest cells. Proc. Natl. Acad. Sci. U.S.A. 85, 5325–5329 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baker C. V., Bronner-Fraser M., Le Douarin N. M., Teillet M. A., Early- and late-migrating cranial neural crest cell populations have equivalent developmental potential in vivo. Development 124, 3077–3087 (1997). [DOI] [PubMed] [Google Scholar]
- 12.Nitzan E., Krispin S., Pfaltzgraff E. R., Klar A., Labosky P. A., Kalcheim C., A dynamic code of dorsal neural tube genes regulates the segregation between neurogenic and melanogenic neural crest cells. Development 140, 2269–2279 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krispin S., Nitzan E., Kassem Y., Kalcheim C., Evidence for a dynamic spatiotemporal fate map and early fate restrictions of premigratory avian neural crest. Development 137, 585–595 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Baggiolini A., Varum S., Mateos J. M., Bettosini D., John N., Bonalli M., Ziegler U., Dimou L., Clevers H., Furrer R., Sommer L., Premigratory and migratory neural crest cells are multipotent in vivo. Cell Stem Cell 16, 314–322 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Lee R. T. H., Nagai H., Nakaya Y., Sheng G., Trainor P. A., Weston J. A., Thiery J. P., Cell delamination in the mesencephalic neural fold and its implication for the origin of ectomesenchyme. Development 140, 4890–4902 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gros J., Tabin C. J., Vertebrate limb bud formation is initiated by localized epithelial-to-mesenchymal transition. Science 343, 1253–1256 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gros J., Hu J. K.-H., Vinegoni C., Feruglio P. F., Weissleder R., Tabin C. J., WNT5A/JNK and FGF/MAPK pathways regulate the cellular events shaping the vertebrate limb bud. Curr. Biol. 20, 1993–2002 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boehm B., Westerberg H., Lesnicar-Pucko G., Raja S., Rautschka M., Cotterell J., Swoger J., Sharpe J., The role of spatially controlled cell proliferation in limb bud morphogenesis. PLOS Biol. 8, e1000420 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zallen J. A., Planar polarity and tissue morphogenesis. Cell 129, 1051–1063 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Gray R. S., Roszko I., Solnica-Krezel L., Planar cell polarity: Coordinating morphogenetic cell behaviors with embryonic polarity. Dev. Cell 21, 120–133 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Amerongen R., Fuerer C., Mizutani M., Nusse R., Wnt5a can both activate and repress Wnt/β-catenin signaling during mouse embryonic development. Dev. Biol. 369, 101–114 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bakker E. R. M., Raghoebir L., Franken P. F., Helvensteijn W., van Gurp L., Meijlink F., van der Valk M. A., Rottier R. J., Kuipers E. J., van Veelen W., Smits R., Induced Wnt5a expression perturbs embryonic outgrowth and intestinal elongation, but is well-tolerated in adult mice. Dev. Biol. 369, 91–100 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Ho H.-Y. H., Susman M. W., Bikoff J. B., Ryu Y. K., Jonas A. M., Hu L., Kuruvilla R., Greenberg M. E., Wnt5a-Ror-Dishevelled signaling constitutes a core developmental pathway that controls tissue morphogenesis. Proc. Natl. Acad. Sci. U.S.A. 109, 4044–4051 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Snippert H. J., van der Flier L. G., Sato T., van Es J. H., van den Born M., Kroon-Veenboer C., Barker N., Klein A. M., van Rheenen J., Simons B. D., Clevers H., Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell 143, 134–144 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Laranjeira C., Sandgren K., Kessaris N., Richardson W., Potocnik A., Vanden Berghe P., Pachnis V., Glial cells in the mouse enteric nervous system can undergo neurogenesis in response to injury. J. Clin. Invest. 121, 3412–3424 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leone D. P., Genoud S., Atanasoski S., Grausenburger R., Berger P., Metzger D., Macklin W. B., Chambon P., Suter U., Tamoxifen-inducible glia-specific Cre mice for somatic mutagenesis in oligodendrocytes and Schwann cells. Mol. Cell. Neurosci. 22, 430–440 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Hari L., Miescher I., Shakhova O., Suter U., Chin L., Taketo M., Richardson W. D., Kessaris N., Sommer L., Temporal control of neural crest lineage generation by Wnt/β-catenin signaling. Development 139, 2107–2117 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Lescroart F., Chabab S., Lin X., Rulands S., Paulissen C., Rodolosse A., Auer H., Achouri Y., Dubois C., Bondue A., Simons B. D., Blanpain C., Early lineage restriction in temporally distinct populations of Mesp1 progenitors during mammalian heart development. Nat. Cell Biol. 16, 829–840 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagase T., Nagase M., Osumi N., Fukuda S., Nakamura S., Ohsaki K., Harii K., Asato H., Yoshimura K., Craniofacial anomalies of the cultured mouse embryo induced by inhibition of sonic hedgehog signaling: An animal model of holoprosencephaly. J. Craniofac. Surg. 16, 80–88 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Favaro R., Valotta M., Ferri A. L. M., Latorre E., Mariani J., Giachino C., Lancini C., Tosetti V., Ottolenghi S., Taylor V., Nicolis S. K., Hippocampal development and neural stem cell maintenance require Sox2-dependent regulation of Shh. Nat. Neurosci. 12, 1248–1256 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Danielian P. S., Muccino D., Rowitch D. H., Michael S. K., McMahon A. P., Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr. Biol. 8, 1323–1326 (1998). [DOI] [PubMed] [Google Scholar]
- 32.Adameyko I., Lallemend F., Furlan A., Zinin N., Aranda S., Kitambi S. S., Blanchart A., Favaro R., Nicolis S., Lübke M., Müller T., Birchmeier C., Suter U., Zaitoun I., Takahashi Y., Ernfors P., Sox2 and Mitf cross-regulatory interactions consolidate progenitor and melanocyte lineages in the cranial neural crest. Development 139, 397–410 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Enshell-Seijffers D., Lindon C., Wu E., Taketo M. M., Morgan B. A., β-Catenin activity in the dermal papilla of the hair follicle regulates pigment-type switching. Proc. Natl. Acad. Sci. U.S.A. 107, 21564–21569 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dinulescu D. M., Cone R. D., Agouti and agouti-related protein: Analogies and contrasts. J. Biol. Chem. 275, 6695–6698 (2000). [DOI] [PubMed] [Google Scholar]
- 35.Saga Y., Miyagawa-Tomita S., Takagi A., Kitajima S., Miyazaki J. i., Inoue T., MesP1 is expressed in the heart precursor cells and required for the formation of a single heart tube. Development 126, 3437–3447 (1999). [DOI] [PubMed] [Google Scholar]
- 36.Yamaguchi T. P., Bradley A., McMahon A. P., Jones S., A Wnt5a pathway underlies outgrowth of multiple structures in the vertebrate embryo. Development 126, 1211–1223 (1999). [DOI] [PubMed] [Google Scholar]
- 37.Schieber M. H., Constraints on somatotopic organization in the primary motor cortex. J. Neurophysiol. 86, 2125–2143 (2001). [DOI] [PubMed] [Google Scholar]
- 38.Méhes E., Vicsek T., Collective motion of cells: From experiments to models. Integr. Biol. 6, 831–854 (2014). [DOI] [PubMed] [Google Scholar]
- 39.David R., Luu O., Damm E. W., Wen J. W. H., Nagel M., Winklbauer R., Tissue cohesion and the mechanics of cell rearrangement. Development 141, 3672–3682 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Kanca O., Caussinus E., Denes A. S., Percival-Smith A., Affolter M., Raeppli: A whole-tissue labeling tool for live imaging of Drosophila development. Development 141, 472–480 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Worley M. I., Setiawan L., Hariharan I. K., TIE-DYE: A combinatorial marking system to visualize and genetically manipulate clones during development in Drosophila melanogaster. Development 140, 3275–3284 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Young N. M., Hu D., Lainoff A. J., Smith F. J., Diaz R., Tucker A. S., Trainor P. A., Schneider R. A., Hallgrímsson B., Marcucio R. S., Embryonic bauplans and the developmental origins of facial diversity and constraint. Development 141, 1059–1063 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Theveneau E., Mayor R., Neural crest delamination and migration: From epithelium-to-mesenchyme transition to collective cell migration. Dev. Biol. 366, 34–54 (2012). [DOI] [PubMed] [Google Scholar]
- 44.Minoux M., Rijli F. M., Molecular mechanisms of cranial neural crest cell migration and patterning in craniofacial development. Development 137, 2605–2621 (2010). [DOI] [PubMed] [Google Scholar]
- 45.Hu D., Young N. M., Li X., Xu Y., Hallgrímsson B., Marcucio R. S., A dynamic Shh expression pattern, regulated by SHH and BMP signaling, coordinates fusion of primordia in the amniote face. Development 142, 567–574 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Foppiano S., Hu D., Marcucio R. S., Signaling by bone morphogenetic proteins directs formation of an ectodermal signaling center that regulates craniofacial development. Dev. Biol. 312, 103–114 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schoenebeck J. J., Hutchinson S. A., Byers A., Beale H. C., Carrington B., Faden D. L., Rimbault M., Decker B., Kidd J. M., Sood R., Boyko A. R., Fondon J. W. III, Wayne R. K., Bustamante C. D., Ciruna B., Ostrander E. A., Variation of BMP3 contributes to dog breed skull diversity. PLOS Genet. 8, e1002849 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.M. Tanaka, Evolution of vertebrate limb development, in Encyclopedia of Life Sciences (eLS) (John Wiley & Sons Ltd., Chichester, UK, 2009), p. 1–9. [Google Scholar]
- 49.Freitas R., Zhang G., Cohn M. J., Evidence that mechanisms of fin development evolved in the midline of early vertebrates. Nature 442, 1033–1037 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Shubin N., Tabin C., Carroll S., Deep homology and the origins of evolutionary novelty. Nature 457, 818–823 (2009). [DOI] [PubMed] [Google Scholar]
- 51.Mongera A., Singh A. P., Levesque M. P., Chen Y.-Y., Konstantinidis P., Nüsslein-Volhard C., Genetic lineage labeling in zebrafish uncovers novel neural crest contributions to the head, including gill pillar cells. Development 140, 916–925 (2013). [DOI] [PubMed] [Google Scholar]
- 52.Pan Y. A., Freundlich T., Weissman T. A., Schoppik D., Wang X. C., Zimmerman S., Ciruna B., Sanes J. R., Lichtman J. W., Schier A. F., Zebrabow: Multispectral cell labeling for cell tracing and lineage analysis in zebrafish. Development 140, 2835–2846 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.M. Westerfield, The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Danio rerio) (University of Oregon Press, Eugene, OR, ed. 4, 2000). [Google Scholar]
- 54.O’Brien G. S., Rieger S., Martin S. M., Cavanaugh A. M., Portera-Cailliau C., Sagasti A., Two-photon axotomy and time-lapse confocal imaging in live zebrafish embryos. J. Vis. Exp. (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/8/e1600060/DC1
fig. S1. Identification of rare double-color and GFP+ clones in neural crest ectomesenchyme in E9.5 to E10.5 embryonic faces.
fig. S2. Clonal mixing and distribution of NCC-derived clones in the embryonic trunk and head through the development.
fig. S3. Defined borders between mesoderm- and neural crest–derived progenies at postnatal and embryonic stages.
fig. S4. Genetic tracing of mesoderm-derived mesenchymal progenies reveals similarities with the neural crest–derived ectomesenchyme.
fig. S5. Live imaging of ectomesenchymal clones and progenitors in the eye shows difference between organized crowd movements and individual migrations.
movie S1. Live imaging of genetically traced neural crest–derived progenies in Sox10-CreERT2/Ubi:zebrabow-S zebrafish larvae between 30 and 56 hpf, ventral view.
movie S2. Live imaging of translocating ectomesenchymal clusters in Sox10-CreERT2/Ubi:zebrabow-S zebrafish larvae between 39 and 52 hpf, ventral view.
movie S3. Live imaging of genetically traced neural crest–derived progenies in Sox10-CreERT2/Ubi:zebrabow-S zebrafish larvae between 30 and 88hpf, ventral view.
movie S4. Live imaging of translocating ectomesenchymal clusters in Sox10-CreERT2/Ubi:zebrabow-S zebrafish larvae between 39 and 52 hpf, ventral view.
movie S5. 3D EdU analysis of Col2a1aBAC:mCherry zebrafish larva’s entire head at 4 dpf corresponding to Fig. 5 (O to Q).
Supplementary Materials and Methods