

Abstract
Ischemic injury is associated with acute myocardial infarction, percutaneous coronary intervention, coronary artery bypass grafting and open heart surgery. The timely re-establishment of blood flow is critical in order to minimize cardiac complications. Reperfusion after a prolonged ischemic period, however, can induce severe cardiomyocyte dysfunction with mitochondria serving as a major target of ischemia/reperfusion (I/R) injury. An increase in the formation of reactive oxygen species (ROS) induces damage to mitochondrial respiratory complexes leading to uncoupling of oxidative phosphorylation. Mitochondrial membrane perturbations also contribute to calcium overload, opening of the mitochondrial permeability transition pore (mPTP) and the release of apoptotic mediators into the cytoplasm. Clinical and experimental studies show that ischemic preconditioning (ICPRE) and postconditioning (ICPOST) attenuate mitochondrial injury and improve cardiac function in the context of I/R injury. This is achieved by the activation of two principal cell survival cascades: 1) the Reperfusion Injury Salvage Kinase (RISK) pathway; and 2) the Survivor Activating Factor Enhancement (SAFE) pathway. Recent data suggest that high density lipoprotein (HDL) mimics the effects of conditioning protocols and attenuates myocardial I/R injury via activation of the RISK and SAFE signaling cascades. In this review, we discuss the roles of apolipoproteinA-I (apoA-I), the major protein constituent of HDL, and sphingosine 1-phosphate (S1P), a lysosphingolipid associated with small, dense HDL particles as mediators of cardiomyocyte survival. Both apoA-I and S1P exert an infarct-sparing effect by preventing ROS-dependent injury and inhibiting the opening of the mPTP.
Keywords: HDL, mitochondrion, ApoA-I, Sphingosine 1-Phosphate, ischemia-reperfusion, myocardium
1. INTRODUCTION
A principal function of HDL is to mediate reverse cholesterol transport, a process by which excess cholesterol is removed from non-hepatic tissues and transferred to the liver for metabolism and excretion into the bile (1,2). HDL also possesses anti-inflammatory and antioxidant properties that are attributed, in large part, to its major protein constituent apoA-I (3–6). Helical regions of apoA-I serve as a platform for the binding of antioxidant proteins, including paraoxonase 1 (PON1) and platelet-activating factor acetylhydrolase (PAF-AH) (7,8). These enzymes play an important role by degrading cholesteryl esters and phospholipids in oxidized lipoproteins. A recent study shows that the HDL proteome consists of more than 85 proteins (9). It follows that HDL is a heterogenous particle and that HDL subspecies may display discreet functional properties.
The lipid composition of HDL is also an important determinant of its function (10,11). Lipid species maintain the structural integrity of HDL and regulate the activities of HDL-associated proteins (12). Among the phospholipids, phosphatidylcholine, sphingomyelin (SM) and sphingosine 1-phosphate (S1P) are well represented. S1P is synthesized in hematopoietic and endothelial cells through the action of sphingosine kinase 1 (SphK1) (13,14). HDL takes up S1P and serves as its principal carrier in plasma (15,16). Anti-inflammatory and antioxidant properties are prominently exhibited by small, dense HDL particles including preβ-HDL and HDL3 (15). This is due, in part, to the increased ratio of S1P to SM in HDL3 particles compared to more buoyant HDL1 and HDL2 particles.
Many of the salutary effects of HDL on cardiac and vascular function have been ascribed to the presence of S1P in the lipoprotein particle while other responses to HDL are S1P–independent (17,18). HDL-bound S1P is significantly reduced in patients with coronary artery disease (CAD) compared to healthy controls (19,20). A corresponding increase in non-HDL-bound S1P is associated with an increase in the severity of CAD symptoms (20). In light of these findings, HDL isolated from CAD patients displays impaired S1P–dependent signaling responses under in vitro conditions (19). Supplementation of HDL from CAD patients with S1P, however, effectively restores the functional properties of HDL-bound S1P (19).
Cardiac ischemia arises in response to pathological events including acute myocardial infarction (AMI), unstable angina and thrombolysis as well as surgical procedures (21). Reperfusion results in the activation of deleterious signaling pathways, with the mitochondrion serving as a critical site of injury (21–23). The prompt re-establishment of coronary blood flow is thus critically required to minimize myocardial infarct size (21,23, 24). In light of the high energy demands of the heart, ischemia/reperfusion (I/R) is associated with degradation of mitochondrial bioenergetics. An increase in reactive oxygen species (ROS) formation and uncoupling of oxidative phosphorylation are early events followed by opening of the mitochondrial permeability transition pore (mPTP) (25). The mPTP is a high conductance channel spanning the inner and outer mitochondrial membranes that remains in a closed state under normal conditions (23,26). mPTP opening occurs in response to I/R injury and is associated with dissipation of the mitochondrial membrane potential (ΔΨm), calcium influx and the release of pro-apoptotic factors (23,26). Minimizing mitochondrial damage is clearly an important strategy for maintaining normal cardiac function. Ischemic pre-conditioning (ICPRE) and post-conditioning (ICPOST) have been shown to reduce myocardial injury upon sustained reperfusion (22,27,28). ICPRE and ICPOST are characterized by repetitive, brief episodes of ischemia and reperfusion performed either prior to or after a prolonged period of ischemia (27,28). Both conditioning procedures protect the heart by activating cell survival pathways that converge at the level of the mitochondrion. HDL has also been shown to improve cardiac function in the context of I/R injury by preventing defects in mitochondrial function. The goal of this review is to discuss survival pathways activated by HDL that preserve myocardial function in the context of I/R injury.
2. MITOCHONDRIA AND CELLULAR BIOENERGETICS
Mitochondria are abundant in tissues with a high metabolic demand including cardiac and skeletal muscle (29). These organelles are characterized by a double-membrane structure separated by an intermembrane space. Respiratory complexes located in the inner mitochondrial membrane utilize oxidative phosphorylation to generate energy in the form of ATP. Nicotinamide adenine dinucleotide (NADH) initially serves as an electron donor for complex I (NADH:ubiquinone oxidoreductase) which transfers electrons to ubiquinone (30). Complex II (succinate dehydrogenase) functions in parallel with complex I and transfers electrons from succinate to ubiquinone. Electrons are subsequently shuttled from complexes I and II to complex III (CoQH2-cytochrome c reductase) via coenzyme Q and the Q cycle (30). Cytochrome c (cyt c) is a protein located in the intermembrane space that supports mitochondrial respiration by shuttling electrons from complex III to complex IV (cytochrome c oxidase) coincident with cyt c reduction (31). As oxygen is consumed at complex IV, cyt c is re-oxidized and water is formed. Throughout this process, hydrogen ions derived from complexes I, III, and IV are pumped from the matrix into the intermembrane space. As hydrogen ions accumulate at this site, a proton gradient is established which gives rise to ΔΨm (32). Finally, complex V (ATP synthase) utilizes the energy stored in the proton gradient to generate ATP. While electron transfer is tightly regulated, some electrons may react with oxygen to form superoxide anion. Deleterious effects of superoxide, however, are minimized by the presence of manganese superoxide dismutase which reduces superoxide to hydrogen peroxide. Under conditions where reactive oxygen species (ROS) are formed in excess, damage to mitochondrial structural components and DNA occurs.
3. MITOCHONDRIAL RESPONSES TO ISCHEMIA/REPERFUSION INJURY
Mitochondrial dysfunction is a hallmark of I/R injury. Damage to mitochondria is initiated during the ischemic period and becomes amplified during reperfusion (33–36). Recent data suggest that the complex II substrate succinate accumulates during ischemia, and, upon reperfusion, electron transport operates in the reverse mode with significant quantities of superoxide being generated at complex I (36). Ischemic injury at the level of complex III also plays an important role in stimulating ROS formation at sites upstream in the electron transport chain (31,37,38). ROS, generated in this manner, induce damage to mitochondrial respiratory complexes, structural components and DNA (35, 39– 41). Cardiolipin and cyt c are critical sites of ROS-dependent injury. Cardiolipin is a phospholipid in the inner mitochondrial membrane that stabilizes respiratory proteins, in part, by forming a complex with cyt c (31). An increase in ROS formation induces the peroxidation of the cardiolipin-cyt c complex resulting in the release of cyt c into the cytosol (31). The loss of cyt c facilitates apoptosis, inhibits respiration at complex IV and stimulates further generation of ROS (37,38,42). It follows that tissue oxygen utilization and ATP formation are severely impaired, and apoptotic/necrotic mechanisms are activated (43).
The ability of mitochondria to respond to fluctuations in cytosolic calcium (Ca2+) concentration is an important indicator of mitochondrial quality (44,45). Healthy mitochondria take up calcium via the uniporter located on the inner membrane and release calcium under normal conditions via the Na+/Ca2+ antiporter. However, this pathway is vulnerable to bioenergetic dysfunction and can result in accumulation of mitochondrial calcium. During reperfusion, mPTP opening is stimulated by numerous factors including ROS, calcium overload and dissipation of ΔΨm (46). Induction of mPTP results in calcium release, mitochondrial swelling and apoptotic cell death (44,46–51). These responses are negatively correlated with cardiomyocyte survival (49).
4. PRE- AND POST-CONDITIONING ATTENUATE ISCHEMIA-REPERFUSION INJURY
ICPRE and ICPOST describe intermittent episodes of I/R prior to sustained ischemia and reperfusion, respectively (27,28). The reduction in cardiomyocyte injury in response to ICPRE and ICPOST has been linked to the activation of two principal survival pathways: The Reperfusion Injury Salvage Kinase (RISK) pathway and the Survivor Activating Factor Enhancement (SAFE) pathway (22,49,52,53). The RISK cascade includes the pro-survival kinases phosphatidylinositol 3-kinase (PI3K), protein kinase B (Akt) and extracellular regulated kinase 1/2 (ERK1/2) (22,49,52,53). These enzymes phosphorylate multiple substrates in the cell that converge to inhibit opening of mPTP (49,52–54). Glycogen synthase kinase 3 beta (GSK3β) is thought to play an important role in RISK-dependent cardiomyocyte survival (49,55). Phosphorylation of GSK3β abolishes its enzymatic activity. In this inactivated form, pGSK3β reduces I/R injury by three mechanisms. First, pGSK3β fails to phosphorylate Bcl-2 resulting in an increase in Bcl-2 anti-apoptotic activity. Second, pGSK3β inhibits the translocation of pro-apoptotic Bax to the outer mitochondrial membrane. Finally, pGSK3β stabilizes the mPTP regulatory protein cyclophilin D, thus preventing mPTP induction (49,56).
The Survivor Activating Factor Enhancement (SAFE) cascade represents a second survival pathway activated by ICPRE and ICPOST (22,57). The SAFE pathway paradoxically utilizes tumor necrosis factor alpha (TNFα) as a cardioprotective mediator. Reperfusion injury, in the absence of conditioning, is associated with the release of a high concentration of TNFα which mediates cardiotoxic effects via the TNF receptor type 1 (TNFR1) (22,58,59). In contrast, release of low amounts of TNFα are associated with ICPRE and ICPOST. Under these conditions, TNFα binds to the TNF receptor type 2 (TNFR2) (22,58). ICPRE in a rodent model of coronary I/R injury reduces infarct size (60). The importance of TNFα in this response is underscored by the observation that administration of soluble TNFα receptor abrogated the effects of ICPRE (60). Lacerda and colleagues showed that administration of TNFα at the beginning of reperfusion mimics the protective effect of ICPOST on infarct size (57). This was characterized by a reduction in infarct size in wildtype and TNFR1−/− mice but not in TNFα−/− and TNFR2−/− mice (57). Similarly, administration of low doses of TNFα to mouse hearts ex vivo mimicked cardioprotective effects of ICPRE (61). Pro-survival mechanisms activated by TNFR2 engagement include the induction of Janus kinase (JAK) which, in turn, phosphorylates/activates the signal transducer and activator of transcription 3 (STAT3) (22). ICPRE in the pig was shown to reduce infarct size by a mechanism involving increased STAT3 phosphorylation and inhibition of mPTP opening (62). While the RISK and SAFE pathways activate distinct signaling cascades in cardiomyocytes, it is likely that the protective mechanisms underlying ICPRE and ICPOST are due to cross-talk between the pathways (22). Convergence of these survival cascades at the level of the mPTP has been suggested (57).
5. STAT3-DEPENDENT MECHANISMS OF CARDIOPROTECTION
The JAK-mediated phosphorylation of STAT3 occurs at two major sites (51, 63). Phosphorylation of tyrosine residue 705 is generally associated with translocation of STAT3 to the nucleus (64). STAT3-mediated nuclear transcription results in up-regulation of anti-apoptotic Bcl-2 and the antioxidant genes MnSOD and metallothionein while inhibiting pro-apoptotic Bax/Bad expression (22,65). Phosphorylation of serine residue 727 directs STAT3 to the mitochondrion where it has been shown to regulate the electron transport chain (64,65). pSTAT3 thus regulates gene expression and directly modulates mitochondrial respiration (65). Data suggest, however, that mitochondrial STAT3-dependent cytoprotection occurs on a time scale that precludes the STAT3-mediated transcription of nuclear genes (64,65). Mitochondrial STAT3 is therefore likely the principal mediator of cell survival in response to I/R injury.
GRIM-19 (gene associated with retinoid interferon induced cell mortality 19) acts as a chaperone to recruit serine phosphorylated STAT3 into mitochondria where it binds to complex I (51). Other data suggest that STAT3 inhibits complex I and II respiration during ischemia and thus inhibits ROS formation (65). This is supported by the observation that overexpression of STAT3 reduces electron flow through complex I, prevents ROS formation and inhibits the release of cyt c (65). Further, ICPRE studies show that mitochondrial STAT3 preserves complex I activity and mitochondrial respiration upon reperfusion. (62). It is proposed that STAT3 protects mitochondria during ischemia by uncoupling electron flow between respiratory complexes and preventing mPTP induction (65,66). Under these conditions, ROS formation is reduced. This uncoupling of electron transport is reversible and, upon reperfusion, mitochondrial respiration can proceed normally.
The importance of the SAFE pathway and STAT3 activation in limiting cardiac injury in I-R models has been revealed by studies using inhibitors of JAK. Treatment with the JAK inhibitor AG-490 reduces STAT3 phosphorylation in rats undergoing coronary ligation/reperfusion (67). Additional consequences of JAK inhibition include a reduction in cardiomyocyte viability and an increase in proapoptotic caspase-3 activity and Bax expression (67). AG-490 similarly reduces the phosphorylation of Akt, ERK2 and GSK3β and cardiac contractility in isolated rodent hearts exposed to anoxia-reoxygenation (68). This observation again supports crosstalk interaction between the RISK and SAFE pathways.
6. CARDIOPROTECTIVE RESPONSES TO HDL
HDL possesses functions that extend beyond its ability to mediate reverse cholesterol transport (69,70). Protein and lipid components of HDL both act as mediators of the cardioprotective response to HDL. The HDL-associated protein PON1 performs an important antioxidant function by hydrolyzing cholesteryl esters and phospholipids in oxidized lipoproteins (71). Glycosylated and oxidized LDL carry peroxides that stimulate ROS formation and impair oxygen consumption at respiratory complexes I, II/III, and IV resulting in mitochondrial dysfunction (71– 73). PON1 may thus indirectly preserve mitochondrial function by degrading oxidized lipid species. In contrast, a direct role for apoA-I and S1P in mediating cardiomyocyte survival has been proposed (63, 74–77). Knockout of apoA-I significantly increases infarct size in mice undergoing coronary artery ligation/reperfusion compared to control C56BL/6 (78). This defect was associated with a reduction in Coenzyme Q (CoQ) in mitochondria isolated from apoA-I−/− mice. CoQ deficiency resulted in a 67% decrease in electron transfer from complex II to complex III (78). Intraperitoneal administration of CoQ restored mitochondrial CoQ levels and reduced infarct size in apoA-I−/− mice (78). Results of this study indicated that apoA-I plays a critical role in maintaining the effective coupling of electron transport proteins. The molecular mechanism by which apoA-I influences the delivery of CoQ to mitochondria is unknown at this time.
HDL has also been shown to mimic cardioprotective effects of ICPRE and ICPOST by inducing RISK and SAFE survival pathways (79). Kalakech and colleagues reported that apoA-I, administered in vivo prior to coronary artery occlusion, attenuates morphologic changes associated with ischemic injury (myofibril tears, interstitial edema and leukocyte infiltration) and reduces infarct size (79). This infarct-sparing response was equivalent to the cardioprotection provided by ICPRE. In related experiments, administration of purified apoA-I was preceded by administration of inhibitors of Akt, ERK1/2 and JAK/STAT. The ability of apoA-I to reduce infarct size was attenuated under these conditions (79). Consistent with known targets of the RISK and SAFE signaling cascades, apoA-I treatment increased the phosphorylation of Akt and GSK3β. These data suggested that the inhibitory effect of apoA-I on myocardial infarct size was due to activation of both RISK and SAFE survival pathways.
7. SPHINGOSINE 1-PHOSPHATE AND CARDIOPROTECTION
Multiple mechanisms have been proposed to explain the cardioprotective effects of S1P. Apolipoprotein M (apoM) is a member of the lipocalin protein family (80,81). It associates with approximately 5% of HDL particles and is thought to play a major role in attenuating atherogenesis by stimulating preβ-HDL formation, facilitating macrophage cholesterol efflux and inhibiting LDL oxidation (81–86). ApoM also binds S1P and facilitates its incorporation into HDL particles (19,80, 86,87). HDL from apoM−/− mice does not contain S1P, while the S1P content of transgenic apoM (apoM-TG) mice is significantly increased (87). Accordingly, HDL from apoM-TG mice stimulates S1P–dependent signaling mechanisms in endothelial cells while HDL from apoM null mice does not (87). The relationship between apoM expression and S1P-mediated cardioprotection has been recently evaluated in wildtype and transgenic mice undergoing coronary artery ligation and reperfusion (88,89). Plasma S1P levels were increased in apoM-TG mice by approximately 250% compared to wildtype mice. This was accompanied by a significant reduction in myocardial infarct size and neutrophil accumulation (88). A link between apoM and S1P as mediators of cardioprotection in vivo was thus established. In vitro analyses of neonatal rat ventricular cardiomyocytes showed that S1P induced phosphorylation of Connexin43 (Cx43) and reduced gap junctional communication between cardiomyocytes (88). It was proposed that S1P protects the heart against I/R injury by inhibiting cell-cell coupling (88,89). In this manner, the passage of death signals through gap junctions is reduced, resulting in attenuation of I/R-induced cardiomyocyte injury (89).
HDL-associated S1P has also been shown to act as an inducer of both the RISK and SAFE pathways (63,90,91). Cardiomyocyte responses to S1P actions are mediated by multiple receptor isoforms (S1P1, S1P2 and S1P3) (18,70,90,92). Exposure of neonatal rat cardiac cardiomyocytes to native HDL or purified S1P activates S1P2 receptors resulting in the phosphorylation of STAT3 on serine 727 followed by tyrosine 705. Reconstituted HDL that was devoid of S1P failed to support STAT3 phosphorylation (93). Inhibition of PI3K with wortmannin did not influence STAT3 phosphorylation induced by either HDL or S1P suggesting the specific activation of the SAFE cascade (93).
HDL treatment prior to hypoxia-reoxygenation improves the viability of mouse cardiomyoctyes (18). HDL activated Akt and ERK1/2 pathways via distinct S1P receptors. S1P1 binding activated ERK1/2 while S1P3 induced Akt activation. The phosphorylation of GSK3β, a known inhibitor of mPTP opening, was shown to be a major downstream effector of HDL. These responses were blocked by treatment with S1P receptor inhibitors as well as the PI3K inhibitor wortmannin. It was concluded that HDL-associated S1P improves cardiomyocyte viability in the context of hypoxia-reoxygenation by activating the RISK pathway (18).
In vivo studies recapitulate cardioprotective effects of S1P in the context of I/R injury and heart failure (17,18,90,94,95). Somers and colleagues have assessed cardioprotective responses to S1P in hearts isolated from wildtype, TNFα−/− and cardiomyocyte-specific STAT3−/− mice. Hearts were subjected to global ischemia followed by reperfusion. While S1P administration at the beginning of reperfusion reduced infarct size in wildtype hearts, this response was absent in hearts from STAT3−/− and TNFα−/− mice (92). Addition of the JAK inhibitor AG-490 to wildtype mice prior to reperfusion reduced the nuclear translocation of STAT3 and abolished the effect of S1P on infarct size, supporting a role for S1P in the induction of the SAFE survival pathway. Pre-treatment of hearts with the PI3K inhibitor wortmannin also abolished the infarct-sparing effect of S1P, suggesting that the RISK pathway may also be activated by S1P (92). Mechanistically, it was shown that the S1P-dependent activation of PI3K/Akt was associated with the phosphorylation/inactivation of GSK3β and inhibition of cyt C release in mitochondria isolated from murine cardiomyocytes (96).
Cardioprotective responses to HDL and S1P are ultimately mediated at the level of the mitochondrion. Frias and colleagues have shown that administration of HDL during the reperfusion period reduces infarct size in a concentration-dependent manner (97). This response was associated with induction of STAT3 phosphorylation in both the cytosol and mitochondria and inhibition of mPTP opening. Survival mechanisms induced by HDL were abolished in TNFα−/− and cardiomyocyte-specific STAT3−/− mice, suggesting induction of the SAFE pathway (97).
Forkhead box O-1 (FOXO-1) is a transcription factor that is known to increase ROS formation and apoptosis in the non-phosphorylated state (98). While the RISK pathway and PI3K/Akt activation are classically associated with the inactivation of FOXO-1, data suggest that the SAFE survival pathway also modulates FOXO-1 activity (92,98). S1P stimulated the nuclear phosphorylation/inactivation of FOXO-1 in a manner that was blocked by both a JAK/STAT3 and PI3K inhibitor (92). This result suggests that the S1P-dependent phosphorylation of FOXO-1 may represent a point of convergence for the RISK and SAFE survival cascades (92,98).
The presence of sphingosine kinase 2 (SphK2) in mitochondria represents an alternate pathway for S1P production and action in cardiomyocytes (63). Ludovic and colleagues demonstrated that ICPRE in mice followed by I/R activated SphK1 to increase cytosolic levels of S1P (99). It was proposed that S1P utilizes an “inside-out” signaling mechanism whereby S1P released from the cell bound to cell surface S1P receptors (99,100). Receptor binding then activated signaling pathways culminating in an increase in mitochondrial SphK2 activity and S1P formation (99). Mitochondrial S1P was shown to regulate complex IV assembly and cellular respiration via interaction with mitochondrial prohibitin-2 (PHB2) (99,101). The principal role of PHB2 is to act as a scaffold providing structural stability for the inner mitochondrial membrane (102). Deletion of mitochondrial SphK2 abolished the cardioprotective response to ICPRE as reflected by a decrease in oxidative phosphorylation and opening of mPTP (99,101). These data suggest that mitochondrial S1P and PHB2 stabilize complex IV thus reducing ROS formation while also supporting oxidative phosphorylation (99).
8. AUTOPHAGY AS A CELL SURVIVAL MECHANISM
ATP depletion, ROS formation and mPTP opening are characteristic responses to I/R that are also associated with the induction of autophagy (103,104). Autophagy is a process by which cell death is minimized via removal of protein aggregates and damaged organelles (105,106). A specialized form of autophagy known as mitophagy arises in response to mPTP induction and a decrease in ΔΨm and is associated with the localization of mitochondria in autophagosomes (107). Ischemia and nutrient deprivation initiate autophagy by inducing the de-phosphorylation and inactivation of the mammalian target of rapamycin (mTOR) which normally acts as a suppressor of autophagy (108). Concurrently, an increase in the ratio of AMP/ATP induces AMP-activated protein kinase (AMPK) which stimulates autophagy through multiple mechanisms. The vacuolar protein sorting-34 (Vps34), a class III PI-3 kinase, plays an important role in the initiation of phagophore formation through its association with beclin1 (108). This pre-autophagosomal structure engulfs cytoplasmic components, including damaged mitochondria. Phosphatidylethanolamine (PE) and microtubule associated protein light chain-3 (LC3 I) interact to form the conjugated product LC3 II. LC3 II and the adaptor protein p62 are recruited to yield the mature autophagosome (108). The autophagosome then fuses with a lysosome where lysosomal hydrolases digest the contents.
Mitochondrial damage induced by I/R injury releases apoptotic factors that, in turn, damage neighboring mitochondria. It has been proposed that autophagy serves a cytoprotective role by clearing damaged mitochondria and limiting potentially deleterious effects on neighboring organelles. Induction of autophagy has been shown to reduce myocardial injury induced by I/R. Decker and Wildenthal reported that cardiac contractility was sustained in reperfused rabbit hearts that were exposed to short periods (20–40 minutes) of hypoxia (109). This was ascribed to induction of a cellular repair process that was characterized by an increase in lysosomal autophagy. In contrast, exposure to hypoxia for longer time periods resulted in irreversible cardiomyocyte injury (109). Other studies show that coronary I/R induces autophagy proteins in the heart that attenuate post-infarction cardiac remodeling (110).
Inhibitors of autophagy promote ROS formation and aggravate mitochondrial injury in response to I/R (111). Treatment of mice with an inhibitor of the autophagy protein Atg5 was shown to ablate the infarct-sparing effect of ICPRE. It follows that cytoprotection associated with ICPRE has been linked to activation of the autophagy pathway (112,113). In this respect, it is thought that autophagy improves cardiomyocyte survival by removing damaged mitochondria while leaving behind a mitochondrial population that is more resistant to mPTP opening (104). The role of S1P in the inhibition of mTOR activity and activation of autophagy has been recently reviewed (114,115). Data suggest that SphK1 plays an important role in this response since pharmacological blockade of the enzyme was shown to reduce S1P levels and inhibit autophagy (116). As the principal carrier of S1P, HDL may induce autophagy as a cell survival mechanism. Recent supportive data show that HDL inhibits mTOR activity, stimulates the expression of LC3 II and induces the formation of autophagosomes in enterocytes (117). To date, however, no studies have specifically tested whether HDL reduces myocardial I/R by activating autophagy.
9. CONCLUSION
A reduction in plasma HDL concentration is a strong, independent predictor of cardiovascular risk (118). A low baseline HDL concentration (<33mg/dL) prior to percutaneous coronary intervention is associated with a significant increase in one-year mortality (119). Similarly, low baseline HDL is a predictor of recurrent cardiovascular events in patients with acute coronary syndrome (120). In addition to changes in HDL levels, the lipoprotein may also undergo changes in its functional properties. Under pathological conditions, HDL can be converted to an acute phase lipoprotein that is depleted of apoA-I and displays pro-inflammatory properties (121–123). The formation of dysfunctional HDL is associated with alterations in both the protein and lipid content of the particle (124,125). With respect to protein composition, reductions in both apolipoproteins and accessory proteins that regulate lipid metabolism have been reported. The loss of apoA-I and PON1 is associated with a decrease in the anti-inflammatory and antioxidant properties of HDL (126). Alterations in the S1P/SM ratio also influence the anti-inflammatory effects of HDL-associated S1P (15). Small, dense HDL3, characterized by an elevated S1P/SM ratio, is anti-inflammatory and anti-apoptotic. In contrast, enrichment of HDL2 with SM and a reduced S1P/SM ratio negatively impact HDL surface fluidity and lecithin-cholesterol acyltransferase (LCAT) activity (15). Lipoprotein oxidation is also associated with a significant reduction in S1P levels and accumulation of the pro-inflammatory lipid species lysophosphatidylcholine in HDL particles (127). It has become clear that HDL is not just a passive mediator of cholesterol transport but is an active signaling particle that, depending on its composition, exerts either anti-inflammatory or pro-inflammatory effects. Therapeutic strategies that raise circulating HDL and its functional properties may therefore play an important role in minimizing cardiac injury.
ApoA-I and reconstituted HDL have been shown to reduce inflammatory tissue injury by multiple mechanisms (128). As discussed in this review, HDL-associated proteins and lipids reduce I/R injury by preserving mitochondrial function. The pro-survival benefits of HDL are dependent on the composition of the lipoprotein particle. PON1 plays a role by reducing the damaging effects of oxidized lipids on mitochondrial respiratory complexes. The protective response to apoA-I per se may be due to multiple factors. ApoA-I stabilizes complex II function by a CoQ-dependent mechanism and inhibits ROS-mediated damage to respiratory complexes. Other data suggest that both apoA-I and S1P induce RISK and SAFE survival cascades that preserve mitochondrial function via activation of Akt, ERK1/2 and JAK/STAT. The effect of S1P is mediated via activation of cell surface S1P receptors while the response to apoA-I is likely related to an interaction with ABCA1 (74,91,129). Data discussed in this review suggest that two effector mechanisms, common to RISK and SAFE cascades, are critically required for cardioprotection. It is proposed that survival kinases phosphorylate/inactivate FOXO-1, thus inhibiting mitochondrial ROS formation and apoptosis. Phosphorylated GSK3β is a second effector that also inhibits apoptotic mechanisms but, importantly, stabilizes the mPTP regulatory protein cyclophilin D and prevents mPTP induction. Major cellular targets of RISK and SAFE cascades are summarized in Figure 1.
Figure 1.
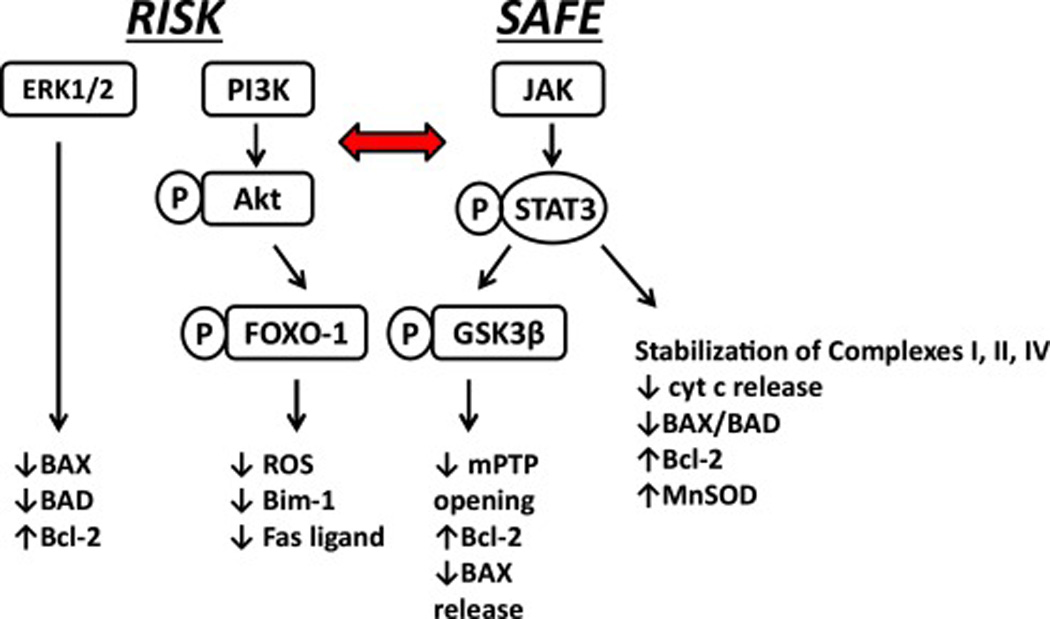
Cellular targets of RISK and SAFE survival pathways. Phosphorylation of multiple target molecules results in suppression of apoptotic mechanisms, stabilization of respiratory complexes and inhibition of mPTP opening. The horizontal arrow indicates crosstalk between each pathway. While the specific site of interaction has not been clearly defined, data suggest that FOXO-1 and GSK3β are terminal effectors of each pathway.
An increase in mitochondrial ROS formation is a principal contributor to I/R injury. Pharmacological strategies that inhibit oxidative stress, however, fail to provide therapeutic benefit (36). The observation that apoA-I and S1P activate signaling components of the RISK and SAFE pathways has encouraged the development of HDL-based therapies to attenuate I/R injury. Several studies suggest that rHDL is effective in reducing post-ischemic cardiac injury. rHDL composed of apoA-I, S1P and phospholipid is as effective as native HDL in reducing infarct size in a rodent model of I/R injury (130). Both native HDL and rHDL containing S1P increased activation of pro-survival proteins Akt, STAT3 and ERK1/2 (130). This study showed that the infarct-sparing effect of rHDL was attenuated in the absence of S1P. Mutant apoA-IMilano has also been used to test cardioprotective responses to rHDL. Incorporation of recombinant apoA-IMilano into rHDL particles reduces post-ischemic cardiac dysfunction in the isolated rabbit heart (131). A complication with apoA-IMilano-based therapy, however, is that a large amount of recombinant protein and phospholipid are required to form rHDL particles and the procedure is costly (132). In light of the prominent cardioprotective effects of S1P, there is strong rationale for the development of S1P receptor agonists. FTY720 is a compound that activates multiple S1P receptors and has been shown to attenuate I/R injury in animal models (17,133–136). The potent immunosuppressive effect of FTY720, however, may limit its therapeutic application (135,136). FTY720 also induces bradycardia, thus its use in patients with compromised cardiac function is contraindicated (137). Newer S1P receptor agonists are currently undergoing clinical evaluation.
ApoA-I mimetic peptides have been extensively studied by us and other laboratories and have been shown to mimic several properties of apoA-I (138–141). 4F is an 18 amino acid peptide and, due to its amphipathic nature, it is capable of forming aggregates which mimic those formed by apoA-I (142). Administration of the apoA-I mimetic peptide 4F to atherosclerosis-sensitive mice inhibits lesion formation without changing plasma cholesterol (143). This has been attributed to formation of preβ HDL particles and scavenging of lipid hydroperoxides and ROS (144). Thus, the beneficial effect of 4F may be due to a modification of HDL properties or to a direct effect on cells, as described above for apoA-I. HDL has been shown to activate JAK in macrophages by binding to the ATP binding cassette transporter subfamily A member 1 (ABCA1) (145). The apoA-I mimetic peptides 2F, 4F, L-37pA, and D-37pA form amphipathic helices that interact with ABCA1 to stimulate JAK2 autophosphorylation (146). Recent data show that L-37pA and D-37pA form HDL-like particles that reduce post-ischemic cardiac contractile dysfunction and creatine kinase release in a rat heart model of I/R (147). L-37pA and D-37pA complexes were as effective as rHDL containing apoA-I in reducing cardiac injury. The cytoprotective response to 4F has also been tested in animal models of I/R. Pre-treatment with L-4F was shown to normalize left ventricular function and increase fractional shortening in diabetic mouse hearts that were subjected to I/R injury (148). A separate study reported that D-4F significantly improves the infarct-sparing effect of anesthetic preconditioning (149). These data suggest that apoA-I mimetic peptides show strong potential for reducing myocardial injury associated with I/R injury.
Highlights.
HDL mimics the cardioprotective effects of ischemic pre- and post-conditioning.
HDL-associated S1P is associated with preservation of mitochondrial function.
HDL/S1P attenuate ischemia-reperfusion injury by activating distinct survival cascades.
RISK and SAFE cascades inhibit opening of the mitochondrial permeability transition pore.
Acknowledgments
Disclosures
Dr. Anantharamaiah is a Principal in Bruin Pharma, Inc. and holds shares in LipimetiX LLC. This work was supported by NIH HL34343, GM115367 and DK108836.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- 1.Lewis GF, Rader DJ. New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circ Res. 2005;96:1221–1232. doi: 10.1161/01.RES.0000170946.56981.5c. [DOI] [PubMed] [Google Scholar]
- 2.Fazio S, Linton MF. Sorting out the complexities of reverse cholesterol transport: CETP polymorphisms, HDL, and coronary disease. J Clin Endocrinol Metab. 2006;91:3273–3275. doi: 10.1210/jc.2006-1264. [DOI] [PubMed] [Google Scholar]
- 3.Ansell BJ, Watson KE, Fogelman AM, Navab M, Fonarow GC. High-density lipoprotein function: recent advances. J Am Coll Cardiol. 2005;46:1792–1798. doi: 10.1016/j.jacc.2005.06.080. [DOI] [PubMed] [Google Scholar]
- 4.Assmann G, Nofer JR. Atheroprotective effects of high-density lipoproteins. Annu Rev Med. 2003;54:321–341. doi: 10.1146/annurev.med.54.101601.152409. [DOI] [PubMed] [Google Scholar]
- 5.Dunbar RL, Rader DJ. Current drug options for raising HDL cholesterol. Curr Treat Options Cardiovasc Med. 2005;7:15–23. doi: 10.1007/s11936-005-0002-6. [DOI] [PubMed] [Google Scholar]
- 6.Navab M, Anantharamaiah GM, Reddy ST, Van Lenten BJ, Hough G, Wagner A, Nakamura K, Garber DW, Datta G, Segrest JP, et al. Human apolipoprotein A-I mimetic peptides for the treatment of atherosclerosis. Current Opinion in Investigational Drugs. 2003;4:1100–1104. [PubMed] [Google Scholar]
- 7.Gu X, Huang Y, Levison BS, Gerstenecker G, DiDonato AJ, Hazen LB, Lee J, Gogonea V, DiDonato JA, Hazen SL. Identification of critical paraoxonase 1 residues involved in high density lipoprotein interaction. J Biol Chem. 2016;291:1890–1904. doi: 10.1074/jbc.M115.678334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bashtovyy D, Jones MK, Anantharamaiah GM, Segrest JP. Sequence conservation of apolipoprotein A-I affords novel insights into HDL structure-function. J Lipid Res. 2011;52:435–450. doi: 10.1194/jlr.R012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shah AS, Tan L, Long JL, Davidson WS. Proteomic diversity of high density lipoproteins: our emerging understanding of its importance in lipid transport and beyond. J Lipid Res. 2013;54:2575–2585. doi: 10.1194/jlr.R035725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ashby D, Gamble J, Vadas M, Fidge N, Siggins S, Rye KA, Barter PJ. Lack of an effect of serum amyloid A (SAA) on the ability of high-density lipoproteins to inhibit endothelial cell adhesion molecular expression. Atherosclerosis. 2001;154:113–121. doi: 10.1016/s0021-9150(00)00437-8. [DOI] [PubMed] [Google Scholar]
- 11.Baker PW, Rye KA, Gamble JR, Vadas MA, Barter PJ. Ability of reconstituted high density lipoproteins to inhibit cytokine-induced expression of vascular cell adhesion molecule-1 in human umbilical vein endothelial cells. J Lipid Res. 1999;40:345–353. [PubMed] [Google Scholar]
- 12.Weisner P, Leidl K, Boettcher A, Schmitz G, Liebisch G. Lipid profiling of FPLC-separated lipoprotein fractions by electrospray ionization tandem mass spectrometry. J Lipid Res. 2009;50:574–585. doi: 10.1194/jlr.D800028-JLR200. [DOI] [PubMed] [Google Scholar]
- 13.Yatomia Y, Ozakia Y, Ohmoria T, Igarashi Y. Sphingosine 1-phosphate: synthesis and release. Prostaglandins & Other Lipid Mediators. 2001;64:107–122. doi: 10.1016/s0090-6980(01)00103-4. [DOI] [PubMed] [Google Scholar]
- 14.Venkataraman K, Lee YM, Michaud J, Thangada S, Ai Y, Bonkovsky HL, Parikh NS, Habrukowich C, Hla T. Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circulation Research. 2008;102:669–676. doi: 10.1161/CIRCRESAHA.107.165845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kontush A, Therond P, Zerrad A, Couturier M, Négre-Salvayre A, de Souza JA, Chantepie S, Chapman MJ. Preferential sphingosine-1-phosphate enrichment and sphingomyelin depletion are key features of small dense HDL3 particles: relevance to antiapoptotic and antioxidative activities. Arterioscler Thromb Vasc Biol. 2007;27:1843–1849. doi: 10.1161/ATVBAHA.107.145672. [DOI] [PubMed] [Google Scholar]
- 16.Yatomi Y, Ozaki Y, Ohmori T, Igarashi Y. Sphingosine 1-phosphate: synthesis and release. Prostaglandins Other Lipid Mediat. 2001;64:107–122. doi: 10.1016/s0090-6980(01)00103-4. [DOI] [PubMed] [Google Scholar]
- 17.Sattler K, Levkau B. Sphingosine-1-phosphate as a mediator of high-density lipoprotein effects in cardiovascular protection. Cardiovasc Res. 2009;82:201–211. doi: 10.1093/cvr/cvp070. [DOI] [PubMed] [Google Scholar]
- 18.Tao R, Hoover HE, Honbo N, Kalinowski M, Alano CC, Karliner JS, Raffai R. High-density lipoprotein determines adult mouse cardiomyocyte fate after hypoxia-reoxygenation through lipoprotein-associated sphingosine 1-phosphate. Am J Physiol. 2010;298:H1022–H1028. doi: 10.1152/ajpheart.00902.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sattler K, Gräler M, Keul P, Weske S, Reimann CM, Jindrová H, Kleinbongard P, Sabbadini R, Bröcker-Preuss M, Erbel R, Heusch G, Levkau B. Defects of High-Density Lipoproteins in Coronary Artery Disease Caused by Low Sphingosine-1-Phosphate Content: Correction by Sphingosine-1-Phosphate-Loading. J Am Coll Cardiol. 2015;66:1470–1485. doi: 10.1016/j.jacc.2015.07.057. [DOI] [PubMed] [Google Scholar]
- 20.Sattler KJ, Elbasan S, Keul P, Elter-Schulz M, Bode C, Gräler MH, Bröcker-Preuss M, Budde T, Erbel R, Heusch G, Levkau B. Sphingosine 1-phosphate levels in plasma and HDL are altered in coronary artery disease. Basic Res Cardiol. 2010;105:821–832. doi: 10.1007/s00395-010-0112-5. [DOI] [PubMed] [Google Scholar]
- 21.Verma S, Fedak PWM, Weisel RD, Butany J, Rao V, Maitland A, Li RK, Dhillon B, Yau TM. Fundamentals of reperfusion injury for the clinical cardiologist. Circulation. 2002;105:2332–2336. doi: 10.1161/01.cir.0000016602.96363.36. [DOI] [PubMed] [Google Scholar]
- 22.Lecour S. Activation of the protective Survivor Activating Factor Enhancement (SAFE) pathway against reperfusion injury: Does it go beyond the RISK pathway? Journal of Molecular and Cellular Cardiology. 2009;47:32–40. doi: 10.1016/j.yjmcc.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 23.Perrelli MG, Pagliaro P, Penna C. Ischemia/reperfusion injury and cardioprotective mechanisms: Role of mitochondria and reactive oxygen species. World J Cardiol. 2011;3:186–200. doi: 10.4330/wjc.v3.i6.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hausenloy DJ, Yellon DM. New directions for protecting the heart against ischaemia-reperfusion injury: targeting the Reperfusion Injury Salvage Kinase (RISK)-pathway. Cardiovascular Research. 2004;61:448–460. doi: 10.1016/j.cardiores.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 25.Hill BG, Benavides GA, Lancaster JR, Ballinger S, Dell’Italia L, Zhang J, Darley-Usmar VM. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol Chem. 2012;393:1485–1512. doi: 10.1515/hsz-2012-0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heusch G, Boengler K, Schulz R. Inhibition of mitochondrial permeability transition pore opening: the holy grail of cardioprotection. Basic Res Cardiol. 2010;105:151–154. doi: 10.1007/s00395-009-0080-9. [DOI] [PubMed] [Google Scholar]
- 27.Vinten-Johansen J, Yellon DM, Opie LH. Postconditioning: a simple, clinically applicable procedure to improve revascularization in acute myocardial infarction. Circulation. 2005;112:2085–2088. doi: 10.1161/CIRCULATIONAHA.105.569798. [DOI] [PubMed] [Google Scholar]
- 28.Vinten-Johansen J, Zhao ZQ, Jiang R, Zatta AJ, Dobson GP. Preconditioning and postconditioning: innate cardioprotection from ischemia-reperfusion injury. Journal of Applied Physiology. 2007;103:1441–1448. doi: 10.1152/japplphysiol.00642.2007. [DOI] [PubMed] [Google Scholar]
- 29.Forner F, Foster LJ, Campanaro S, Valle G, Mann M. Quantitative proteomic comparison of rat mitochondria from muscle, heart, and liver. Molecular & Cellular Proteomics. 2006;5:608–619. doi: 10.1074/mcp.M500298-MCP200. [DOI] [PubMed] [Google Scholar]
- 30.Sazanov LA. A giant molecular proton pump: structure and mechanism of respiratory complex I. Nature Reviews Molecular Cell Biology. 2015;16:375–388. doi: 10.1038/nrm3997. [DOI] [PubMed] [Google Scholar]
- 31.Chen Q, Yin G, Stewart S, Hu Y, Lesnefsky EJ. Isolating the segment of the mitochondrial electron transport chain responsible for mitochondrial damage during cardiac ischemia. Biochem Biophys Res Commun. 2010;397:656–660. doi: 10.1016/j.bbrc.2010.05.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amo T, Sato S, Saiki S, Wolf AM, Toyomizu M, Gautier CA, Shen J, Ohta S, Hattori N. Mitochondrial membrane potential decrease caused by loss of PINK1 is not due to proton leak, but to respiratory chain defects. Neurobiol Dis. 2011;41:111–118. doi: 10.1016/j.nbd.2010.08.027. [DOI] [PubMed] [Google Scholar]
- 33.Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol. 2012;298:229–317. doi: 10.1016/B978-0-12-394309-5.00006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Q, Camara AK, Stowe DF, Hoppel CL, Lesnefsky EJ. Modulation of electron transport protects cardiac mitochondria and decreases myocardial injury during ischemia and reperfusion. Am J Physiol. 2007;292:C137–C147. doi: 10.1152/ajpcell.00270.2006. [DOI] [PubMed] [Google Scholar]
- 35.Becker LB. New concepts in reactive oxygen species and cardiovascular reperfusion physiology. Cardiovasc Res. 2004;61:461–470. doi: 10.1016/j.cardiores.2003.10.025. [DOI] [PubMed] [Google Scholar]
- 36.Pell VR, Chouchani ET, Murphy MP, Brookes P, Krieg T. Moving forwards by blocking back-flow: the yin and yang of MI therapy. Circ Res. 2016;118:898–906. doi: 10.1161/CIRCRESAHA.115.306569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Q, Hoppel CL, Lesnefsky EJ. Blockade of electron transport before cardiac ischemia with the reversible inhibitor amobarbital protects rat heart mitochondria. J Pharmacol Exp Ther. 2006;316:200–207. doi: 10.1124/jpet.105.091702. [DOI] [PubMed] [Google Scholar]
- 38.Lesnefsky EJ, Chen Q, Slabe TJ, Stoll MS, Minkler PE, Hassan MO, Tandler B, Hoppel CL. Ischemia, rather than reperfusion, inhibits respiration through cytochrome oxidase in the isolated, perfused rabbit heart: role of cardiolipin. Am J Physiol. 2004;287:H258–H267. doi: 10.1152/ajpheart.00348.2003. [DOI] [PubMed] [Google Scholar]
- 39.Casillas-Ramirez A, Mosbah IB, Ramalho F, Rosello-Catafau J, Peralta C. Past and future approaches to ischemia-reperfusion lesion associated with liver transplantation. Life Sci. 2006;79:1881–1894. doi: 10.1016/j.lfs.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 40.Jaeschke H, Mitchell JR. Mitochondria and xanthine oxidase both generate reactive oxygen species in isolated perfused rat liver after hypoxic injury. Biochem Biophys Res Commun. 1989;160:140–147. doi: 10.1016/0006-291x(89)91632-x. [DOI] [PubMed] [Google Scholar]
- 41.Singer M, Brealey D. Mitochondrial dysfunction in sepsis. Biochem Soc Symp. 1999;66:149–166. doi: 10.1042/bss0660149. [DOI] [PubMed] [Google Scholar]
- 42.Lemasters JJ, Holmuhamedov E. Voltage-dependent anion channel (VDAC) as mitochondrial governator-thinking outside the box. Biochim Biophys Acta. 2006;1762:181–190. doi: 10.1016/j.bbadis.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 43.Fink MP. Cytopathic hypoxia. Mitochondrial dysfunction as mechanism contributing to organ dysfunction in sepsis. Crit Care Clin. 2001;17:219–237. doi: 10.1016/s0749-0704(05)70161-5. [DOI] [PubMed] [Google Scholar]
- 44.Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochimica et biophysica acta. 2009;1787:1395–1401. doi: 10.1016/j.bbabio.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rodriguez-Enriquez S, He L, Lemasters JJ. Role of mitochondrial permeability transition pores in mitochondrial autophagy. Int J Biochem Cell Biol. 2004;36:2463–2472. doi: 10.1016/j.biocel.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 46.Wong R, Steenbergen C, Murphy E. Mitochondrial permeability transition pore and calcium handling. Methods Mol Biol. 2012;810:235–242. doi: 10.1007/978-1-61779-382-0_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bopassa JC, Michel P, Gateau-Roesch O, Ovize M, Ferrera R. Low-pressure reperfusion alters mitochondrial permeability transition. Amer J Physiol. 2005;288:H2750–H2755. doi: 10.1152/ajpheart.01081.2004. [DOI] [PubMed] [Google Scholar]
- 48.Kim JS, Nitta T, Mohuczy D, O’Malley KA, Moldawer LL, Dunn WA, Behrns KE. Impaired autophagy: a mechanism of mitochondrial dysfunction in anoxic rat hepatocytes. Hepatology. 2008;47:1725–1736. doi: 10.1002/hep.22187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Juhaszova M, Zorov DB, Yaniv Y, Nuss HB, Wang S, Sollott SJ. Role of glycogen synthase kinase-3in cardioprotection. Circ Res. 2009;104:1240–1252. doi: 10.1161/CIRCRESAHA.109.197996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chopra M, Golden HB, Mullapudi S, Dowhan W, Dostal DE, Sharma AC. Modulation of myocardial mitochondrial mechanisms during severe polymicrobial sepsis in the rat. PLoS One. 2011;6(6):e21285. doi: 10.1371/journal.pone.0021285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tammineni P, Anugula C, Mohammed F, Anjaneyulu M, Larner AC, Sepuri NB. The import of the transcription factor STAT3 into mitochondria depends on GRIM-19, a component of the electron transport chain. J Biol Chem. 2013;288:4723–4732. doi: 10.1074/jbc.M112.378984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hausenloy DJ, Yellon DM. Reperfusion injury salvage kinase signalling: taking a RISK for cardioprotection. Heart Fail Rev. 2007;12:217–234. doi: 10.1007/s10741-007-9026-1. [DOI] [PubMed] [Google Scholar]
- 53.Hausenloy DJ, Tsang A, Yellon DM. The reperfusion injury salvage kinase pathway: a common target for both ischemic preconditioning and postconditioning. Trends Cardiovasc Med. 2005;15:69–75. doi: 10.1016/j.tcm.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 54.Hausenloy DJ, Maddock HL, Baxter GF, Yellon DM. Inhibiting mitochondrial permeability transition pore opening: a new paradigm for myocardial preconditioning? Cardiovasc Res. 2002;55:534–543. doi: 10.1016/s0008-6363(02)00455-8. [DOI] [PubMed] [Google Scholar]
- 55.Li J, Xuan W, Yan R, Tropak MB, Jean-St-Michel E, Liang W, Gladstone R, Backx PH, Kharbanda RK, Redington AN. Remote preconditioning provides potent cardioprotection via PI3K/Akt activation and is associated with nuclear accumulation of β-catenin. Clin Sci (Lond) 2011;120:451–62. doi: 10.1042/CS20100466. [DOI] [PubMed] [Google Scholar]
- 56.Miura T, Tanno M. Mitochondria and GSK-3beta in cardioprotection against ischemia/reperfusion injury. Cardiovasc Drugs Ther. 2010;24:255–263. doi: 10.1007/s10557-010-6234-z. [DOI] [PubMed] [Google Scholar]
- 57.Lacerda L, Somers S, Opie LH, Lecour S. Ischaemic postconditioning protects against reperfusion injury via the SAFE pathway. Cardiovascular Research. 2009B;84:201–208. doi: 10.1093/cvr/cvp274. [DOI] [PubMed] [Google Scholar]
- 58.Kleinbongard P, Schulz R, Heusch G. TNFα in myocardial ischemia/reperfusion, remodeling and heart failure. Heart Fail Rev. 2011;16:49–69. doi: 10.1007/s10741-010-9180-8. [DOI] [PubMed] [Google Scholar]
- 59.Stadler J, Bentz BG, Harbrecht BG, Di Silvio M, Curran RD, Billiar TR, Hoffman RA, Simmons RL. Tumor necrosis factor alpha inhibits hepatocyte mitochondral respiration. Ann. Surg. 1992;216:539–546. doi: 10.1097/00000658-199211000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Deuchar GA, Opie LH, Lecour S. TNFalpha is required to confer protection in an in vivo model of classical ischaemic preconditioning. Life Sci. 2007;80:1686–1691. doi: 10.1016/j.lfs.2007.01.040. [DOI] [PubMed] [Google Scholar]
- 61.Lecour S, Suleman N, Deuchar GA, Somers S, Lacerda L, Huisamen B, Opie LH. Pharmacological preconditioning with tumor necrosis factor-alpha activates signal transducer and activator of transcription-3 at reperfusion without involving classic prosurvival kinases (Akt and extracellular signal-regulated kinase) Circulation. 2005;112:3911–3918. doi: 10.1161/CIRCULATIONAHA.105.581058. [DOI] [PubMed] [Google Scholar]
- 62.Heusch G, Musiolik J, Gedik N, Skyschally A. Mitochondrial STAT3 activation and cardioprotection by ischemic postconditioning in pigs with regional myocardial ischemia/reperfusion. Circ Res. 2011;109:1302–1308. doi: 10.1161/CIRCRESAHA.111.255604. [DOI] [PubMed] [Google Scholar]
- 63.Frias MA, Lecour S, James RW, Pedretti S. High density lipoprotein/sphingosine-1-phosphate-induced cardioprotection: role of STAT3 as part of the SAFE pathway. JAK-STAT. 2012;1:92–100. doi: 10.4161/jkst.19754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M, Gornicka A, Moh A, Moghaddas S, Chen Q, Bobbili S, Cichy J, Dulak J, Baker DP, Wolfman A, Stuehr D, Hassan MO, Fu XY, Avadhani N, Drake JI, Fawcett P, Lesnefsky EJ, Larner AC. Function of mitochondrial Stat3 in cellular respiration. Science. 2009;323:793–797. doi: 10.1126/science.1164551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Szczepanek K, Chen Q, Derecka M, Salloum FN, Zhang Q, Szelag M, Cichy J, Kukreja RC, Dulak J, Lesnefsky EJ, Larner AC. Mitochondrial-targeted signal transducer and activator of transcription 3 (STAT3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species. J Biol Chem. 2011;286:29610–29620. doi: 10.1074/jbc.M111.226209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Boengler K, Hilfiker-Kleiner D, Heusch G, Schulz R. Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res Cardiol. 2010;105:771–785. doi: 10.1007/s00395-010-0124-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Negoro S1, Kunisada K, Tone E, Funamoto M, Oh H, Kishimoto T, Yamauchi-Takihara K. Activation of JAK/STAT pathway transduces cytoprotective signal in rat acute myocardial infarction. Cardiovasc Res. 2000;47:797–805. doi: 10.1016/s0008-6363(00)00138-3. [DOI] [PubMed] [Google Scholar]
- 68.Pedretti S, Raddatz E. STAT3a interacts with nuclear GSK3beta and cytoplasmic RISK pathway and stabilizes rhythm in the anoxic-reoxygenated embryonic heart. Basic Res Cardiol. 2011;106:355–369. doi: 10.1007/s00395-011-0152-5. [DOI] [PubMed] [Google Scholar]
- 69.James RW, Frias MA, Lecour S. Lipid-induced modulation of protective signalling pathways in cardiovascular disease: the role of high density lipoproteins. Current Signal Transduction Therapy. 2012;7:96–103. [Google Scholar]
- 70.Kimura T, Sato K, Malchinkhuu E, Tomura H, Tamama K, Kuwabara A, Murakami M, Okajima F. High-density lipoprotein stimulates endothelial cell migration and survival through sphingosine 1-phosphate and its receptors. Arterioscler Thromb Vasc Biol. 2003;23:1283–1288. doi: 10.1161/01.ATV.0000079011.67194.5A. [DOI] [PubMed] [Google Scholar]
- 71.Huang Y, Wu Z, Riwanto M, Gao S, Levison BS, Gu X, Fu X, Wagner MA, Besler C, Gerstenecker G, Zhang R, Li XM, DiDonato AJ, Gogonea V, Tang WHW, Smith JD, Plow EF, Fox PL, Shih DM, Lusis AJ, Fisher EA, DiDonato JA, Landmesser U, Hazen SL. Myeloperoxidase, paraoxonase-1, and HDL form a functional ternary complex. J Clin Invest. 2013;123:3815–3828. doi: 10.1172/JCI67478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shen GX. Mitochondrial dysfunction, oxidative stress and diabetic cardiovascular disorders. Cardiovasc Hematol Disord. Drug Targets. 2012;12:106–112. doi: 10.2174/1871529x11202020106. [DOI] [PubMed] [Google Scholar]
- 73.Sangle Ganesh V, Roy Chowdhury Xueping Xie Subir K, Gerald LStelmack Gerald L, Halayko Andrew J, Shen Garry X. Impairment of mitochondrial respiratory chain activity in aortic endothelial cells induced by glycated low-density lipoprotein. Free Radical Biology and Medicine. 2010;48:781–790. doi: 10.1016/j.freeradbiomed.2009.12.017. [DOI] [PubMed] [Google Scholar]
- 74.Keul G, Schmidt C, Herrmann J, Keul P, Schäfers M, Herrgott I, Mersmann J, Larmann J, Hermann S, Stypmann J, Schober O, Hildebrand R, Schulz R, Heusch G, Haude M, von Wnuck Lipinski K, Herzog C, Schmitz M, Erbel R, Chun J, Levkau B. High-density lipoproteins and their constituent, sphingosine-1-phosphate, directly protect the heart again ischemia/reperfusion injury in vivo via the S1P3 lysophospholipid receptor. Circulation. 2006;114:1403–1409. doi: 10.1161/CIRCULATIONAHA.105.607135. [DOI] [PubMed] [Google Scholar]
- 75.Cockerill GW, McDonald MC, Mota-Filipe H, Cuzzocrea S, Miller NE, Thiemermann C. High density lipoproteins reduce organ injury and organ dysfunction in a rat model of hemorrhagic shock. FASEB J. 2001;15:1941–1952. doi: 10.1096/fj.01-0075com. [DOI] [PubMed] [Google Scholar]
- 76.Rossoni G, Gomaraschi M, Berti F, Sirtori CR, Franceschini G, Calabresi L. Synthetic high-density lipoproteins exert cardioprotective effects in myocardial ischemia/reperfusion injury. J Pharmacol Exp Ther. 2004;308:79–84. doi: 10.1124/jpet.103.057141. [DOI] [PubMed] [Google Scholar]
- 77.Marchesi M, Booth EA, Davis T, Bisgaier CL, Lucchesi BR. Apolipoprotein A-IMilano and 1-palmitoyl-2-oleoyl phosphatidylcholine complex (ETC-216) protects the in vivo rabbit heart from regional ischemia-reperfusion injury. J Pharmacol Exp Ther. 2004;311:1023–1031. doi: 10.1124/jpet.104.070789. [DOI] [PubMed] [Google Scholar]
- 78.Dadabayev AR, Yin G, Latchoumycandane C, McIntyre TM, Lesnefsky EJ, Penn MS. Apolipoprotein A1 regulates coenzyme Q10 absorption, mitochondrial function, and infarct size in a mouse model of myocardial infarction. J Nutr. 2014;144:1030–1036. doi: 10.3945/jn.113.184291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kalakech H, Hibert P, Prunier-Mirebeau D, Tamareille S, Letournel F, Macchi L, Pinet F, Furber A, Prunier F. RISK and SAFE signaling pathway involvement in apolipoprotein A-I-induced cardioprotection. PLoS ONE. 2014;9(9):e107950. doi: 10.1371/journal.pone.0107950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Duan J, Dahlbäck B, Villoutreix BO. Proposed lipocalin fold for apolipoprotein M based on bioinformatics and site-directed mutagenesis. FEBS Lett. 2001;499:127–132. doi: 10.1016/s0014-5793(01)02544-3. [DOI] [PubMed] [Google Scholar]
- 81.Dahlbäck B, Nielsen LB. Apolipoprotein M affecting lipid metabolism or just catching a ride with lipoproteins in the circulation? Cell Mol Life Sci. 2009;66:559–564. doi: 10.1007/s00018-009-8764-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Christoffersen C, Nielsen LB, Axler O, Andersson A, Johnsen AH, Dahlbäck B. Isolation and characterization of human apolipoprotein M-containing lipoproteins. J Lipid Res. 2006;47:1833–1843. doi: 10.1194/jlr.M600055-JLR200. [DOI] [PubMed] [Google Scholar]
- 83.Elsøe S, Christoffersen C, Luchoomun J, Turner S, Nielsen LB. Apolipoprotein M promotes mobilization of cellular cholesterol in vivo. Biochim Biophys Acta. 2013;1831:1287–1292. doi: 10.1016/j.bbalip.2013.04.009. [DOI] [PubMed] [Google Scholar]
- 84.Wolfrum C, Poy MN, Stoffel M. Apolipoprotein M is required for prebeta-HDL formation and cholesterol efflux to HDL and protects against atherosclerosis. Nature Med. 2005;11:418–422. doi: 10.1038/nm1211. [DOI] [PubMed] [Google Scholar]
- 85.Christoffersen C, Jauhiainen M, Moser M, Porse B, Ehnholm C, Boesl M, Dahlback B, Nielsen LB. Effect of apolipoproteinM on high density lipoprotein metabolism and atherosclerosis in low density lipoprotein receptor knock-out mice. J Biol Chem. 2008;283:1839–1847. doi: 10.1074/jbc.M704576200. [DOI] [PubMed] [Google Scholar]
- 86.Blaho VA, Hla T. An update on the biology of sphingosine 1-phosphate receptors. J Lipid Res. 2014;55:1596–1608. doi: 10.1194/jlr.R046300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Christoffersen C, Obinata H, Kumaraswamy SB, Galvani S, Ahnström J, Sevvana M, Egerer-Sieber C, Muller YA, Hla T, Nielsen LB, Dahlbäck B. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc Natl Acad Sci USA. 2011;108:9613–9618. doi: 10.1073/pnas.1103187108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Morel S, Christoffersen C, Rochemont V, Montecucco F, Frias M, Pelli G, Mach F, James RW, Nielsen LB, Kwak BR. Molecular insight in apoM-S1P–induced cardioprotection against ischemia/reperfusion injury. Cardiovascular Research. 2014;103:S120. [Google Scholar]
- 89.Morel S, Christoffersen C, Axelsen LN, Montecucco F, Rochemont V, Frias MA, Mach F, James RW, Naus CC, Chanson M, Lampe PD, Nielsen MS, Nielsen LB, Kwak BR. Sphingosine-1-phosphate reduces ischaemia-reperfusion injury by phosphorylating the gap junction protein Connexin43. Cardiovascular Research. 2016;109:385–396. doi: 10.1093/cvr/cvw004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Theilmeier G, Schmidt C, Herrmann J, Keul P, Schäfers M, Herrgott I, Mersmann J, Larmann J, Hermann S, Stypmann J, Schober O, Hildebrand R, Schulz R, Heusch G, Haude M, von Wnuck LK, Herzog C, Schmitz M, Erbel R, Chun J, Levkau B. High-density lipoproteins and their constituent, sphingosine-1-phosphate, directly protect the heart again ischemia/reperfusion injury in vivo via the S1P3 lysophospholipid receptor. Circulation. 2006;114:1403–1409. doi: 10.1161/CIRCULATIONAHA.105.607135. [DOI] [PubMed] [Google Scholar]
- 91.Keul P, Sattler K, Levkau B. HDL and its sphingosine-1-phosphate content in cardioprotection. Heart Fail Rev. 2007;12:301–306. doi: 10.1007/s10741-007-9038-x. [DOI] [PubMed] [Google Scholar]
- 92.Somers SJ, Frias M, Lacerda L, Opie LH, Lecour S. Interplay between SAFE and RISK pathways in sphingosine-1-phosphate-induced cardioprotection. Cardiovasc Drugs Ther. 2012;26:227–237. doi: 10.1007/s10557-012-6376-2. [DOI] [PubMed] [Google Scholar]
- 93.Frias MA, James RW, Gerber-Wicht C, Lang U. Native and reconstituted HDL activate STAT3 in ventricular cardiomyocytes via ERK1/2: Role of sphingosine-1-phosphate. Cardiovascular Research. 2009;82:313–323. doi: 10.1093/cvr/cvp024. [DOI] [PubMed] [Google Scholar]
- 94.Deshpande GP, McCarthy J, Mardikar H, Lecour S, Opie L. Effects of sphingosine-1-phosphate on acute contractile heart failure (ACHF) Cardiovasc Drugs Ther. 2010;24:459–460. doi: 10.1007/s10557-010-6258-4. [DOI] [PubMed] [Google Scholar]
- 95.Jin ZQ, Zhou HZ, Zhu P, Honbo N, Mochly-Rosen D, Messing RO, Goetzl EJ, Karliner JS, Gray MO. Cardioprotection mediated by sphingosine-1-phosphate and ganglioside GM-1 in wild-type and PKC epsilon knockout mouse hearts. Am J Physiol. 2002;282:H1970–H1977. doi: 10.1152/ajpheart.01029.2001. [DOI] [PubMed] [Google Scholar]
- 96.Zhang J, Honbo N, Goetzl EJ, Chatterjee K, Karliner JS, Gray MO. Signals from type 1 sphingosine 1-phosphate receptors enhance adult mouse cardiac myocyte survival during hypoxia. Am J Physiol. 2007;293:H3150–H3158. doi: 10.1152/ajpheart.00587.2006. [DOI] [PubMed] [Google Scholar]
- 97.Frias MA, Pedretti S, Hacking D, Somers S, Lacerda L, Opie LH, James RW, Lecour S. HDL protects against ischemia reperfusion injury by preserving mitochondrial integrity. Atherosclerosis. 2013;228:110–116. doi: 10.1016/j.atherosclerosis.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 98.Zhang X, Tang N, Hadden TJ, Rishi AK. Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta. 2011;1813:1978–1986. doi: 10.1016/j.bbamcr.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 99.Ludovic Gomez L, Paillard M, Price M, Chen Q, Teixeira G, Spiegel S, Lesnefsky EJ. A novel role for mitochondrial sphingosine-1-phosphate produced by sphingosine kinase-2 in PTP-mediated cell survival during cardioprotection. Basic Res Cardiol. 2011;106:1341–1353. doi: 10.1007/s00395-011-0223-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Karliner JS. Sphingosine kinase and sphingosine 1-phosphate in cardioprotection. J Cardiovasc Pharmacol. 2009;53:189–197. doi: 10.1097/FJC.0b013e3181926706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Strub GM, Paillard M, Liang J, Gomez L, Allegood JC, Hait NC, Maceyka M, Price MM, Chen Q, Simpson DC, Kordula T, Milstien S, Lesnefsky EJ, Spiegel S. Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 2011;25:600–612. doi: 10.1096/fj.10-167502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Merkwirth C, Langer T. Prohibitin function within mitochondria: Essential roles for cell proliferation and cristae morphogenesis. Biochimica et Biophysica Acta. 2009;1793:27–32. doi: 10.1016/j.bbamcr.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 103.Gustafsson AB, Gottlieb RA. Autophagy in ischemic heart disease. Circulation Research. 2009;104:150–158. doi: 10.1161/CIRCRESAHA.108.187427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gottlieb RA, Gustafsson AB. Mitochondrial turnover in the heart. Biochimica et Biophysica Acta. 2011;1813:1295–1301. doi: 10.1016/j.bbamcr.2010.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang C, Chen K, Xia Y, Dai W, Wang F, Shen M, Cheng P, Wang J, Lu J, Zhang Y, Yang J, Zhu R, Zhang H, Li J, Zheng Y, Zhou Y, Guo C. N-Acetylcysteine attenuates ischemia-reperfusion-induced apoptosis and autophagy in mouse liver via regulation of the ROS/JNK/Bcl-2 pathway. PLoS ONE. 2014;9(9):e108855. doi: 10.1371/journal.pone.0108855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kirkegaard K, Taylor MP, Jackson WT. Cellular autophagy: surrender, avoidance and subversion by microorganisms. Nature Reviews Microbiology. 2004;2:301–314. doi: 10.1038/nrmicro865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kim I, Rodríguez-Enríquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462:245–253. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dutta D, Calvani R, Bernabei R, Leeuwenburgh C, Marzetti E. Contribution of impaired mitochondrial autophagy to cardiac aging mechanisms and therapeutic opportunities. Circulation Research. 2012;110:1125–1138. doi: 10.1161/CIRCRESAHA.111.246108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Decker RS, Wildenthal K. Lysosomal alterations in hypoxic and reoxygenated hearts. I. Ultrastructural and cytochemical changes. Am J Pathol. 1980;98:425–444. [PMC free article] [PubMed] [Google Scholar]
- 110.Kanamori H, Takemura G, Goto K, Maruyama R, Tsujimoto A, Ogino A, Takeyama T, Kawaguchi T, Watanabe T, Fujiwara T, Fujiwara H, Seishima M, Minatoguchi S. The role of autophagy emerging in postinfarction cardiac remodelling. Cardiovasc Res. 2011;91:330–339. doi: 10.1093/cvr/cvr073. [DOI] [PubMed] [Google Scholar]
- 111.Sun K, Xie X, Liu Y, Han Z, Zhao X, Cai N, Zhang S, Song J, Wei L. Autophagy lessens ischemic liver injury by reducing oxidative damage. Cell & Bioscience. 2013;3:26. doi: 10.1186/2045-3701-3-26. http://www.cellandbioscience.com/content/3/1/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yan WJ, Dong HL, Xiong LZ. The protective roles of autophagy in ischemic preconditioning. Acta Pharmacologica Sinica. 2013;34:636–643. doi: 10.1038/aps.2013.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Huang C, Yitzhaki S, Perry CN, Liu W, Giricz Z, Mentzer RM, Gottlieb RA. Autophagy induced by ischemic preconditioning is essential for cardioprotection. J Cardiovasc Trans Res. 2010;3:365–373. doi: 10.1007/s12265-010-9189-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Li Y, Li S, Qin X, Hou W, Dong H, Yao L, Xiong L. The pleiotropic roles of sphingolipid signaling in autophagy. Cell Death and Disease. 2014;5:e1245. doi: 10.1038/cddis.2014.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Young MM, Kester M, Wang HG. Sphingolipids: regulators of crosstalk between apoptosis and autophagy. J Lipid Res. 2013;54:5–19. doi: 10.1194/jlr.R031278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lavieu G, Scarlatti F, Sala G, Carpentier S, Levade T, Ghidoni R, Botti J, Codogno P. Regulation of autophagy by sphingosine kinase 1 and its role in cell survival during nutrient starvation. J Biol Chem. 2006;281:8518–8527. doi: 10.1074/jbc.M506182200. [DOI] [PubMed] [Google Scholar]
- 117.Gerster R, Eloranta JJ, Hausmann M, Ruiz PA, Cosin-Roger J, Terhalle A, Ziegler U, Kullak-Ublick GA, von Eckardstein A, Rogler G. Anti-inflammatory function of high-density lipoproteins via autophagy of IkB kinase. Cellular and Molecular Gastroenterology and Hepatology. 2015;1:171–187. doi: 10.1016/j.jcmgh.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Boden WE. High-density lipoprotein cholesterol as an independent risk factor in cardiovascular disease: assessing the data from Framingham to the Veterans Affairs High- Density Lipoprotein Intervention Trial. Am J Cardiol. 2000;86:19L–22L. doi: 10.1016/s0002-9149(00)01464-8. [DOI] [PubMed] [Google Scholar]
- 119.Ghazzal ZB, Dhawan SS, Sheikh A, Douglas JS, Veledar E, Mavromatis K, Pohlel FK, Vaccarino V. Usefulness of serum high-density lipoprotein cholesterol level as an independent predictor of one-year mortality after percutaneous coronary interventions. Am J Cardiol. 2009;103:902–906. doi: 10.1016/j.amjcard.2008.11.053. [DOI] [PubMed] [Google Scholar]
- 120.Anders GOlsson Anders G, Schwartz Michael Szarek Gregory G, Sasiela William J, Ezekowitz Peter Ganz Michael D, Oliver David Waters Michael F, Zeiher Andreas. High-density lipoprotein, but not low-density lipoprotein cholesterol levels influence short-term prognosis after acute coronary syndrome: results from the MIRACL trial. European Heart Journal. 2005;26:890–896. doi: 10.1093/eurheartj/ehi186. [DOI] [PubMed] [Google Scholar]
- 121.Wu A, Hinds CJ, Thiemermann C. High-density lipoproteins in sepsis and septic shock: metabolism, actions, and therapeutic applications. Shock. 2004;21:210–221. doi: 10.1097/01.shk.0000111661.09279.82. [DOI] [PubMed] [Google Scholar]
- 122.van Leeuwen HJ, Heezius EC, Dallinga GM, van Strijp JA, Verhoef J, van Kessel KP. Lipoprotein metabolism in patients with severe sepsis. Crit Care Med. 2003;31:1359–1366. doi: 10.1097/01.CCM.0000059724.08290.51. [DOI] [PubMed] [Google Scholar]
- 123.Shah PK, Kaul S, Nilsson J, Cercek B. Exploiting the vascular protective effects of high-density lipoprotein and its apolipoproteins: an idea whose time for testing is coming, part I. Circulation. 2001;104:2376–2383. doi: 10.1161/hc4401.098467. [DOI] [PubMed] [Google Scholar]
- 124.Schultz JR, Verstuyft JG, Gong EL, Nichols AV, Rubin EM. Protein composition determines the anti-atherogenic properties of HDL in transgenic mice. Nature. 1993;365:762–764. doi: 10.1038/365762a0. [DOI] [PubMed] [Google Scholar]
- 125.Assmann G, Gotto AM. HDL cholesterol and protective factors in atherosclerosis. Circulation. 2004:III–8. doi: 10.1161/01.CIR.0000131512.50667.46. [DOI] [PubMed] [Google Scholar]
- 126.Navab M, Anantharamaiah GM, Fogelman AM. The role of high-density lipoprotein in inflammation. Trends Cardiovasc Med. 2005;15:158–161. doi: 10.1016/j.tcm.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 127.Kimura T, Sato K, Kuwabara A, Tomura H, Ishiwara M, Kobayashi I, Ui M, Okajima F. Sphingosine 1-phosphate may be a major component of plasma lipoproteins responsible for the cytoprotective actions in human umbilical vein endothelial cells. J Biol Chem. 2001;276:31. doi: 10.1074/jbc.M104353200. 780–31,785. [DOI] [PubMed] [Google Scholar]
- 128.Wu BJ, Ong KL, Shrestha S, Chen K, Tabet F, Barter PJ, Rye KA. Inhibition of arthritis in the Lewis rat by apolipoprotein A-I and reconstituted high-density lipoproteins. Arterioscler Thromb Vasc Biol. 2014;34:543–551. doi: 10.1161/ATVBAHA.113.302832. [DOI] [PubMed] [Google Scholar]
- 129.Tang C, Liu Y, Kessler PS, Vaughan AM, Oram JF. The macrophage cholesterol exporter ABCA1 functions as an anti-inflammatory receptor. J Biol Chem. 2009;284:32336–32343. doi: 10.1074/jbc.M109.047472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Brulhart-Meynet MC, Braunersreuther V, Brinck J, Montecucco F, Prost J-C, Thomas A, Galan K, Pelli G, Pedretti S, Vuilleumier N, Mach F, Lecour S, James RW, Frias MA. Improving reconstituted HDL composition for efficient post-ischemic reduction of ischemia reperfusion injury. PLoS ONE. 2015;10(3):e0119664. doi: 10.1371/journal.pone.0119664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Marchesi M, Booth EA, Rossoni G, García RA, Hill KR, Sirtori CR, Bisgaier CL, Lucchesi BR. Apolipoprotein A-IMilano/POPC complex attenuates post-ischemic ventricular dysfunction in the isolated rabbit heart. Atherosclerosis. 2008;197:572–578. doi: 10.1016/j.atherosclerosis.2007.08.028. [DOI] [PubMed] [Google Scholar]
- 132.Nissen SE, Tsunoda T, Tuzcu EM, Schoenhagen P, Cooper CJ, Yasin M, Eaton GM, Lauer MA, Sheldon WS, Grines CL, Halpern S, Crowe T, Blankenship JC, Kerensky R. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA. 2003;290:2292–2300. doi: 10.1001/jama.290.17.2292. [DOI] [PubMed] [Google Scholar]
- 133.Tolle M, Levkau B, Keul P, Brinkmann V, Giebing G, Schonfelder G, Schafers M, von Wnuck Lipinski K, Jankowski J, Jankowski V, Chun J, Zidek W, Van der Giet M. Immunomodulator FTY720 induces eNOS-dependent arterial vasodilatation via the lysophospholipid receptor S1P3. Circ Res. 2005;96:913–920. doi: 10.1161/01.RES.0000164321.91452.00. [DOI] [PubMed] [Google Scholar]
- 134.Brinkmann V, Cyster JG, Hla T. FTY720: sphingosine 1-phosphate receptor-1 in the control of lymphocyte egress and endothelial barrier function. Am J Transplant. 2004;4:1019–1025. doi: 10.1111/j.1600-6143.2004.00476.x. [DOI] [PubMed] [Google Scholar]
- 135.Frink M, Kaudel CP, Hildebrand F, Pape HC, Klempnauer J, Winkler M, Krettek C, van Griensven M. FTY720 improves survival after transient ischemia and reperfusion of the hind limbs. J Trauma. 2007;63:263–267. doi: 10.1097/TA.0b013e3180d0a6fc. [DOI] [PubMed] [Google Scholar]
- 136.Kaudel CP, Frink M, van Griensven M, Schmiddem U, Probst C, Bergmann S, Krettek C, Klempnauer J, Winkler M. FTY720 application following isolated warm liver ischemia improves long-term survival and organ protection in a mouse model. Transplant Proc. 2007;39:493–498. doi: 10.1016/j.transproceed.2007.01.021. [DOI] [PubMed] [Google Scholar]
- 137.Roberts E, Guerrero M, Urbano M, Rosen H. Sphingosine 1-phosphate receptor agonists: a patent review (2010–2012) Expert Opin Ther Pat. 2013;23:817–841. doi: 10.1517/13543776.2013.783022. [DOI] [PubMed] [Google Scholar]
- 138.Navab M, Anantharamaiah GM, Hama S, Garber DW, Chaddha M, Hough G, Lallone R, Fogelman AM. Oral administration of an Apo A-I mimetic Peptide synthesized from D-amino acids dramatically reduces atherosclerosis in mice independent of plasma cholesterol. Circulation. 2002;105:290–292. doi: 10.1161/hc0302.103711. [DOI] [PubMed] [Google Scholar]
- 139.Navab M, Anantharamaiah GM, Reddy ST, Van Lenten BJ, Hough G, Wagner A, Nakamura K, Garber DW, Datta G, Segrest JP, et al. Human apolipoprotein A-I mimetic peptides for the treatment of atherosclerosis. Current Opinion in Investigational Drugs. 2003;4:1100–1104. [PubMed] [Google Scholar]
- 140.Ditiatkovski M, D’Souza W, Kesani R, Chin-Dusting J, de Haan JB, Remaley A, Sviridov D. An apolipoprotein A-I mimetic peptide designed with a reductionist approach stimulates reverse cholesterol transport and reduces atherosclerosis in mice. PLoS One. 2013;8:e68802. doi: 10.1371/journal.pone.0068802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Getz GS, Reardon CA. The structure/function of apoprotein A-I mimetic peptides: an update. Curr Opin Endocrinol Diabetes Obes. 2014;21:129–133. doi: 10.1097/MED.0000000000000045. [DOI] [PubMed] [Google Scholar]
- 142.Datta G, Chaddha M, Hama S, Navab M, Fogelman AM, Garber DW, Mishra VK, Epand RM, Epand RF, Lund-Katz S, Phillips MC, Segrest JP, Anantharamaiah GM. Effects of increasing hydrophobicity on the physical-chemical and biological properties of a class A amphipathic helical peptide. J Lipid Res. 2001;42:1096–1104. [PubMed] [Google Scholar]
- 143.Garber DW, Datta G, Chaddha M, Palgunachari MN, Hama S, Navab M, Fogelman AM, Segrest JP, Anantharamaiah GM. A new synthetic class A amphipathic peptide analogue protects mice from diet-induced atherosclerosis. J Lipid Res. 2001;42:545–552. [PubMed] [Google Scholar]
- 144.Vakili L, Hama S, Kim JB, Tien D, Safarpoor S, Ly N, Vakili G, Hough G, Navab M. The Effect of HDL Mimetic Peptide 4F on PON1. Advances in Experimental Medicine and Biology. 2009;660:167–172. doi: 10.1007/978-1-60761-350-3_15. [DOI] [PubMed] [Google Scholar]
- 145.Shao B, Tang C, Heinecke JW, Oram JF. Oxidation of apolipoprotein A-I by myeloperoxidase impairs the initial interactions with ABCA1 required for signaling and cholesterol export. J Lipid Res. 2010;51:1849–1858. doi: 10.1194/jlr.M004085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tang C, Vaughan AM, Anantharamaiah GM, Oram JF. Janus kinase 2 modulates the lipid-removing but not protein-stabilizing interactions of amphipathic helices with ABCA1. J Lipid Res. 2006;47:107–114. doi: 10.1194/jlr.M500240-JLR200. [DOI] [PubMed] [Google Scholar]
- 147.Gomaraschi M, Calabresi L, Rossoni G, Iametti S, Franceschini G, Stonik JA, Remaley AT. Anti-inflammatory and cardioprotective activities of synthetic high-density lipoprotein containing apolipoprotein A-I mimetic peptides. J Pharmacol Exp Ther. 2008;324:776–783. doi: 10.1124/jpet.107.129411. [DOI] [PubMed] [Google Scholar]
- 148.Vecoli C, Cao J, Neglia D, Inoue K, Sodhi K, Vanella L, Gabrielson KK, Bedja D, Paolocci N, L’Abbate A, Abraham NG. Apolipoprotein A-I mimetic peptide L-4F prevents myocardial and coronary dysfunction in diabetic mice. J Cell Biochem. 2011;112:2616–2626. doi: 10.1002/jcb.23188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Baotic I, Ge ZD, Sedlic F, Coon A, Weihrauch D, Warltier DC, Kersten JR. Apolipoprotein A-1 mimetic D-4F enhances isoflurane-induced eNOS signaling and cardioprotection during acute hyperglycemia. Am J Physiol. 2013;305:H219–H227. doi: 10.1152/ajpheart.00850.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]