

Abstract
Apoptosis is a form of active cell death engaged by developmental cues as well as many different cellular stresses in which the dying cell essentially ‘packages’ itself for removal. The process of apoptotic cell death, as defined at the molecular level, is unique to the Metazoa (animals). Yet active cell death exists in non-animal organisms, and in some cases molecules involved in such death show some sequence similarities to those involved in apoptosis, leading to extensive speculation regarding the evolution of apoptosis. Here, we examine such speculation from the perspective of the functional properties of molecules of the mitochondrial apoptotic cell death pathway. We suggest scenarios for the evolution of one pathway of apoptosis, the mitochondrial pathway, and consider how they might be tested. We conclude with a ‘Just So Story’ of how the mitochondrial pathway of apoptosis might have evolved during eukaryotic evolution.
Introduction: Just So Stories
“‘Explain! Explain! Explain!’” How the First Letter Was Written1
Since Darwin, evolutionary biologists have engaged in rigorous, and often not-so-rigorous, speculation into the selective events that favored the emergence of characteristics associated with living things. More recently, these have been extended to molecular events that function at the cellular level. With the elucidation of genome sequences and the development of analytic tools to compare these sequences, many biologists with only a passing knowledge of evolutionary theory have felt free to elaborate on how molecular pathways and processes evolved, often with minimal information beyond protein sequence. In the discussion presented herein, we make no claims regarding the robustness of the analysis or the rigor of the speculation, as we are firmly seated among those with, at best, only a superficial appreciation of evolutionary concepts. Those with a deeper understanding should feel free to criticize or disregard these ideas, but for those who, like us, have an interest in cell death mechanisms and wonder about their origins, the following discussion might be viewed as a joint exploration into possibilities.
A ‘Just So Story’ is an untestable and thus unfalsifiable idea, such as a scenario for the evolution of a biological trait or process. It is just that, a story. But, as suggested elsewhere (http://epjournal.net/blog/2012/09/just-so-stories-are-bad-explanations-functions-are-much-better-explanations/), “the goal should not be to expel stories from science, but rather to identify the stories that are also good explanations”. Here, we will explore some stories specifically relating to the evolution of one particular type of cell death – apoptosis – and how they might be investigated (if not falsified). In the process, we discuss at least one bewildering paradox that lies at the heart of the evolution of a major pathway of apoptosis in animals and consider a possible resolution.
It is important to note that this is not the first foray into the evolution of cell death, and several excellent reviews should be examined by the interested reader [1–3].
First Principles
“‘Ah!’ said Tegumai. ‘Will that do to begin with?’” How the Alphabet Was Made
Much of the speculation into the evolution of molecular pathways is based on the principle of homology and the relationships between the sequences of genes or proteins. As discussed below, there are caveats to drawing conclusions from such limited information without more detailed knowledge of how the proteins encoded by these genes interact and function. This is the position taken herein: a consideration of protein function beyond sequence similarity can alter our views (and our Just So Stories) about how a pathway may have evolved. Indeed, by such exploration of protein function, we can begin to test our stories.
An important (and, in retrospect, obvious) precept in any discussion of evolution is that all currently existing organisms and, by extension, biological molecules have ‘evolved’ for precisely the same period of time. In considering genes from two different organisms, we do not state that ‘X evolved from Y’, but rather can speculate that ‘X and Y had a common ancestor’. As discussed below, this is a critical point that allows us to speculate that a function of a particular protein present in one organism may have been lost in another, despite similarity in their sequences. It is also important to point out that sequence similarity is not proof of homology, and that similar genes may be homologs, orthologs, or paralogs, with different evolutionary relationships.
One further principle relates specifically to our consideration of apoptosis. This cell death process has been broadly defined by morphology; animal (metazoan) cells that die by apoptosis lose volume, display condensation of nuclear chromatin, sustain structures of intracellular organelles (such as mitochondria and endoplasmic reticulum), often show protrusions from the plasma membrane (blebbing), and frequently fragment the nucleus and/or cell body. This has led to a search for similar morphologies in dying non-metazoan cells, with subsequent evolutionary speculation. More recently, however, a molecular definition of apoptosis has been proposed [4], and it is this definition (elaborated below) that is employed herein.
Apoptosis, with a Focus on the Mitochondrial Pathway
“Hear and attend and listen; for this befell and behappened and became and was.” The Cat that Walked by Himself
Apoptosis, as a defined molecular process, is a type of cell death unique to metazoans. The morphological characteristics of apoptosis are brought about by the actions of a set of cysteine proteases, the executioner caspases, which are only found in animals [5]. When activated, the executioner caspases cleave approximately a thousand substrates, including several that have been identified as mediating the characteristic changes in the dying cell, including membrane blebbing, DNA fragmentation, phosphatidylserine externalization (which promotes clearance by phagocytes), loss of electron transport function in mitochondria, and other events [6–8].
Executioner caspases, in their inactive form, are dimers that become activated upon cleavage at a site located between the resulting large and small subunits of the active protease [9]. In the majority of cases (the exception being cleavage by the serine protease granzyme B, which is introduced into cells by cytotoxic lymphocytes [10]), this activating cleavage is mediated by initiator caspases. Unlike the executioner caspases, initiator caspases are monomeric and are activated by enforced dimerization (‘induced proximity’ [9,11]), rather than by cleavage. (Although initiator caspases undergo auto-cleavage upon activation, this is not the activating event, and cleavage of an initiator caspase is not proof of its activation [12,13].)
The enforced dimerization of initiator caspases, leading to their activation, occurs by the action of adapter proteins, which form an ‘activation platform’ that binds to interaction regions in the prodomain of the caspase. An apoptotic pathway is defined by the mode of formation of the caspase activation platform and the identity of the initiator caspase recruited by the adapter. Here, we focus on the so-called intrinsic or mitochondrial pathway of apoptosis.
In the mitochondrial pathway (Figure 1), the initiator caspase, caspase-9, is activated when it binds to its activation platform (the apoptosome), composed of an oligomer of the adapter protein APAF1. Inactive APAF1 is a monomer that oligomerizes upon interaction with cytochrome c, which is normally sequestered in the mitochondrial intermembrane space. During apoptosis, the proteins of the intermembrane space are released to the cytosol upon mitochondrial outer membrane permeabilization (MOMP). These include cytochrome c, as well as proteins (including Smac/Diablo and Omi/Htra2) that function to inactivate an inhibitor of active executioner caspases, X-IAP [14]. The latter role for MOMP links the mitochondrial pathway to other apoptotic pathways. It will be important to note that, while animal cytochrome c activates APAF1 (indeed, it appears that any animal cytochrome c will activate mammalian APAF1 [15]), apocytochrome c, which lacks the heme group, does not activate APAF1, nor does cytochrome c from non-animal sources, such as yeast.
Figure 1. Variations on a theme: the mitochondrial pathway of apoptosis.
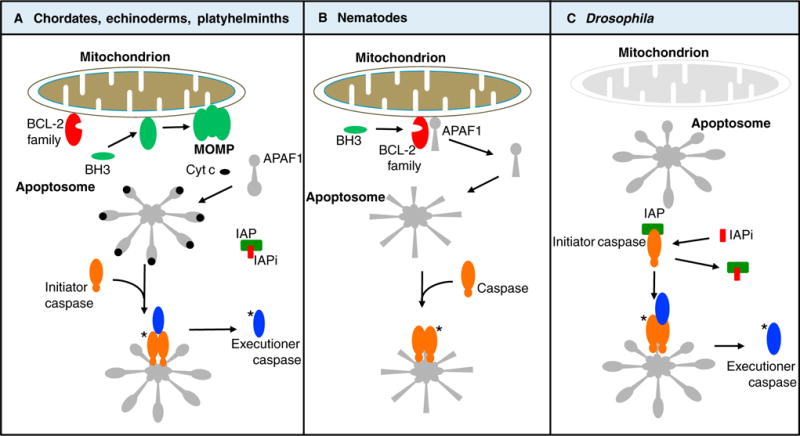
(A) In chordates, echinoderms, platyhelminths, and possibly other phyla, apoptotic conditions engage the members of the Bcl-2 family, which either promote (green) or inhibit (red) MOMP in response to activation of BH3-only proteins. MOMP results in the release of cytochrome c (cyt c) and other proteins of the intermembrane space. Cytochrome c binds to APAF1, which oligomerizes to create the apoptosome that in turn binds to and activates an initiator caspase. The initiator caspase cleaves and thereby activates executioner caspases. Both the initiator and executioner caspases are inhibited by an inhibitor of apoptosis protein (IAP). MOMP releases antagonists of the IAP (IAPi), permitting the executioner caspases to orchestrate apoptosis. Asterisks (*) refer to active caspases. (B) In nematodes, the anti-apoptotic Bcl-2 protein sequesters the APAF1 homolog. Apoptotic conditions induce expression of BH3-only proteins, which bind to the Bcl-2 protein, releasing the APAF1 homolog to form an apoptosome. This binds and thereby activates the caspase to promote apoptosis. (C) In Drosophila, the APAF1 homolog appears to spontaneously form an apoptosome, but the initiator caspase is inhibited by an IAP. Apoptotic conditions promote expression of IAPi, permitting the apoptosome to now activate the initiator caspase, which in turn cleaves and thereby activates executioner caspases, promoting apoptosis. Note that MOMP, upstream of caspase activation, occurs only in the pathway shown in (A).
The process of MOMP is mediated by the action of members of the BCL-2 protein family. These include the effectors of MOMP (e.g. BAX and BAK), which upon activation cause the permeabilization of the mitochondrial outer membrane. Anti-apoptotic BCL-2 proteins (e.g., BCL-2, BCL-xL, and MCL-1) prevent MOMP, binding to the activated effectors or to the proteins that activate them. A third sub-family, the BH3-only proteins, either activate the effectors (e.g., BID, BIM, and PUMA) and/or neutralize the anti-apoptotic proteins (e.g., BAD, BMF, HRK, PUMA, and NOXA) [16]. During cellular stress (or other pro-apoptotic conditions) changes in the expression and/or function of the BH3-only proteins initiate the process of MOMP via such interactions. Even if caspase activation downstream of MOMP is blocked or disrupted, the process of MOMP can condemn cells to death (albeit non-apoptotic death) [17], although cells can survive MOMP in some situations [14,18].
All of the components of the mitochondrial pathway of apoptosis have homologs throughout the animals, but, to date, functional homologs of these proteins have not been convincingly demonstrated outside of metazoans. Before engaging in a discussion of these homologs and their functions, however, it may be useful to examine claims regarding apoptosis in non-metazoans.
Caspases versus Metacaspases: Non-Animal Apoptosis?
“Before the High and Far-Off Times, O my Best Beloved, came the Time of the Very Beginnings.” The Crab That Played with the Sea
Active cell death processes are not unique to animals and have been described in plants [19], yeast [20], protozoa [21], and bacteria [22]. In some cases, attempts have been made to relate these events to apoptosis, sometimes implying an evolutionary relationship. While that view will be challenged here, we contend that the study of such non-animal cell death has value in its own right, both as a way to understand the life and death of those organisms (and cells in the case of plants) and perhaps to elucidate general principles of active cell death.
Caspase proteases are not found outside of the animals; therefore, if apoptosis is strictly defined as involving the activation and function of caspases, apoptosis does not exist outside the animals. However, a family of proteases that are related to caspases – the metacaspases – have been implicated in cell death in plants and yeast [23]. (Another caspase-like protease type – the paracaspases, present in animals as well as in Dictyostelium – is not involved in cell death in any organism, including Dictyostelium, which lacks an apoptosis-like cell death [24].) Further, in plants, some cell death processes appear to involve proteins related to the adapter APAF1 [25]. While metacaspases cleave substrates after an arginine residue, unlike caspases, which cleave after aspartate, a substrate has been identified that is cleaved by either protease and has been suggested to participate in cell death [26]. Such findings have led to the idea that apoptosis and these death processes in non-metazoan organisms have a common ancestor [27].
There is a difficulty with this idea, however. Despite some sequence and structural similarity between caspases and metacaspases, their biochemistry is fundamentally different. The principle of induced proximity, discussed above, is central to the way in which initiator caspases are activated by the platforms composed of their adapters, but metacaspases are monomers and are not activated by dimerization [5]. Therefore, recruitment to an adapter protein, even one similar to APAF1, should not activate a metacaspase. The functional pathways of apoptosis, involving the interactions between adapters, initiator caspases, and executioner caspases, are therefore unlikely to be paralleled by the putative homologs in plants or yeast. If caspases and metacaspases have a common molecular ancestor, there is no evidence that their mechanisms of activation are the same. Of course this position could change if we were to gain an understanding of whether and how molecular relatives of APAF1 might activate metacaspases in plant cell death (an idea for which there is currently no supporting evidence) [19].
Functional Similarities in the Mitochondrial Pathway of Apoptosis
“Make me different from all other animals by five this afternoon.” The Sing-Song of Old Man Kangaroo
The key molecular components of the mitochondrial pathway of apoptosis in mammals, as outlined above, are proteins of the BCL-2 family, cytochrome c, APAF1, an initiator caspase (caspase-9), and executioner caspases (Figure 1). Attempts to identify homologs of these proteins have met with success throughout many animal phyla. Homologs of chordate BCL-2 proteins have been identified in poriferans (sponges) [28], cnidarians (Hydra) [29], platyhelminths (planaria) [15], arthropods (flies) [30], and nematodes [31], among others [1]. Cytochrome c is among the most highly conserved proteins, found in all animals (and eukaryotes, as well as bacteria). APAF1 homologs have been described for representatives of several animal phyla, and can even be found in the placozoans (Trichoplax) (see Figures S1–S3 in the Supplemental Information), which may represent the most basal animal phylum (although this is debatable [32]). And a great many caspases, including caspases with pro-domains containing protein interaction motifs similar to the pro-domain of caspase-9, have been widely identified in animal phyla. Based on these considerations, it is widely held that the mitochondrial pathway of apoptosis, or at least the proteins that comprise it, arose with the animals.
It is when we consider the biochemical functions of these proteins in apoptosis that the picture becomes much more murky. One example is the relationships between cytochrome c, APAF1, and caspase-9. In those cases that have been examined functionally, the interaction between APAF1-like molecules and caspase-9-like molecules results in activation of the caspase, as observed in arthropods [33], nematodes [34], and chordates [35] (Figure 1). The role of cytochrome c is more complex. While in chordates cytochrome c is required to activate APAF1 to form the activation platform for caspase-9, this is not the case for nematodes, where the APAF1 homolog, CED-4, lacks the region of the molecule required for cytochrome c interaction [34,36]. In arthropods (e.g. Drosophila), the APAF1 homolog has this region, but the evidence that cytochrome c activates this APAF1 homolog is contentious; some studies suggest that cytochrome c mutants can be identified that are defective in apoptosis [37], while in vitro studies in Drosophila cells failed to find a role for cytochrome c in caspase activation [38–41]. In contrast, cytosolic extracts from platyhelminths (planaria) and echinoderms (sea urchins and sand dollars) displayed robust caspase activation in response to cytochrome c addition [15], suggesting that APAF1 in such animals is activated by cytochrome c.
In chordates, the BCL-2 proteins regulate and effect MOMP, which is required for the release of cytochrome c into the cytosol to activate APAF1 (as discussed above). Although BCL-2 proteins have been found throughout the animals, as noted, the involvement of MOMP is less clear. During apoptosis in platyhelminths, cytochrome c release (indicative of MOMP) can be observed [15], but no MOMP is seen during apoptosis in nematodes [42]. Consistent with these observations, a platyhelminth BCL-2 family protein from planaria has MOMP activity [15], while the single nematode BCL-2 protein in Caenorhabditis elegans does not [12], and instead has an anti-apoptotic function unrelated to MOMP (see below). In Drosophila, cytochrome c release is observed during apoptosis, but this release depends upon the function of caspases [43], unlike most cases of MOMP in chordate cells.
Arthropod BCL-2 proteins, to date, have not been observed to regulate apoptosis directly [44], although they may have indirect effects [45,46]. Instead, it appears that apoptosis in the arthropods takes a different path. The Drosophila APAF1 homolog, ARK, appears to be constitutively active, but the activation of the initiator caspase, DRONC, is held in check by an inhibitor, DIAP1 (related to X-IAP in mammals, see above). Upon apoptosis induction, proteins that antagonize DIAP1 are expressed, releasing active DRONC to proceed with executioner caspase activation and cell death [47]. These proteins share a functional motif with mammalian antagonists of X-IAP, but, unlike the mammalian proteins, they are not sequestered in the mitochondrial intermembrane space.
A paradox emerges when we compare the functions of BCL-2 proteins in nematodes and chordates. In C. elegans, the anti-apoptotic BCL-2 protein, CED-9, directly binds to the APAF1 homolog, CED-4, preventing its function [48]. The BH3-only protein Egl-1 disrupts this interaction, permitting CED-4 to bind and activate the caspase CED-3. In contrast, anti-apoptotic BCL-2 proteins in chordates do not inhibit APAF1 or the activation of caspase-9, but instead prevent MOMP, which is needed for cytochrome c and APAF1 activation. Despite the similarities between the proteins and the overall effect (i.e., in both cases anti-apoptotic BCL-2 proteins prevent APAF1 activation), the biochemistry is distinct. It is important to note that there is nothing paradoxical about a protein assuming different functions in different organisms; what we suggest is surprising is that a superficial function (inhibition of caspase activation by BCL-2 proteins) appears to be achieved via fundamentally different biochemical mechanisms (inhibition of MOMP versus direct inhibition of APAF1).
Comparing APAF1 homologs from different animal phyla, it seems likely that in nematodes the ability of the APAF1 protein to interact with cytochrome c may have been ‘lost’. This may also be the case in arthropods, some of which maintain the cytochrome c-binding (WD) domain (Figures S1–S3) but have an APAF1-like molecule that does not appear to interact with cytochrome c. This idea only has validity if the common ancestor of platyhelminths, nematodes, and arthropods possessed an APAF1 that was activated by holocytochrome c; the alternative proposal is that cytochrome c-mediated APAF1 activation arose only after such a ‘split’.
Can we reconcile these differences, which, despite sequence similarity and the same ultimate effect, suggest very different molecular pathways? That is, can we tell a Just So Story to explain how apoptosis evolved in the animals? And can it be tested? To approach this, it may first help to explore a form of cell death that is related to apoptosis, but different. This is the process of pyroptosis.
Pyroptosis, a Caspase-Dependent Cell Death Distinct from Apoptosis
“It was all his ’satiable curiosity.” The Elephant’s Child
Cell death by pyroptosis is a non-apoptotic cell death, more closely allied to necrosis. Like apoptosis, however, it is caspase-dependent, involving the activation of either caspase-1 or a related caspase (caspase-11 in rodents, caspase-4 or -5 in primates) (Figure 2). In general, pyroptosis occurs in response to infection.
Figure 2. Variations on a theme: caspase activation platforms.
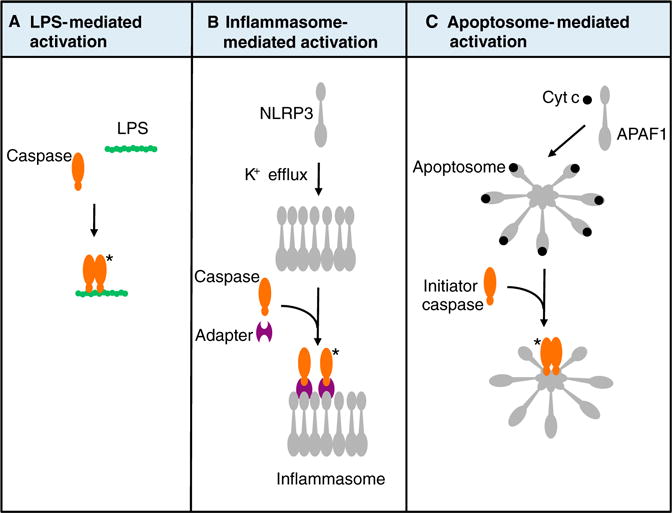
(A) Some caspases (caspase-4, caspase-5, caspase-11) bind directly to intracellular lipopolysaccharides (LPS), causing caspase dimerization and, hence, activation. (B) An example of an inflammasome is shown. NLRP3 is activated to oligomerize under conditions, for example, in response to a decrease in intracellular K+ concentrations. NLRP3 binds to an adaptor protein, which in turn binds to caspase-1, resulting in its activation. (C) The activation of APAF1 by cytochrome c, as described in Figure 1A. Asterisks (*) refer to active caspases.
The activation of caspases-4, -5, and -11 is distinct from that of other monomeric caspases in that the adapter responsible for activation by induced proximity is not encoded by the host genome, but rather by intracellular bacteria. Lipopolysaccharide derived from the outer membrane of Gram-negative bacteria directly engages the prodomains of these caspases, causing their activation [49]. The caspase then cleaves a protein called Gasdermin d, which in its cleaved form kills the cell (by a mechanism that is yet to be elucidated) [50,51].
Intracellular DNA viruses can trigger pyroptosis via another mechanism, involving the engagement of a cytosolic DNA sensor, AIM2, which oligomerizes to create an activation platform for caspase-1. Such activation platforms for caspase-1 are termed inflammasomes because caspase-1 cleaves the proforms of the inflammatory cytokines interleukin-1 and interleukin-18, as well as Gasdermin d. While the cleavage of Gasdermin d by caspase-1 contributes to rapid cell death, cells lacking Gasdermin d nevertheless are still able to die in response to caspase-1 activation [50,51].
Unlike other monomeric caspases, caspase-1 has several activation platforms. Most of these involve proteins of the Nod-like receptor (NLR) family, which includes several involved in caspase-1 activation, such as NLRP1, NLRP3, and NLRC4. These proteins are activated to oligomerize in response to different bacterial products and form a complex with another adapter, ASC, which then binds to and activates caspase-1 (Figure 2). In addition, extracellular bacterial products, and other conditions produced by bacterial infection, can trigger the activation of NLRP3 via other mechanisms, leading to caspase-1 activation and pyroptosis.
Many NLR proteins are described in mammals, but most have unknown functions, or known functions that do not include activation of caspase-1. Strikingly, APAF1 is often considered a member of the NLR family, on the basis of sequence similarity and domain structure. While it is possible that APAF1 arose independently of the NLRs found in inflammasomes (e.g., by convergent evolution), for the purposes of our Just So Story below we will consider APAF1 and NLRs to be evolutionarily related molecules.
NLR proteins are found not only throughout the animals, but also in plants: these are the plant APAF1-related proteins mentioned above. In many animals, there is a bewildering number of NLR proteins, far beyond the large numbers identified in chordates.
A cell death response to intracellular infection is an obvious way in which obligate intracellular parasites can be controlled by removing the infected cell before the invader can replicate. Indeed, this can help to explain active cell death in single-celled organisms. We have previously argued [52] that an active cell death mechanism that might be envisioned to allow single-celled organisms, as a group, to survive limited nutrient availability (a scenario that is often invoked in discussions of the evolution of cell death) is not an evolutionarily stable strategy, as ‘cheaters’ that lose the cell death mechanism will inevitably be favored by selection. However, if we consider a pathogen that has adapted to infect a single-celled organism and therefore, by extension, its clone mates, death of the infected cell is now stable; an individual that loses the cell death mechanism is no longer favored. These considerations and the observations above lead us to our Just So Story.
How the Animal’s Cells Learned to Die
“‘I don’t think it was at all like that,’ said Painted Jaguar, but he felt a little puzzled; ‘but, please, say it again more distinctly.’” The Beginning of the Armadillos
While it is tempting to begin our story with ‘once upon a very long time ago’, we will spare the reader this indignity. Nevertheless, what follows is only a story, by necessity. We begin with an assumption that, at some early point in the evolution of eukaryotes, a mechanism existed for the recognition of intracellular infection, leading to the death of the cell. Given the nature of the pyroptotic caspases-4, -5, and -11, we can envision that the mechanism was a simple one, based on the ‘invention’ of a monomeric protease that was activated upon binding to a bacterial product not found in the Archaea. If infected, this cell died, thereby sparing its clone mates the subsequent infection by the replicating pathogen. This would lead to a Red Queen scenario, in which the infecting organism evolves mechanisms to avoid the Archaea’s cell death mechanism and, in turn, the Archaea evolves strategies to counter these evasions. These counter-strategies included the emergence of adapters that would recognize a range of bacterial products, lending increased surveillance to the mechanism. This might have been an ancestor of the inflammasomes. We will return to this escalation, below.
Among the unique bacterial products recognized by these adapters was cytochrome c and the proteins needed for its maturation, present in bacteria and absent in many of the Archaea [53]. Here it may be important to note that, while a cytochrome c-like protein may have been present, the adapter in our story was specialized to recognize the mature form of cytochrome c, complete with heme. Most Archaea lack the machinery to construct this holocytochrome c [53]. (As an aside, it is worth remembering that APAF1 in animals recognizes only holocytochrome c.)
At some point, an α-purple bacterium resembling Rickettsia [54] invaded a member of the Archaea, possibly of the ‘TACK’ superphylum [55], and, rather than the latter dying, a symbiosis arose that led to the formation of mitochondria. This symbiosis hypothesis for the evolution of mitochondria has remarkable support and is widely accepted [56].
But for our purposes we might suppose that it was not a ‘perfect union’. While under some circumstances the proto-mitochondria and the Archaea that housed it might both gain tremendous selective advantages, in other situations dissolution of the symbiosis might favor the proto-mitochondria, at least those that had sustained their ability to replicate outside the host. In that setting, the proto-mitochondria might engage a strategy common to bacteria – the production of pore-forming toxins. These would target the nearest membrane, i.e. that surrounding the bacteria, and induce lysis. Proteins that exist between the inner (bacterial) and outer (host) membrane would then be released to the cytosol. Some of these (perhaps including holocytochrome c) might then trigger the proto-inflammasome response in the host, resulting in its death.
A component of the symbiosis hypothesis is that genes present in the symbiont somehow ‘transfer’ to the nucleus of the host, coming under the host’s control and thus enforcing the symbiosis [56]. Among these might be the toxins, normally produced by the bacteria. In our story, these would now be host proteins, and would function as the BCL-2 protein family effector proteins, capable of permeabilizing the mitochondrial outer membranes to effect MOMP, release cytochrome c, and activate a specialized ‘inflammasome’ (the APAF1 apoptosome) that engages caspase-9.
The BCL-2 family effectors oligomerize via their BH3 regions to cause MOMP [57]. Anti-apoptotic BCL-2 proteins bind to this region in activated effectors, thereby blocking MOMP. The BH3-only proteins possess just the BH3 domain (hence the name) and bind to the BH3-binding pockets of the effectors to activate them [57,58] or to the anti-apoptotic proteins to neutralize them. Therefore, for our Just So Story we propose that the common ancestor of the BCL-2 proteins was a toxin resembling a pro-apoptotic BCL-2 effector of MOMP.
Before moving on to discuss how this imaginary primordial pathway of apoptosis diverged in the animals, we can ask if there is any evidence in support of our story, whether there are fundamental problems with it, and/or propose ways to test it. The first structural studies of anti-apoptotic BCL-2 proteins noted similarities in structure with bacterial pore-forming toxins [59], raising the possibility that the proto-BCL-2 effector may have been such a toxin. If so, we have not found it in existing bacteria nor in the exceptionally large mitochondrial genomes of the Jakobid protozoans (which most closely resemble proteobacterial genomes [60]).
Our story also features an APAF1-like sensor that creates the proto-caspase activation platform. Although NLR proteins are found in non-metazoans (especially plants), the non-metazoan caspase-like proteins do not have the property of being activated on such platforms (as discussed above). Therefore, if our story is to have any relationship to reality, we must postulate that our proto-apoptotic pathway arose with a common ancestor of the Metazoa. Choanoflagellates are thought to be the closest living relatives of the Metazoa and, while their genomes encode several proteins with similarities to caspases (https://www.broadinstitute.org/annotation/genome/multicellularity_project/MultiHome.html), to date we do not know if any functional caspases exist in these organisms, nor whether these might have the property of activation by induced proximity.
Our Just So Story, however, posits that our primordial pathway would have existed well before the origins of the Choanoflagellates and, indeed, during the emergence of eurkaryotes. If so, why do multicellular kingdoms other than the Metazoa appear to lack a mitochondrial pathway of apoptosis? At this point, compelling evidence for such a pathway does not exist outside the animals, as we discussed. This may be a fatal flaw for our story. But then again, it is only a story. It may be that vestiges of such a pathway do exist outside the Metazoa, and remain to be found. Notwithstanding our previously discussed issues with metacaspases, further investigation of their functions in cell death may elaborate such vestiges.
Once we examine animals, however, we see not only caspases and APAF1-like proteins, but often a radiation of the genes encoding them (so that an organism may have many). An example is in the basal Placozoa (Figure S4), or in the echinoderm Strongylocentrotus purpuratus, where a cursory search revealed 76 possible caspases (not shown). Based on our considerations above, it is tempting to suggest that many of these are involved in the recognition of intracellular invaders, although at this time we do not know if any caspases (beyond mammalian caspases-4, -5, and -11) are activated upon binding to bacterial products.
At this point, we have only our Just So Story, and the presence of the pathway we have described throughout the animals. In the relatively basal platyhelminths, different BCL-2 proteins promote [15] and prevent [61] MOMP and cell death, and cytochrome c promotes caspase activation [15]. Again, though, we have a problem. If anti-apoptotic BCL-2 proteins function by blocking effector oligomerization and MOMP, how did the situation in C. elegans arise, where the anti-apoptotic BCL-2 protein acts by binding the APAF1 protein, which does not itself interact with cytochrome c (and no MOMP is required for its activation)? Like the pro-apoptotic effector proteins, where binding to an activating BH3-only protein induces a conformational change in the former [57,58], the binding of the nematode BH3-only protein to the anti-apoptotic BCL-2 protein also induces a conformational change, in this case releasing the APAF1-like protein [48]. How can these different functions for a similar molecular mechanism be reconciled?
One possibility is that a common ancestor of a BCL-2 protein had both activities and could directly inhibit APAF1 as well as regulate MOMP. If so, we can posit that one or the other function might have been lost in the lineage leading to nematodes. The BCL-2 proteins of basal phyla, such as Cnidaria, Porifera, and Placozoa (Figure S5), have been identified, and it would be interesting to determine whether any of these actually bind to and inhibit the corresponding APAF1-like proteins. Of course, this is not a simple analysis, although it might be possible to determine the likelihood of such interactions using in silico approaches, examining whether the binding interactions determined structurally in the nematode proteins [48] might be possible in the proteins from other organisms. The same analyses might be carried out for interactions between holocytochrome c and APAF1, which have also been elucidated at the structural level [36].
Intriguingly, the BCL-2 protein of nematodes [62], and those of arthropods [43] and chordates [63], regulate mitochondrial dynamics, i.e., the fission and fusion of mitochondria. Further, mitochondrial shape has been implicated in the induction of MOMP by mammalian BCL-2 effectors [64]. A common mechanism may thus exist for the function of these proteins, whether to cause MOMP or to sequester (and release) APAF1. At this point, however, such a unified model is lacking.
Conclusions and Perspectives
“They are quite contented as they are.” How the Leopard Got His Spots
Our discussion of the evolution of cell death in animals focuses on the mitochondrial pathway of apoptosis, how its presence throughout the Metazoa takes different forms, and how it might have evolved. In doing so, we have promoted one set of scenarios that does not take into account possible alternatives. For example, the apparent conservation of the death receptor pathway of apoptosis in Cnidaria [65] raises the possibility that the evolution of this pathway predated that of the mitochondrial pathway, which was then co-opted during the evolution of the mitochondrial pathway. Further, other forms of cell death, such as forms of programmed necrosis (active molecular mechanisms leading to necrotic cell death), which include mitochondrial mechanisms, are not considered in our discussion and may well have had a prominent role in the emergence of apoptosis.
Most investigations into the evolution of cell death in the animals rely on sequence similarities at the protein level, with the assumption that form specifies function. As we have discussed, however, the biochemical mechanisms whereby a molecule may promote or inhibit cell death can be distinct in different animal phyla. In some cases we have proposed that this represents a paradox, such that the superficial function of a protein (e.g., the function of anti-apoptotic BCL-2 proteins to prevent apoptosis) can take different forms (e.g., inhibition of MOMP versus direct inhibition of APAF1). While this might be ‘explained’ by gene duplication and divergence of biochemical function, the source of the paradox is not why two similar proteins may function differently (which is not unexpected), but why the differing functions have the same superficial effect. Ultimately, the resolution of such paradoxes will reside in the detailed analysis of similar proteins from many phyla. While intellectually satisfying, such studies face challenges, not least of all in how they will be regarded by funding agencies.
The reader should not take our scenario for the evolution of the mitochondrial pathway of apoptosis too seriously. It is proposed as an exercise in how one might frame a story to raise questions, and where the answers to those questions may lie. Ultimately, our story will likely remain a Just So Story. But in the meantime, we hope that the tale has been thought provoking, or at least entertaining. In the end, that is what stories are for.
Supplementary Material
Footnotes
All quotes are from Rudyard Kipling, Just So Stories.
References
- 1.Zmasek CM, Godzik A. Evolution of the animal apoptosis network. Cold Spring Harb Perspect Biol. 2013;5:a008649. doi: 10.1101/cshperspect.a008649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Munoz-Pinedo C. Signaling pathways that regulate life and cell death: evolution of apoptosis in the context of self-defense. Adv Exp Med Biol. 2012;738:124–143. doi: 10.1007/978-1-4614-1680-7_8. [DOI] [PubMed] [Google Scholar]
- 3.Ameisen JC. On the origin, evolution, and nature of programmed cell death: a timeline of four billion years. Cell Death Differ. 2002;9:367–393. doi: 10.1038/sj.cdd.4400950. [DOI] [PubMed] [Google Scholar]
- 4.Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, ElDeiry WS, Fulda S, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McLuskey K, Mottram JC. Comparative structural analysis of the caspase family with other clan CD cysteine peptidases. Biochem J. 2015;466:219–232. doi: 10.1042/BJ20141324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crawford ED, Wells JA. Caspase substrates and cellular remodeling. Annu Rev Biochem. 2011;80:1055–1087. doi: 10.1146/annurev-biochem-061809-121639. [DOI] [PubMed] [Google Scholar]
- 7.Green DR. Means to an End: Apoptosis and other Cell Death Mechanisms. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press; 2011. [Google Scholar]
- 8.Toda S, Nishi C, Yanagihashi Y, Segawa K, Nagata S. Clearance of apoptotic cells and pyrenocytes. Curr Top Dev Biol. 2015;114:267–295. doi: 10.1016/bs.ctdb.2015.07.017. [DOI] [PubMed] [Google Scholar]
- 9.Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM, Ricci JE, Edris WA, Sutherlin DP, Green DR, et al. A unified model for apical caspase activation. Mol Cell. 2003;11:529–541. doi: 10.1016/s1097-2765(03)00051-0. [DOI] [PubMed] [Google Scholar]
- 10.Ewen CL, Kane KP, Bleackley RC. A quarter century of granzymes. Cell Death Differ. 2012;19:28–35. doi: 10.1038/cdd.2011.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salvesen GS, Dixit VM. Caspase activation: the induced-proximity model. Proc Natl Acad Sci USA. 1999;96:10964–10967. doi: 10.1073/pnas.96.20.10964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delivani P, Adrain C, Taylor RC, Duriez PJ, Martin SJ. Role for CED-9 and Egl-1 as regulators of mitochondrial fission and fusion dynamics. Mol Cell. 2006;21:761–773. doi: 10.1016/j.molcel.2006.01.034. [DOI] [PubMed] [Google Scholar]
- 13.McStay GP, Salvesen GS, Green DR. Overlapping cleavage motif selectivity of caspases: implications for analysis of apoptotic pathways. Cell Death Differ. 2008;15:322–331. doi: 10.1038/sj.cdd.4402260. [DOI] [PubMed] [Google Scholar]
- 14.Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- 15.Bender CE, Fitzgerald P, Tait SW, Llambi F, McStay GP, Tupper DO, Pellettieri J, Sanchez Alvarado A, Salvesen GS, Green DR. Mitochondrial pathway of apoptosis is ancestral in metazoans. Proc Natl Acad Sci USA. 2012;109:4904–4909. doi: 10.1073/pnas.1120680109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010;37:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chipuk JE, Green DR. Do inducers of apoptosis trigger caspase-independent cell death? Nat Rev Mol Cell Biol. 2005;6:268–275. doi: 10.1038/nrm1573. [DOI] [PubMed] [Google Scholar]
- 18.Lopez J, Tait SW. Mitochondrial apoptosis: killing cancer using the enemy within. Br J Cancer. 2015;112:957–962. doi: 10.1038/bjc.2015.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coll NS, Epple P, Dangl JL. Programmed cell death in the plant immune system. Cell Death Differ. 2011;18:1247–1256. doi: 10.1038/cdd.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strich R. Programmed cell death initiation and execution in budding yeast. Genetics. 2015;200:1003–1014. doi: 10.1534/genetics.115.179150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reece SE, Pollitt LC, Colegrave N, Gardner A. The meaning of death: evolution and ecology of apoptosis in protozoan parasites. PLoS Pathog. 2011;7:e1002320. doi: 10.1371/journal.ppat.1002320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Engelberg-Kulka H, Amitai S, Kolodkin-Gal I, Hazan R. Bacterial programmed cell death and multicellular behavior in bacteria. PLoS Genet. 2006;2:e135. doi: 10.1371/journal.pgen.0020135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salvesen GS, Hempel A, Coll NS. Protease signaling in animal and plant regulated cell death. FEBS J. 2015 doi: 10.1111/febs.13616. http://dx.doi.org/10.1111/febs.13616. [DOI] [PMC free article] [PubMed]
- 24.Giusti C, Luciani MF, Golstein P. A second signal for autophagic cell death? Autophagy. 2010;6:823–824. doi: 10.4161/auto.6.6.12750. [DOI] [PubMed] [Google Scholar]
- 25.Jacob F, Vernaldi S, Maekawa T. Evolution and conservation of plant NLR functions. Front Immunol. 2013;4:297. doi: 10.3389/fimmu.2013.00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sundstrom JF, Vaculova A, Smertenko AP, Savenkov EI, Golovko A, Minina E, Tiwari BS, Rodriguez-Nieto S, Zamyatnin AA, Jr, Valineva T, et al. Tudor staphylococcal nuclease is an evolutionarily conserved component of the programmed cell death degradome. Nat Cell Biol. 2009;11:1347–1354. doi: 10.1038/ncb1979. [DOI] [PubMed] [Google Scholar]
- 27.Wilkinson D, Ramsdale M. Proteases and caspase-like activity in the yeast Saccharomyces cerevisiae. Biochem Soc Trans. 2011;39:1502–1508. doi: 10.1042/BST0391502. [DOI] [PubMed] [Google Scholar]
- 28.Wiens M, Diehl-Seifert B, Muller WE. Sponge Bcl-2 homologous protein (BHP2-GC) confers distinct stress resistance to human HEK-293 cells. Cell Death Differ. 2001;8:887–898. doi: 10.1038/sj.cdd.4400906. [DOI] [PubMed] [Google Scholar]
- 29.Lasi M, Pauly B, Schmidt N, Cikala M, Stiening B, Kasbauer T, Zenner G, Popp T, Wagner A, Knapp RT, et al. The molecular cell death machinery in the simple cnidarian Hydra includes an expanded caspase family and pro- and anti-apoptotic Bcl-2 proteins. Cell Res. 2010;20:812–825. doi: 10.1038/cr.2010.66. [DOI] [PubMed] [Google Scholar]
- 30.Doumanis J, Dorstyn L, Kumar S. Molecular determinants of the subcellular localization of the Drosophila Bcl-2 homologues DEBCL and BUFFY. Cell Death Differ. 2007;14:907–915. doi: 10.1038/sj.cdd.4402082. [DOI] [PubMed] [Google Scholar]
- 31.Hengartner MO, Horvitz HR. C. elegans cell survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2. Cell. 1994;76:665–676. doi: 10.1016/0092-8674(94)90506-1. [DOI] [PubMed] [Google Scholar]
- 32.Dohrmann M, Worheide G. Novel scenarios of early animal evolution–is it time to rewrite textbooks? Integr Comp Biol. 2013;53:503–511. doi: 10.1093/icb/ict008. [DOI] [PubMed] [Google Scholar]
- 33.Pang Y, Bai XC, Yan C, Hao Q, Chen Z, Wang JW, Scheres SH, Shi Y. Structure of the apoptosome: mechanistic insights into activation of an initiator caspase from Drosophila. Genes Dev. 2015;29:277–287. doi: 10.1101/gad.255877.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang W, Jiang T, Choi W, Qi S, Pang Y, Hu Q, Xu Y, Gong X, Jeffrey PD, Wang J, et al. Mechanistic insights into CED-4-mediated activation of CED-3. Genes Dev. 2013;27:2039–2048. doi: 10.1101/gad.224428.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 36.Zhou M, Li Y, Hu Q, Bai XC, Huang W, Yan C, Scheres SH, Shi Y. Atomic structure of the apoptosome: mechanism of cytochrome c- and dATP-mediated activation of Apaf-1. Genes Dev. 2015;29:2349–2361. doi: 10.1101/gad.272278.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arama E, Bader M, Srivastava M, Bergmann A, Steller H. The two Drosophila cytochrome C proteins can function in both respiration and caspase activation. EMBO J. 2006;25:232–243. doi: 10.1038/sj.emboj.7600920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Means JC, Muro I, Clem RJ. Lack of involvement of mitochondrial factors in caspase activation in a Drosophila cell-free system. Cell Death Differ. 2006;13:1222–1234. doi: 10.1038/sj.cdd.4401821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dorstyn L, Mills K, Lazebnik Y, Kumar S. The two cytochrome c species, DC3 and DC4, are not required for caspase activation and apoptosis in Drosophila cells. J Cell Biol. 2004;167:405–410. doi: 10.1083/jcb.200408054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dorstyn L, Read S, Cakouros D, Huh JR, Hay BA, Kumar S. The role of cytochrome c in caspase activation in Drosophila melanogaster cells. J Cell Biol. 2002;156:1089–1098. doi: 10.1083/jcb.200111107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zimmermann KC, Ricci JE, Droin NM, Green DR. The role of ARK in stress-induced apoptosis in Drosophila cells. J Cell Biol. 2002;156:1077–1087. doi: 10.1083/jcb.20112068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jagasia R, Grote P, Westermann B, Conradt B. DRP-1-mediated mitochondrial fragmentation during EGL-1-induced cell death in C. elegans. Nature. 2005;433:754–760. doi: 10.1038/nature03316. [DOI] [PubMed] [Google Scholar]
- 43.Abdelwahid E, Yokokura T, Krieser RJ, Balasundaram S, Fowle WH, White K. Mitochondrial disruption in Drosophila apoptosis. Dev Cell. 2007;12:793–806. doi: 10.1016/j.devcel.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 44.Galindo KA, Lu WJ, Park JH, Abrams JM. The Bax/Bak ortholog in Drosophila, Debcl, exerts limited control over programmed cell death. Development. 2009;136:275–283. doi: 10.1242/dev.019042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tanner EA, Blute TA, Brachmann CB, McCall K. Bcl-2 proteins and autophagy regulate mitochondrial dynamics during programmed cell death in the Drosophila ovary. Development. 2011;138:327–338. doi: 10.1242/dev.057943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Monserrate JP, Chen MY, Brachmann CB. Drosophila larvae lacking the bcl-2 gene, buffy, are sensitive to nutrient stress, maintain increased basal target of rapamycin (Tor) signaling and exhibit characteristics of altered basal energy metabolism. BMC Biol. 2012;10:63. doi: 10.1186/1741-7007-10-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Steller H. Regulation of apoptosis in Drosophila. Cell Death Differ. 2008;15:1132–1138. doi: 10.1038/cdd.2008.50. [DOI] [PubMed] [Google Scholar]
- 48.Yan N, Chai J, Lee ES, Gu L, Liu Q, He J, Wu JW, Kokel D, Li H, Hao Q, et al. Structure of the CED-4-CED-9 complex provides insights into programmed cell death in Caenorhabditis elegans. Nature. 2005;437:831–837. doi: 10.1038/nature04002. [DOI] [PubMed] [Google Scholar]
- 49.Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187–192. doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- 50.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 51.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–671. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 52.James ER, Green DR. Infection and the origins of apoptosis. Cell Death Differ. 2002;9:355–357. doi: 10.1038/sj.cdd.4400986. [DOI] [PubMed] [Google Scholar]
- 53.Kletzin A, Heimerl T, Flechsler J, van Niftrik L, Rachel R, Klingl A. Cytochromes c in Archaea: distribution, maturation, cell architecture, and the special case of Ignicoccus hospitalis. Front Microbiol. 2015;6:439. doi: 10.3389/fmicb.2015.00439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Andersson SG, Zomorodipour A, Andersson JO, Sicheritz-Ponten T, Alsmark UC, Podowski RM, Naslund AK, Eriksson AS, Winkler HH, Kurland CG. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature. 1998;396:133–140. doi: 10.1038/24094. [DOI] [PubMed] [Google Scholar]
- 55.Guy L, Ettema TJ. The archaeal ‘TACK’ superphylum and the origin of eukaryotes. Trends Microbiol. 2011;19:580–587. doi: 10.1016/j.tim.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 56.Ku C, Nelson-Sathi S, Roettger M, Sousa FL, Lockhart PJ, Bryant D, Hazkani-Covo E, McInerney JO, Landan G, Martin WF. Endosymbiotic origin and differential loss of eukaryotic genes. Nature. 2015;524:427–432. doi: 10.1038/nature14963. [DOI] [PubMed] [Google Scholar]
- 57.Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, Lee EF, Yao S, Robin AY, Smith BJ, et al. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell. 2013;152:519–531. doi: 10.1016/j.cell.2012.12.031. [DOI] [PubMed] [Google Scholar]
- 58.Moldoveanu T, Grace CR, Llambi F, Nourse A, Fitzgerald P, Gehring K, Kriwacki RW, Green DR. BID-induced structural changes in BAK promote apoptosis. Nat Struct Mol Biol. 2013;20:589–597. doi: 10.1038/nsmb.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Muchmore SW, Sattler M, Liang H, Meadows RP, Harlan JE, Yoon HS, Nettesheim D, Chang BS, Thompson CB, Wong SL, et al. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature. 1996;381:335–341. doi: 10.1038/381335a0. [DOI] [PubMed] [Google Scholar]
- 60.Burger G, Gray MW, Forget L, Lang BF. Strikingly bacteria-like and gene-rich mitochondrial genomes throughout jakobid protists. Genome Biol Evol. 2013;5:418–438. doi: 10.1093/gbe/evt008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pellettieri J, Fitzgerald P, Watanabe S, Mancuso J, Green DR, Sanchez Alvarado A. Cell death and tissue remodeling in planarian regeneration. Dev Biol. 2010;338:76–85. doi: 10.1016/j.ydbio.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lu Y, Rolland SG, Conradt B. A molecular switch that governs mitochondrial fusion and fission mediated by the BCL2-like protein CED-9 of Caenorhabditis elegans. Proc Natl Acad Sci USA. 2011;108:E813–E822. doi: 10.1073/pnas.1103218108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hoppins S, Edlich F, Cleland MM, Banerjee S, McCaffery JM, Youle RJ, Nunnari J. The soluble form of Bax regulates mitochondrial fusion via MFN2 homotypic complexes. Mol Cell. 2011;41:150–160. doi: 10.1016/j.molcel.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Renault TT, Floros KV, Elkholi R, Corrigan KA, Kushnareva Y, Wieder SY, Lindtner C, Serasinghe MN, Asciolla JJ, Buettner C, et al. Mitochondrial shape governs BAX-induced membrane permeabilization and apoptosis. Mol Cell. 2015;57:69–82. doi: 10.1016/j.molcel.2014.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Quistad SD, Stotland A, Barott KL, Smurthwaite CA, Hilton BJ, Grasis JA, Wolkowicz R, Rohwer FL. Evolution of TNF-induced apoptosis reveals 550 My of functional conservation. Proc Natl Acad Sci USA. 2014;111:9567–9572. doi: 10.1073/pnas.1405912111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.