

SUMMARY
Small molecule BET bromodomain inhibitors (BETi) are actively being pursued in clinical trials for the treatment of a variety of cancers, however, the mechanisms of resistance to BETi remain poorly understood. Using a mass spectrometry approach that globally measures kinase signaling at the proteomic level, we evaluated the response of the kinome to targeted BETi treatment in a panel of BRD4-dependent ovarian carcinoma (OC) cell lines. Despite initial inhibitory effects of BETi, OC cells acquired resistance following sustained treatment with the BETi, JQ1. Through application of Multiplexed Inhibitor Beads (MIBs) and mass spectrometry, we demonstrate that BETi resistance is mediated by adaptive kinome reprogramming, where activation of compensatory pro-survival kinase networks overcomes BET protein inhibition. Furthermore, drug combinations blocking these kinases may prevent or delay the development of drug resistance and enhance the efficacy of BETi therapy.
eTOC blurb
BET inhibitors are currently being evaluated in clinical trials for a number of cancers, including ovarian. Kurimchak et al. demonstrate that BET inhibitors may have limited success as single agents in ovarian cancer due to adaptive kinome reprogramming and will require combination therapies targeting kinases and BET bromodomain proteins
INTRODUCTION
Epigenetic proteins involved in chromatin remodeling, such as the BET bromodomain protein BRD4, have emerged as an exciting new class of targets for the treatment of cancer. Importantly, it has been proposed that BRD4 overexpression can promote tumor growth through the enhancement of transcription of key oncogenes such as MYC (Chapuy et al., 2013; Lovén et al., 2013). Consequently, BET bromodomain inhibitors (BETi) were developed that interfere with acetyl lysine recognition, displacing BET bromodomain proteins from transcriptional complexes and disrupting gene transcription (Filippakopoulos and Knapp, 2014). Recent studies have shown that inhibition of BRD4 blocks transcription of oncogenes in a number of cancer models resulting in tumor regression and apoptosis (Mertz et al., 2011; Puissant et al., 2013; Segura et al., 2013; Shimamura et al., 2013). Therefore, BETi are actively being pursued in clinical trials for a number of cancers, including numerous solid cancers.
Although BETi show great promise as cancer therapeutics, studies have demonstrated that the anti-proliferative effects of BETi are quite variable (Lockwood et al., 2012; Mertz et al., 2011). In addition, emerging studies have shown that cancer cells can acquire resistance to BETi, signifying that single agent therapies targeting BRD4 may not provide durable therapeutic response (Fong et al., 2015; Kumar et al., 2015; Rathert et al., 2015). Recent work from our and other laboratories shows that tumor cells can acquire resistance to targeted kinase inhibitor therapies through “adaptive kinome reprogramming,” a process characterized by system-wide changes in kinase signaling networks (Chandarlapaty et al., 2011; Duncan et al., 2012; Nazarian et al., 2010; Sun et al., 2014). Specifically, tumor cells react to inhibitor treatment by sending signals that cause activation of protein kinase-driven survival pathways that ultimately by-pass the specific drug action, allowing the tumor to escape targeted therapies. These findings highlight the plasticity of the cancer kinase network, but whether kinase-mediated resistance mechanisms are involved in the resistance to BETi is largely unknown.
In the present study, we evaluated the consequence of BET protein inhibition on kinase signaling and explored the role of kinome reprogramming in the acquisition of resistance to BETi using a chemical proteomics approach. A recent study showing that BRD4 activity was essential for growth and survival of epithelial ovarian cancer (EOC) patient-derived tumor models (Baratta et al., 2015) prompted us to explore the consequence of targeted BETi on kinome dynamics in OC cell lines. Using MIB/MS technologies, we uncovered a mechanism of resistance to BETi involving the activation of RTKs and downstream signaling by PI3K, AKT, and ERK. Ovarian cancer cells chronically exposed to JQ1 acquire exquisite sensitivity to combination therapies targeting RTKs, PI3K or MEK-ERK pathways. These critical findings suggests that BETi therapies may have limited success as single agents in some OC due to adaptive kinome reprogramming and that combination strategies involving inhibitors targeting kinases and BET bromodomain proteins may be required to maximize clinical benefit.
RESULTS
Dynamic Reprogramming of the Kinome in Response to Targeted BET Protein Inhibition
To explore the effect of BET protein inhibition on cancer kinome signaling, we used a chemical proteomics approach that couples multiplexed inhibitor beads with quantitative mass spectrometry (MIB/MS) to monitor global changes in protein kinase signaling (Figure 1A) (Cooper et al., 2013; Duncan et al., 2012; Sos et al., 2014; Stuhlmiller et al., 2015). MIBs consists of a layered mixture of immobilized ATP-competitive pan-kinase inhibitors (Purvalanol B, VI16832, PP58 and CTx-0249885) that enriches protein kinases from lysates based on the affinity of individual kinases for the different immobilized inhibitors, their kinase abundance, and the activation state of the kinase. Monitoring both changes in kinase binding to MIBs through quantitative proteomics and kinase expression via RNAseq provides a comprehensive approach to define the fraction of the kinome that is altered in response to therapy. Growth and survival functions of MIBs-defined kinases are then interrogated by RNAi strategies, allowing rational prediction of kinase inhibitor combination therapies.
Figure 1. Dynamic Reprogramming of the Kinome in Response to Targeted BET Protein Inhibition.

(A) Flowchart of experimental design. Combining MIB/MS and RNA sequencing to define the proteogenomic response of the kinome to BETi in OC cells. RNAi and small molecule inhibitors are used to define kinase survival functions. Kinome tree reproduced courtesy of Cell Signaling Technology.
(B) RAS/PI3K mutation analysis of OC cell lines used in MIB/MS kinome studies.
(C) Knockdown of BRD4 causes growth arrest in a number of OC cell lines.
(D) Growth inhibition of OC cells in response to JQ1 treatment.
(E) JQ1 induces apoptosis in OC cells. OC cells were treated with escalating doses of JQ1 or a single dose of JQ1 (500 nM) and cleaved PARP protein levels determined by blot.
(F–G) Genome-wide transcriptome analysis of JQ1-treated of OC cells determined by RNAseq. Volcano plots of gene expression differences for A1847 and OVCAR5 cells treated with JQ1 (500 nM) for 48 h.
(H) JQ1-treatment reduces MYC and/or FOSL1 protein levels. OC cells were treated with escalating doses of JQ1 or a single dose of JQ1 (500 nM), and MYC and FOSL1 protein levels determined by blot.
(I) MIB/MS-defined kinome response profiles of 8 OC cell lines following 48 h JQ1 (500nM) treatment. Heat map depicts SILAC-determined Log2fold changes in MIB binding as a ratio of JQ1/DMSO. ed is JQ1-sensitive and green is JQ1-resistant based on GI50.
Data presented in (C) and (D) are triplicate experiments SEM. *p ≤0.05 by student’s t-test. Also see Figure S1 and Tables S1, and S2.
Initially, to identify OC cells that require BRD4 activity for growth and survival, we determined the effect of siRNA-mediated knockdown of BRD4 or small molecule BET inhibition using JQ1 in a panel of widely used and established OC cell lines. Notably, the OC cell line panel harbors many of the PI3K/RAS alterations observed in ovarian carcinomas (Hanrahan et al., 2012), as well as a number that overexpress the BRD4 target proteins MYC and/or FOSL1 (Figures 1B and S1A). Knockdown of BRD4 caused significant growth inhibition in a number of OC cell lines, demonstrating that A2780, A2780CP, OVCAR5, OVCAR3, A1847, IGROV1 and SKOV3 exhibit dependency for BRD4 activity (Figure 1C). As previously reported (Marcotte et al., 2016; Shi and Vakoc, 2014; Zhang et al., 2016), we observed differential sensitivity to small molecule inhibition of BRD4 with BETi JQ1, IBET151 or PF1 across the OC cells (Figures 1D and S1B–D). Based on recent studies evaluating JQ1 across cancer cell lines, we categorized the OC cell lines as sensitive (GI50 ≤ 0.75 μM, A2780, A1847, PEO1 and OVCAR3) or resistant (GI50 ≥ 0.75 μM, SKOV3, IGROV1, A2780CP and OVCAR5) (Marcotte et al., 2016; Puissant et al., 2013). BETi-mediated apoptosis also varied amongst the OC cells, where JQ1 induced PARP cleavage in A2780, A1847, PEO1, OVCAR3 and SKOV3 cells but not in IGROV1, A2780CP and OVCAR5 cells (Figures 1E and S1E). Analysis of global gene expression changes, using RNA sequencing in A1847 and OVCAR5 cells following JQ1 therapy, showed that BET protein inhibition reduced RNA levels of previously established BRD4 targets, including, IL7R, FOXM1, CDK4, CDK6, MYC and FOSL1, as well as induced HEXIM1 expression, a recent biomarker of BET protein inhibition (Figures 1F, 1G and Table S1) (Bartholomeeusen et al., 2012; Lockwood et al., 2012; Ott et al., 2012; Zhang et al., 2016). Subsequent analysis by western blot showed that JQ1 treatment resulted in reduced protein levels of BRD4-targets FOSL1 and/or MYC across the OC cell lines (Figures 1H and S1F). Collectively, the response of OC cells to BETi is variable, where some cell lines exhibited potent growth inhibition and apoptosis, while other OC cells displayed inherent resistance to increasing doses of the BETi.
To investigate the role of the protein kinome in promoting resistance to BETi, the cell line panel was treated with JQ1 for 48 h. and alterations of nearly 70% of the kinome was measured using MIB/MS (Figures 1I and Table S2). Consistent with the role of BRD4 in regulation of a large number of genes, we observed changes in MIB-binding of kinases across all kinome subfamilies in response to JQ1 treatment. The kinome response profile of I-BET151 treated SKOV3 or A1847 cells showed considerable overlap with JQ1 therapy (SKOV3 r=0.73, A1847 r=0.72), demonstrating that targeting BET proteins with structurally distinct small molecules elicit similar kinome alterations (Figures S1G, S1H and Table S2). Comparative analysis of kinome expression levels by RNA sequencing in A1847, A2780 and OVCAR5 cells, following 48 h JQ1 treatment, showed that many of the kinase changes detected by MIBs were also induced or repressed at the RNA level (≥1.5-fold or ≤ 1.5-fold change), demonstrating the marked effect of BET protein inhibition on kinase transcription (Figures 2A–C and Table S1). Notably, MIB-binding analysis revealed a select group of kinases altered at the protein level independent of RNA level suggesting that inhibition of BET proteins can also promote changes in translation, degradation and/or activity of protein kinases (Figures S2A–C).
Figure 2. BET Protein Inhibition Induces Variable Kinome Responses Across Ovarian Cancer Cells.

(A–C) MIB-seq scatterplot shows overlap of kinases (in red) induced (≥1.5-fold) or repressed (≤1.5-fold) by 48 h JQ1 (500 nM) treatment analyzed by MIB/MS and RNAseq analysis.
(D) Boxplot of statistically significant MIB-binding changes after 48 h JQ1 treatment commonly induced or repressed across 8 OC cell lines based on stepup-adjusted p values at FDR of <0.05.
(E–F) Activation of pro-apoptotic kinase signaling and inhibition of proliferative kinase signaling network in JQ1 treated OC cells. Kinase network induced or repressed by 48 h JQ1 (500nM) treatment across 8 OC cell lines determined by STRING, Go Ontology and Pathway Enrichment Analysis.
(G) Downregulation of kinases by JQ1 treatment currently being explored as therapeutic targets in OC. Heat map depicts SILAC-determined Log2fold changes in MIB binding as a ratio of JQ1/DMSO.
(H) JQ1-induced Hippo/MST1 signaling. OC cells treated with JQ1 for 48 h and protein levels determined by blot.
(I) JQ1-induced RIP and TGF-beta signaling. Cells treated with JQ1 for 48 h and protein levels determined by blot.
(J) JQ1 treatment blocks kinase survival signaling in OC cells. Cells were treated with JQ1 (500 nM) for 48 h and activating phosphorylation or total protein levels of kinases determined by blot.
(K) Principal component analysis of MIBs-defined kinome response profiles across OC cell lines treated with JQ1 (500nM) for 48 h.
Analysis of the MIB-binding data revealed a subset of kinases commonly induced or repressed by 48 h JQ1 treatment across the 8 OC cells lines independent of JQ1-responsivness (Figures 2D and Table S2). Kinases universally induced in response to JQ1 treatment across the OC cells were predominately involved in positive regulation of apoptosis and function as key nodes in the RIP signaling, Hippo signaling, TGF-beta signaling and stress response signaling pathways (Figures 2E and S2D). Kinases repressed by JQ1 were largely involved in cell cycle progression, proliferation, cell survival and RTK signaling (Figures 2F and S2D). Importantly, we observed decreased MIB-binding of a number of kinases currently being explored as drug targets for OC (Figure 2G). Activation of the pro-apoptotic kinase STK4 (MST1) and its subsequent downstream hippo-signaling pathway was confirmed by western blot in PEO1 and A2780 cells treated with JQ1 (Figure 2H). Additionally, activation of RIPK1, JNK2 (MAPK9), TGFBR1 and MAP3K3 components of RIP and TGF-beta signaling were shown by blot in OC cells corroborating MIBs-binding data (Figure 2I). Reduced activating phosphorylation of ERK1, AKT, and SRC, as well as decreased AURKA and ERK1 protein levels, was observed by western blot following 48 h JQ1 treatment supporting a role for BET proteins in promoting many of the essential cancer kinase pathways (Figure 2J).
Principle component analysis (PCA) and R-squared regression analysis was then used to explore the variation in MIBs-determined kinome response to JQ1 amongst the OC cell line panel. PCA analysis revealed that A2780, IGROV1, OVCAR3 and PEO1 OC cells exhibited the most distinctive kinome response profiles relative to the other OC cells, while the kinome MIB-binding changes were most similar amongst the SKOV3 and OVCAR5 or the A1847, A2780 and A2780CP OC cells (Figures 2K and S2E). Although no clear separation between JQ1-senstivie and resistant populations was observed by PCA, linear regression analysis showed JQ1-resistant SKOV3 and OVCAR5 cells exhibited the most similar response to JQ1 with the highest r2 value, 0.398, while JQ1-sensitive A1847 and A2780 cells recorded the second highest r2 value of 0.345 (Figure S2F–H).
BET Protein Inhibition Rewires Receptor Tyrosine Kinase (RTK) Activity in OC Cells
Recent studies investigating BETi therapies in breast and other cancers demonstrated that BRD4 promotes the transcription of a number of RTKs, establishing a role for BET proteins in the regulation of RTK signaling (Stratikopoulos et al., 2015; Stuhlmiller et al., 2015). Consistent with these findings, PCA analysis of all kinase MIB-binding ratios following JQ1 treatment across the 8 OC cell lines demonstrated RTKs (red) were amongst the most prominent responders to BET protein inhibition (Figure 3A). These changes were also observed by RNAseq, confirming a role for BRD4 in the regulation of RTK expression in OC cells (Figures S3A–C). A heat map depiction of RTK MIB-binding ratios (isolated from Figure 1I) highlights the marked effect and heterogeneity of the RTK-reprogramming response to BET protein inhibition across the OC cell line panel (Figure 3B). Importantly, PCA analysis of the RTK MIBs response in the 8 OC cells showed more similar responses in JQ1-sensitive OC cells (A2780, A1847, PEO1 and OVCAR3) than those of JQ1-resistant OC cells (Figure 3C). These findings demonstrate that JQ1-sensitive and resistant cells rewire their RTK networks uniquely to BET protein inhibition.
Figure 3. Rewiring of the Receptor Tyrosine Kinome Following BET Bromodomain Inhibition.

(A) RTK response to BET inhibition in OC cells. Principal component analysis of all MIBs-defined kinases across OC cell lines treated with JQ1 (500nM) for 48 h. RTKs depicted in red.
(B) RTK response of OC cells to JQ1 (500 nM) 48 h as determined by MIB/MS. Heat map depicts SILAC-determined Log2fold changes in MIB binding as a ratio of JQ1/DMSO.
(C) Differential response of RTKs to JQ1 treatment between JQ1-sensitive and-resistant OC cells. Principal component analysis of all MIBs-defined RTKs across OC cell lines treated with JQ1 (500nM) for 48 h.
(D–E) Dose dependent decrease in RTK protein levels and downstream survival signaling following JQ1 (500 nM) treatment for 48 h in A1847 cells. Tyrosine phosphorylation inhibited by JQ1 treatment as shown by RTK array.
(F–G) JQ1-mediated repression of RTKs and AKT signaling in A2780 and PEO1 cells following JQ1 (500 nM) treatment for 48 h.
(H–I) Dose dependent activation of RTKs and subsequent AKT and ERK signaling in response to JQ1 48 h treatment in JQ1-resistant OVCAR5 and SKOV3 cells depicted by western blot.
(J–K) Induced RTKs in response to JQ1 48 h treatment in IGROV1 and A2780CP cells depicted by western blot.
(L–M) Knockdown of FGFRs sensitizes OVCAR5 and SKOV3 cells to BET protein inhibition. Growth inhibition of OC cells in response to escalating doses of JQ1 with or without FGFR siRNA knockdown. Triplicate experiments SEM. *p ≤0.05 by student’s t-test.
Also see Figure S3.
A dose-dependent depletion of RTKs PDGFRB, DDR1, AXL, ERBB3 and EGFR protein levels was observed by western blot in JQ1-sensitive A1847 cells following JQ1 therapy. This was accompanied by inhibition of downstream RAF-MEK-ERK and AKT signaling (Figure 3D). Analysis of global RTK tyrosine phosphorylation in these cells revealed that JQ1 treatment reduces ERBB3, AXL, PDGFRB, MET and DDR1 phosphorylation, corroborating our MIBs studies and demonstrating the inhibitory properties of JQ1 on RTK activity (Figure 3E). Knockdown of PDGFRB, AXL or DDR1 in A1847 cells blocked cell growth, suggesting that the growth inhibitory properties of BET protein inhibition in A1847 cells are, in part, caused by repression of multiple pro-survival RTKs (Figure S3D). Reduced RTK activity and downstream AKT signaling were also observed in JQ1-sensitive A2780, PEO1 and OVCAR3 cells (Figures 3F, 3G, and S3E).
Conversely, JQ1-resistant OVCAR5 and SKOV3 cell lines showed a dose dependent increase in expression and activity of numerous RTKs and their downstream survival signaling pathways (Figures 3H and 3I). Induced expression and/or activity of RTKs, FGFR1–3, EGFR and IGF1R were detected by blot in OVCAR5 and SKOV3 in response to JQ1 treatment that was accompanied by a dose-dependent increase in downstream P70S6 kinase, AKT and RAF-MEK-ERK activity. Conversely, while treatment of IGROV1 cells with JQ1 resulted in induction of RTKs, ERBB4, FGFR2 and FGFR3, reduced downstream signaling by P70S6K, AKT and ERK was observed (Figure 3J). A2780CP cells showed a pronounced induction of FGFR3 protein levels, while AKT and ERK activity remained unaltered by JQ1 treatment (Figure 3K). Collectively, BET protein inhibition reduces levels of RTKs and downstream AKT signaling in OC cells sensitive to JQ1, whereas in a subset of OC cells inherently resistant to JQ1, BET inhibition activates RTKs and their downstream survival signaling.
Rapid activation of RTKs and their downstream AKT and/or ERK signaling has been shown to promote intrinsic resistance to a variety of targeted kinases inhibitors (Chandarlapaty et al., 2011; Duncan et al., 2012; Nazarian et al., 2010). Combination therapies blocking these drug-induced RTKs enhance tumor growth inhibition and apoptosis, providing durable therapeutic response. Importantly, RNAi-mediated knockdown of FGFR1–3, IGF1R or ERK2 sensitized OVCAR5 cells to JQ1, while knockdown of FGFR2 or AKT1 sensitized SKOV3 cells to BET protein inhibition (Figures 3L, 3M, S3F, S3G and S3H). These findings support a role for JQ1-induced RTK activation as a mechanism of resistance to BET inhibition therapies.
Ovarian Cancer Cell Lines Acquire Resistance to the BET Inhibitor JQ1 Through Adaptive Kinome Reprogramming
BET inhibitors are currently being evaluated in clinical trials for a number of cancers including solid tumors (Filippakopoulos and Knapp, 2014). Acquired drug resistance occurs to the majority of therapies in cancer, and emerging studies suggest that BETi therapies may not be sufficient as single agents to provide long-term therapeutic benefit (Rathert et al., 2015; Shu et al., 2016). To understand the mechanistic basis for BETi resistance, we assessed kinome-wide signaling alterations in OC cell line models that had adapted to BETi after chronic exposure to the drug. The chronically exposed OC cell lines (JQ1-R) were more resistant to BETi than treatment naïve (i.e., parental cells), where they showed a rightward shift in JQ1 dose-response cell viability curves (Figure 4A). Additionally, A1847 and OVCAR5 JQ1-R cells showed cross-resistance to other selective BETi, I-BET151 and OTX-015 in dose response curves (Figures 4SA and 4SB). Following chronic JQ1 exposure, A2780 and PEO1 failed to induce apoptosis to the extent of parental cells following JQ1 exposure (Figures 4B and 4C). In contrast to parental A1847 cells, treatment of A1847-R cells with increasing doses of JQ1 was insufficient to reduce the BRD4 target gene FOSL1 RNA or protein levels (Figures 4D and 4E). Similarly, although JQ1 treatment resulted in the loss of BRD4 target MYC protein in OVCAR5 parental cells, dose escalation of JQ1 in OVCAR5-R cells was insufficient to reduce MYC protein levels (Figure 4F).
Figure 4. Adaptation of the OC Kinome to Chronic JQ1 Exposure.

(A) OC cell lines acquire resistance to JQ1. Parental and JQ1 resistant cells were treated with escalating doses of JQ1. JQ1-R treated cell viabilities normalized to DMSO treated JQ1-R cells. Viability assessed by CellTiter-Glo.
(B) JQ1 treatment fails to induce apoptosis in PEO1-R and A2780-R cells as shown by PARP blot.
(C) Treatment of A2780-R cells fails to induce apoptosis to the extent of parental A2780 cells as determined by Caspase-3-Glo.
(D) A1847 parental or A1847-R cells were treated with escalating doses of JQ1 for 48 h. JQ1-mediated FOSL1/MYC protein changes determined by western blot.
(E) RNA levels of FOSL1 in A1847 following 4 h, 48 h or chronic exposure to JQ1 (500 nM) treatment as determined by qRT-PCR.
(F) OVCAR5 and OVCAR5-R cells were treated with escalating doses of JQ1 for 48 h JQ1-mediated FOSL1/MYC protein changes determined by western blot.
(G) MIB/MS-defined kinome resistance profiles of OC cell lines following chronic exposure to JQ1 (500 nM) treatment. Heat map depicts SILAC-determined Log2fold changes in MIB binding as a ratio of JQ1-R/ parental.
(H) Principal component analysis of MIBs-defined kinome resistance profiles across OC cell lines chronically treated with JQ1 (500nM). Different views of the 3D PCA plots are shown to illustrate the grouping of kinome responses.
(I–J) MIB-seq scatterplot shows overlap of kinases (in red) induced (≥1.5-fold) or repressed (≤1.5-fold) following chronic exposure to JQ1 (500 nM) treatment analyzed by MIB/MS and RNAseq analysis.
Data presented in (A) and (C) are triplicate experiments SEM. Also see Figure S4 and Tables S1 and S2.
We next compared baseline kinome profiles of OC JQ1-R cells relative to parental cells using MIB/MS analysis to define the role of kinase signaling in acquisition of JQ1 resistance, (Figures 4G and Table S2). OVCAR3-R cells were not analyzed by MIB/MS due to slow growth rate of resistant cells. PCA analysis revealed that MIB-binding ratios of A1847, PEO1, IGROV1, A2780CP and A2780 JQ1-R cells reprogrammed their kinome(s) similarly, while SKOV3-R and OVCAR5-R MIBs profiles were more similar to one another but distinct from the other JQ1-R cells (Figure 4H). R-squared regression studies of JQ1-R cells revealed that the response of the kinome to chronic JQ1 exposure exhibited a high degree of heterogeneity with no r2 values ≥ 0.3 with the exception of IGROV1-R and A2780-R cells (r2=0.387) (Figure S4C). Comparative analysis of kinase expression of A1847-R and A2780-R cells relative to parental cells by RNAseq showed changes in RNA expression of many of the kinases identified by MIBs, highlighting the continued effect of chronic JQ1 treatment on kinase transcription (Figures 4I, 4J, and Table S1).
Chronic BET Protein Inhibition Promotes Activation of RTKs and Their Downstream PI3K/RAS Signaling in OC Cells
Analysis of MIB-binding data of JQ1-R cells showed chronic exposure to JQ1 promoted alterations in an assortment of cancer signaling pathways relative to parental cells. Increased MIB-binding of a diverse network of pro-survival RTKs were observed amongst the JQ1-R OC cells relative to parental cells (Figure 5A). Follow up confirmation by blot, showed increased levels and/or corresponding activating phosphorylation of RTKs FGFR1–4, IGF1R and/or EGFR amongst the JQ1-R cells, while reduced PDGFRB levels were observed in A1847-R and A2780-R cells relative to parental cells (Figures 5A–5C and S5A). Notably, activating phosphorylation of PDGFRA or EGFR was detected in A2780-R or A1847-R cells, respectfully, despite reduced protein levels relative to parental cells. Analysis of RTK ligand expression by RNAseq in A1847-R and A2780-R cells showed increased expression of PDGFA (A2780-R) or TGFA (A1847-R) relative to parental cells (Figures S5B, S5C, and Table S1). These findings suggest that JQ1-R cells may promote RTK activity through increased ligand production, despite transcriptional repression of RTKs by BET protein inhibition.
Figure 5. Chronic Exposure to JQ1 Activates an Assortment of Kinase Signaling Cascades That Are Reversible.

(A) Continuous JQ1 treatment of OC cell lines promotes activation of a variety of pro-survival kinase-signaling pathways. Heat maps depict SILAC-determined Log2fold changes in MIB binding as a ratio of JQ1-R/parental.
(B–D) Common activation of RTK signaling and their downstream AKT and ERK signaling pathways in JQ1-R cells shown by western blot.
(E–H) MIB-binding kinases induced (≥1.5-fold) or repressed (≤1.5-fold) following 48 h or chronic JQ1 therapy. Scatterplot depicts common and/or unique (in red) SILAC-determined Log2fold changes in kinase MIB binding as a ratio of JQ1 48-h/DMSO or JQ1-R/ parental.
(I) Reversible nature of JQ1-mediated kinome response in OC cells. MIB/MS-defined kinome profiles of OC cell lines following chronic JQ1 (500 nM) treatment or 48 h post JQ1 removal (wash-out, WO). Heat map depicts SILAC-determined Log2fold changes in MIB binding as a ratio of JQ1-R/ parental or JQ1-R-WO / JQ1-R.
Increased MIB-binding of the established downstream RTK signaling MEK-ERK2 kinase signaling cascade was detected by MIBs analysis and activity of RAF, MEK and ERK2 confirmed by blot amongst JQ1-R cells (Figures 5A–5D). Induced MIB-binding of PI3K/mTOR/AKT1 pathway was detected in JQ1-R OC cells relative to parental cells and activating phosphorylation of AKT (T308) confirmed by blot, with the exception of SKOV3-R cells, which showed reduced AKT (T308) levels. Notably, elevated TGFBR1 protein levels were detected in JQ1-R cells relative to parental cells by western blot, and increased JAK1 levels and corresponding STAT3 phosphorylation were observed in A2780-R. Overall, these findings demonstrate significant congruence between MIBs-binding and pathway activation as assayed by western blotting. Furthermore, they indicate that elevated signaling downstream of RTKs is a common feature in JQ1-R OC cells.
PCA analysis was applied to search for similarities in response to 48 h and chronic exposure to JQ1 amongst the OC cell line panel (Figures 5SD and 5SE). No strong similarities were observed, suggesting the initial response of the kinome to JQ1 is overall distinct from adaptation of the kinome to continuous JQ1 exposure. However, a subset of kinases induced by 48 h JQ1 treatment remained elevated in the JQ1-R cells relative to parental cells, while other kinases were uniquely altered only in short-term or chronic exposure (Figures 5E–5H and 5SF–J). For example, PDGFRB remained inhibited in A1847-R and A2780-R cells, while PRKCA, JAK1/3 and EPHB3 were induced in both the JQ1–48h treatment and chronically exposed A2780-R cells. Unique changes in MIB-binding were also observed in OC cells chronically exposed to JQ1, including increased MIB-binding of PDGFRA in A2780-R cells, which was initially dramatically reduced following JQ1–48h treatment and elevated DDR1 and TYRO3 in A1847-R cells which were both inhibited by short-term JQ1 treatment. Elevated MIB-binding of FGFRs was commonly observed in both parental and JQ1-R OVCAR5 and SKOV3 cells (Figures 5G and 5H).
Removal of JQ1 from chronically exposed OVCAR5-R, A1847-R and A2780-R cells resulted in the reversal of many of the kinase changes observed by sustained JQ1 treatment (Figure 5I). Moreover, removal of JQ1 from OVCAR5-R cells resulted in loss of FGFR1, FGFR3 and IGF1R MIB-binding demonstrating the requirement for continuous JQ1 to maintain the RTK resistance network (Figure S5K). Consistent with the reversal of the JQ1-induced kinome response, we observed that removal of JQ1 from A1847-R cells re-sensitizes them to JQ1 similarly to parental A1847 cells (Figure S5L). Collectively, these findings suggest that many of the JQ1-mediated kinome changes involved in the adaptive kinome reprogramming response are directly dependent on the presence of the BETi JQ1.
Chronic BET Protein Inhibition Sensitizes OC Cells to Targeted Kinase Inhibitors
Induced protein levels and/or activation of FGFR1, 2, 3 or 4 were observed in JQ1-R cells. Therefore, we evaluated the effect of the pan-FGFR inhibitor AZD4547 on long-term colony formation in OC cells chronically exposed to JQ1. Blockade of the FGFR signaling pathway resulted in a greater reduction of colony formation in JQ1-R cells relative to parental cells, demonstrating the acquired dependency of FGFR signaling in JQ1-resistant OC cells (Figure 6A). Moreover, RNAi-mediated depletion of FGFR2 resulted in a greater growth inhibition in SKOV3-R, OVCAR5-R and OVCAR3-R cells relative to parental cells (Figures S6A–S6C). Treatment of SKOV3-R with AZD4547 inhibits JQ1-mediated activation of FGFR, AKT and CRAF signaling and reduces MYC protein levels (Figure S6D). In addition to FGFR dependency, enhanced sensitivity of A1847-R, OVCAR5-R and OVCAR3-R to siRNA-mediated knockdown or small molecule inhibition of IGF1R was observed relative to parental cells (Figures 6B and 6SA–6SG). A1847-R cells also exhibited enhanced sensitivity to EGFR knockdown and small molecule inhibition of EGFR relative to parental cells (Figures 6C and S6E). Treatment of A1847-R cells with either lapatinib or GSK1904529A inhibited JQ1-mediated activation of AKT, STAT3 and MEK-ERK signaling (Figure 6D). These findings demonstrate that JQ1-R cells acquire dependency on a variety of RTKs following chronic exposure to JQ1 sensitizing these cells to RTK inhibition. However, designing combination therapies involving RTKs and BETi may present a challenge as each tumor cell may require blocking unique sets of JQ1-induced RTKs in individualized therapies.
Figure 6. Ovarian Cancer Cells Chronically Exposed to JQ1 Acquire Dependency on RTK, PI3K and/or ERK Signaling and Exhibit Exquisite Sensitivity to Kinase Inhibitors Targeting These Pathways.

(A) Inhibition of FGFR signaling blocks growth in JQ1-R OC cells to a greater extent than parental cells in colony formation assays. Long-term 14-day colony formation assay of JQ1-R or parental cells treated with FGFR inhibitor AZD4547 (1 μM) or DMSO.
(B–C) A1847-R cells show enhanced sensitivity to IGF1R inhibitor (GSK1904529A) or EGFR inhibitor (lapatinib) relative to parental cells. JQ1-R treated cell viabilities normalized to DMSO treated JQ1-R cells. Viability assessed by CellTiter-Glo.
(D) Blockade of RTKs EGFR or IGF1R in A1847-R cells inhibits downstream kinase survival signaling as shown by blot. Cells were treated with lapatinib (1 or 3 μM) or GSK1904529A (1 or 3 μM) for 48 h.
(E–F) A1847-R cells show enhanced sensitivity to trametinib or GDC-0941 relative to parental cells. JQ1-R treated cell viabilities normalized to DMSO treated JQ1-R cells. Cell viability was assessed by CellTiter-Glo.
(G) Acquired dependency on AKT and ERK signaling in A1847-R cells. Parental A1847 cells or JQ1-R A1847 cells were transfected with siRNAs targeting PI3Ks, AKTs and ERK2 and cultured for 72 h. JQ1-R knockdown cells were normalized to JQ1-R cells transfected with non-targeting siRNA.
(H) Treatment of A1847-R cells with trametinib or GDC-0941 represses FOSL1 and MYC protein levels to a greater extent than JQ1 treatment. Parental and JQ1 resistant cells were treated with escalating doses of JQ1, GDC-0941 or trametinib for 48 h and protein levels determined by western blot.
(I–J) Escalating doses of GDC-0941 or trametinib for 48 h induces apoptosis and blocks RNA pol II phosphorylation in A1847-R cells. Protein levels determined by western blot.
(K) Enhanced sensitivity to PI3K inhibition across JQ1-R cells. Parental or JQ1-resistant OC cells were treated with escalating doses of GDC-0941. Viability assessed by CellTiter-Glo.
(L) Escalating doses of GDC-0941 for 48 h induces apoptosis in JQ1-R cells to a greater extent than parental cells. Caspase activity determined by Caspase-Glo 3/7 assay according to manufacturer.
(M) Blockade of PI3K signaling inhibits colony formation of JQ1-R OC cells. Long-term 14-day colony formation assay of JQ1-R OC cells treated with DMSO (500 nM, JQ1), I-BET151 (2 μM) or GDC-0941 (500 nM). The dose of (500 nM) GDC-0941 was selected, as it is sufficient to reduce downstream AKT activity.
Data presented in (B–C), (E–F), (G), (K) and (L) are triplicate experiments SEM. *p ≤0.05 by student’s t-test. Also see Figure S6.
To further explore kinase vulnerabilities in the JQ1-R cells, we carried out a kinase inhibitor screen comparing differential inhibitor sensitivities between A1847 parental and resistant cells (Figure S6H) using a collection of small molecule kinase inhibitors of well defined target selectivity (Anastassiadis et al., 2011). Collectively, the A1847 JQ1-R cells were shown to be more sensitive than parental cells to inhibitors targeting EGFR, MEK, PI3K/mTOR/AKT, CDK, IKK and SRC kinases. Treatment of A1847-R cells with the clinically relevant PI3K/mTOR inhibitor, GDC-0941, or MEK1/2 inhibitor, trametinib demonstrated that JQ1-R cells were more sensitive to these kinase inhibitor therapies than parental cells (Figures 6E and 6F). Additionally, knockdown of PIK3CG, AKT1, or ERK2 resulted in greater growth inhibition in the A1847 JQ1-R cells relative to the parental cells, signifying that the resistant cells acquired greater dependency on the specific isoforms AKT1 and ERK2 for survival in the presence of BET protein inhibition (Figure 6G).
Previous studies have demonstrated that phosphorylation of either FOSL1 on S265 or MYC on S62 by ERK1/2 promotes protein stability and prevents their degradation by the proteasome (Basbous et al., 2007; Sears R, 2000). Importantly, trametinib or GDC-0941 treatment in the JQ1-R cells resulted in the inhibition of ERK-mediated phosphorylation of FOSL1 (S265) and MYC (S62), leading to the degradation of FOSL1 and MYC protein levels to a greater extent than JQ1 treatment (Figure 6H). Furthermore, knockdown of FOSL1 or MYC decreased cell viability to a greater extent in the A1847-R cells than in parental cells, signifying the acquired dependency of JQ1-R cells on FOSL1/MYC signaling (Figure S6I). A more pronounced induction of apoptosis was observed in the A1847 JQ1-R cells relative to the parental cells following treatment with either the MEK or PI3K inhibitor, demonstrating the increased dependence for ERK and/or AKT activity for survival in the presence of BET protein inhibition (Figures 6I and 6J). Notably, we observed a potent reduction in phosphorylation of RNA polymerase II at S2 in the A1847-R cells in response to MEK or PI3K inhibition. Importantly, this phosphorylation event is required for transcriptional elongation of mRNA (Ahn et al., 2004), suggesting that ERK and AKT activity may promote transcription in the absence of BET protein function.
Acquired dependency on PI3K signaling was also observed in the other JQ1-R OC cells, where treatment of JQ1-R cells with GDC-0941 showed greater growth inhibition than in parental cells (Figure 6K). RNAi-mediated knockdown of PI3K (PIK3CA, PIK3CB and PIK3CG) in SKOV3-R cells resulted in growth inhibition to a greater extent than parental cells, while treatment of JQ1-R cells with GDC-0941 inhibited AKT activity reduced MYC/FOSL1 levels and blocked RNA polymerase II (S2) phosphorylation (Figures S6J–S6N). Importantly, treatment of JQ1-R cells with GDC-0941 resulted in enhanced caspase-dependent apoptosis relative to parental cells (Figure 6L). Evaluation of long-term colony formation, following treatment of JQ1-R cells with GDC-0941, showed an enhanced growth inhibition over a 14-day period relative to parental cells (Figure 6M). MEK inhibition via trametinib treatment blocked colony formation in the A1847-R, OVCAR5-R, PEO1-R and A2780-R cells, while AKT inhibition was ineffective (Figure S6O). Notably, non-tumorigenic human immortalized ovarian surface epithelial (HIO) cell lines formed colonies in the presence of AZD4547, trametinib or GDC-0941, highlighting the increased sensitivity of the JQ1-R cells to FGFR, MEK or PI3K inhibition compared to both parental and normal ovarian epithelial cells (Figure S6P). Taken together, treatment of OC JQ1-R cells with GDC-0941 was sufficient to greatly reduce colony formation and induce apoptosis universally in JQ1-R cells, demonstrating the commonly acquired dependency on PI3K signaling for survival in the presence of BET protein inhibition.
Combination Therapies Targeting RTK, PI3K or ERK Signaling Concurrently with BET Protein Inhibitors Enhance Growth Inhibition in OC Cell Lines
To test whether co-targeting RTK, PI3K/AKT, or MEK/ERK signaling pathways concurrently with JQ1 can block or delay BETi resistance, we analyzed long-term colony formation of parental cells in response to single agent or combination therapies (Figure 7A). The combination of JQ1 and AZD4547 prevented JQ1 resistant colony formation in SKOV3 and reduced colonies in A2780 relative to single agent therapies, while co-treatment of JQ1 and trametinib or ERK inhibitor, Vx-11e, repressed JQ1-resistant colony formation in A1847, OVCAR5, A2780 and PEO1 cells. Consistent with the observed activation of AKT in OVCAR5 and SKOV3 following short-term BETi exposure (Figures 3H and 3I), MK2206 in combination with JQ1 blocked colony formation to a greater extent than individual agents. Importantly, blockade of the PI3K pathway with GDC-0941, concurrently with BET protein inhibition, prevents the development of colonies in cells initially sensitive to JQ1 (A1847, A2780 and PEO1) and those inherently resistant to JQ1 (SKOV3 and OVCAR5). Treatment of HIO117 non-tumorigenic ovarian epithelial cells with JQ1 reduced colony formation and to a greater extent when combined with AZD4547, however; no enhancement of growth inhibition was observed when JQ1 was combined with GDC-0941 or trametinib (Figure S7A). Additionally, FGFR or MEK inhibition increased JQ1-mediated growth arrest in a subset of OC cells in 72 h viability assays, while the combined treatment with JQ1 and GDC-0941 blocked cell growth to a greater extent than either agent alone across all of the OC cell lines (Figures 7B and S7B).
Figure 7. Combination Therapies Targeting RTK, PI3K or ERK Signaling Concurrently With BET Protein Inhibitors Enhance Growth Inhibition in OC Cells.

(A) Combination therapies of AZD4547 (1 μM), GDC-0941 (500 nM), MK2206 (500 nM), trametinib (10nM) or Vx-11e (500 nM) with JQ1 (200 nM) synergistically block cell growth of OC cells in 14-day crystal violet colony formation assay.
(B) Co-targeting BET bromodomain proteins and FGFRs, MEK or PI3K enhances growth inhibition of parental OC cells. Cells were treated for 72 h with JQ1 (500 nM) and AZD4547 (1 μM), BGJ398 (1 μM), trametinib (50 nM), or GDC-0941 (1 μM). Viability assessed by CellTiter-Glo.
(C) Co-targeting BET proteins with either GDC-0941 (500 nM) or trametinib (10 nM) reduce MYC and/or FOSL1 levels to a greater extent than single agents in OVCAR5 cells intrinsically resistant to JQ1 as depicted by blot.
(D–H) Ovarian cancer cells generated from HGS-OvCa patient-derived xenografts (PDX) grown on J2 feeder cells were treated with escalating doses of JQ1, GDC-0941 or the combination of GDC-0941 for 72 h and cell viability assessed by CellTiter-Glo. Chou–Talalay analysis indicated synergy (CI ≤0.5) between GDC-0941 and JQ1 in 4 of 5 HGS-OvCa PDX cells, Combination index (CI) values shown.
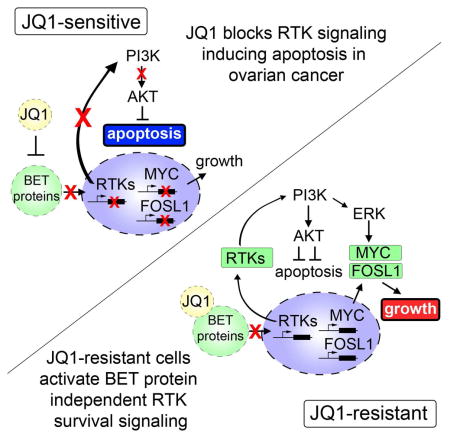
(I) Resistance to BET bromodomain inhibition mediated by kinome reprogramming in OC cells. Activation of RTK-PI3K-ERK signaling promotes resistance to JQ1 supporting combination therapies co-targeting BET proteins and RTKs, PI3K or MEK pathways.
Data present in (B) and (D–H) are triplicate experiments SEM. *p ≤0.05 by student’s t-test. Also see Figure S7 and Table S3.
Molecular analysis of OVCAR5 and SKOV3 cells treated with JQ1 and either GDC-0941 or trametinib demonstrates the effectiveness of these combination therapies at blocking JQ1-mediated activation of ERK and/or AKT signaling in OC cells inherently resistant to JQ1 (Figures 7C and S7C). Furthermore, the combination therapies reduced levels of pMYC (S62) and pFOSL1 (S265) leading to degradation of BRD-targets MYC and FOSL1 protein levels to a greater extent than single agent therapies, as well as blocked RNApol II (S2) phosphorylation. These studies demonstrate that blockade of PI3K pathway with GDC-0941, concurrently with BET protein inhibition, provides universal enhancement of JQ1 efficacy in OC cells. To test the generality of this response, we next examined this combination therapy in patient-derived EOC cell lines grown on J2 feeder cells. Importantly, the combination of JQ1 and GDC-0941 blocks cell growth to a greater extent than single agent treatments in patient-derived EOC cell line models, with some cells (OC-1, OC-20 and OC-16) showing drug synergy to the combination therapy (CI ≤0.5) (Figures 7D–7H and Table S3). These findings further demonstrate the potential of targeting both BET proteins and PI3K signaling for the treatment of OC.
DISCUSSION
In this study, we utilized MIB/MS to explore the consequence of both short-term and chronic BET protein inhibition on global kinase signaling in BRD4-dependent OC cells. Our proteomic analysis of the response of the kinome to the BETi revealed an essential role for BET proteins in the regulation of RTKs and their downstream signaling in OC (Figure 7I). Short-term BET protein inhibition resulted in reduced kinase expression of many growth-promoting RTKs that have established roles in tumorigenesis, as well as many kinases essential for cancer cell growth. Additionally, we observed activation of a cell death inducing kinase network following JQ1 treatment consistent with the growth arrest and apoptosis observed in OC cell lines sensitive to JQ1 therapy. To our surprise, JQ1 treatment induced a diverse network of RTKs and their downstream AKT and ERK survival signaling in a subset of OC cells, representing an intrinsic mechanism of resistance to BET protein inhibition. Activation of these RTKs and their downstream PI3K/ERK signaling pathways were also observed in OC cells that acquired resistance to BET protein inhibition following chronic exposure. Importantly, blockade of JQ1-induced RTK-PI3K-ERK signaling re-sensitized the OC cells to BET protein inhibition resulting in potent growth arrest and apoptosis. Our findings suggest that combination therapies co-targeting BET proteins and kinases will overcome resistance to BETi and provide durable responses for the treatment of OC.
Emerging studies have shown that BRD4 is enriched at a number of RTK promoters in a variety of cancer models, and that BET protein inhibition displaces BRD4 resulting in reduced expression of the kinases (Stratikopoulos et al., 2015; Stuhlmiller et al., 2015). Furthermore, BET protein inhibition has been shown to block the induction of RTKs in response to targeted kinase inhibitors overcoming drug resistance due to adaptive kinome reprogramming in both PI3K-altered and/or HER2-driven cancers. Consistent with these findings, we demonstrate that BET protein inhibition in OC models harboring a variety of PI3K/RAS alterations reduces transcription of many RTKs, as well as other key kinase oncogenes such as AURKA, SRC, MAPK3 and AKT2. Moreover, we demonstrate that chronic exposure to BETi can maintain transcriptional repression of a number of kinases with established roles in feedback-mediated resistance to kinase inhibitors. These findings suggest that pre-treatment of cancer cells with BETi may sensitize them to a wide variety of clinically relevant kinase inhibitors representing a general strategy to overcome resistance mediated by kinome reprogramming. Interestingly, our kinome profiling studies also revealed that BET protein inhibition induces the expression and activation of RTKs and their downstream PI3K signaling in a subset of OC cells as an intrinsic resistance mechanism to BETi. Contrary to previous reports (Stratikopoulos et al., 2015; Stuhlmiller et al., 2015), we observed activation of a variety of RTKs following BETi treatment, suggesting that enhancement of RTK transcription by BET proteins may be cell-type specific. The dose-dependent induction of RTKs, in response to JQ1 treatment in a subset of OC cells, suggests that BET proteins may also participate in the negative regulation of RTKs; particularly FGFRs, which were induced by JQ1 in the majority of OC cells. Collectively, our findings suggest that differential sensitivity of OC cells to JQ1 may be, in part, due to differences in BET protein regulation of intrinsic RTK programs.
Ovarian cancer cells chronically exposed to JQ1 acquire sensitivity to PI3K inhibitors, and inhibition of this pathway results in dramatic cell growth arrest and apoptosis. Importantly, the PI3K/AKT pathway has well-established roles in the negative regulation of apoptosis in cancer through direct phosphorylation of pro-apoptotic proteins (Kurokawa and Kornbluth, 2009). Recently, studies have shown that tumor cells harboring PIK3CA mutations are intrinsically resistant to BETi and require the combined blockade of PI3K and BET proteins to induce tumor apoptosis (Marcotte et al., 2016; Stratikopoulos et al., 2015). Consistent with these findings, our analysis of kinome reprogramming following short-term or chronic JQ1 exposure in PIK3CA (H1047R) mutant SKOV3 cells revealed that BET protein inhibition activated an intrinsic resistance program consisting of upregulation of FGFRs and their downstream PI3K signaling protecting these cells from apoptosis. Targeting the BETi-induced RTKs or PI3K sensitized these cells to JQ1 treatment, further demonstrating the utility of combining PI3K and BET proteins inhibitors to circumvent BETi resistance in PI3K altered cancers. Additionally, we demonstrate that an OC cell line harboring a G12V KRAS mutation also engages RTKs and PI3K signaling to overcome short-term BET protein inhibition, suggesting cells harboring KRAS mutations might exhibit intrinsic resistance to JQ1 through feedback-mediated RTK reprogramming. Further studies investigating the consequence of KRAS mutations on kinome responses to BET protein inhibition will be required to determine whether KRAS status is predictive of BETi response.
Recent work investigating BRD4 as a target in EOC showed that primary human xenografts, derived from MYCN and c-MYC overexpressing strains, exhibited sensitivity to BRD4 inhibitors (Baratta et al., 2015). In the present studies, we discovered that MYC and/or FOSL1 were initially downregulated by short-term JQ1 treatment in MYC and/or FOSL1 overexpressing OC cells, however, MYC and FOSL1 protein levels returned to baseline following chronic JQ1 exposure. Previous studies have demonstrated that MYC and FOSL1 protein stability is regulated by phosphorylation by ERK (Sears et al., 2000; Basbous et al., 2007). Importantly, we observed degradation of MYC and FOSL1 proteins in response to MEK or PI3K inhibitor treatment in JQ1-R cells but not in response to high dose JQ1 therapy. Thus, the elevated ERK and AKT signaling acquired following continuous JQ1 therapy promotes MYC and FOSL1 protein stability, permitting cell growth and survival in the presence of BETi. Intriguingly, elevated MYC and FOSL1 RNA levels were detected in A1847 JQ1-R cells suggesting an alternative mechanism for regulation of MYC and/or FOSL1 transcription in the absence of BET protein function. Recent studies investigating resistance mechanisms of BETi in leukemia cancers showed that activation of WNT/β-catenin and TGF-β signaling could promote MYC signaling following chronic BET protein inhibition and that blockade of these pathways restored sensitivity to BETi (Fong et al., 2015; Rathert et al., 2015). In our MIB/MS studies of JQ1-R cells, we also identified upregulation of a number of kinases within the WNT/β-catenin and TGF-β signaling including ACVR1, ACVR2 and TGFBR1, as well as CK1γ1 in A1847-R cells, which showed enhanced sensitivity to CK1 inhibitors relative to parental cells. Based on these findings, an alternative transcription network, such as WNT/β-catenin, could conceivably regulate the increased MYC and FOSL1 RNA expression detected in A1847 JQ1-R cells; however, further transcriptional studies will be required to identify epigenetic mechanisms of resistance to JQ1 in OC cells. Nevertheless, our studies demonstrate that treatment of JQ1-R cells with MEK or PI3K inhibitors promotes the degradation of MYC and/or FOSL1 at the protein level, rapidly inducing apoptosis, representing a highly translational therapeutic strategy to overcome BETi resistance in OC.
Collectively, we demonstrate that BET proteins promote expression and activity of many kinases essential for cancer cell proliferation and survival. Using MIB/MS kinome profiling, we show that in a subset of OC cells inherently resistant to BETi, treatment with JQ1 results in induced expression and activation of a diverse network of RTKs and their downstream kinase survival pathways, overcoming JQ1 drug effects. Furthermore, OC cell lines that acquire resistance to JQ1 utilize these RTK networks to promote downstream PI3K/ERK signaling to block JQ1-mediated apoptosis. Importantly, OC cells chronically exposed to JQ1 were exquisitely sensitive to combination therapies targeting RTKs, PI3K or MEK-ERK pathways. These studies suggest that single agent BETi therapies may not provide durable therapeutic response in OC and will likely require combination therapies targeting protein kinase signaling.
EXPERIMENTAL PROCEDURES
MIB Chromatography and LC/MS
Experiments using MIB/MS were performed as previously described (Duncan et al., 2012; Stuhlmiller et al., 2015). For SILAC quantitation, equal volumes of SILAC-labeled heavy and light lysates were mixed and endogenous kinases isolated by MIB columns. SILAC-labeled peptides were separated on a Thermo RSLC Ultimate 3000 UPLC through a Thermo Easy-Spray 75μm x 50cm C-18 column on a 235 min gradient (4–25% acetonitrile with 0.1% formic acid at 300 nL/min). Thermo Q-Exactive plus ESI mass spectrometers were used. For details, see Supplementary Experimental Procedures.
For cell growth assays, apoptotic assays, western blotting, qRT-PCR, siRNA, RNAseq, drug synergy, see Supplemental Experimental Procedures.
Supplementary Material
Highlights.
Inhibition of BET proteins reprograms kinome activity in ovarian cancer cells.
Receptor tyrosine kinase activation overcomes BET inhibition causing resistance.
Elevated PI3K/ERK activity stabilizes MYC/FOSL1 proteins in JQ1-resistant cells.
Co-targeting BET proteins and RTK or PI3K signaling enhances BET inhibitor therapy.
Acknowledgments
Funded by NIH CORE Grant CA06927 (Fox Chase Cancer Center), R01 GM083025 (J.R.P), R01 CA142928 (J.C.), NIH T32 CA009035 (L.F., A.M.K). Kinome profiling studies were funded by the Cancer Kinome Initiative (CKI) at FCCC, which was established by a donation from Don Morel. MIBs supplies generously provided by Gary L Johnson at the University of North Carolina. Development of patient-derived ovarian carcinoma cell lines was supported in part by a grant with the Pennsylvania Department of Health (SAP#4100068716) and a generous donation from the Roberta Dubrow Fund to D.C.C.
Footnotes
ACCESION NUMBERS
The GEO accession number for the mRNA sequencing data reported in this paper is GSE82329.
Supplemental information includes Supplemental Experimental Procedures, seven figures and three tables and can be found with this article online at
AUTHOR CONTRIBUTIONS
J.S.D. and J.R.P. wrote the manuscript. C.S. and A.M.K. performed all siRNA, growth and survival analysis, drug response studies and western blots. K.E.D., J.S.D., J.B., K.J. and A.M.K. performed all MIB/MS experiments. K.E.D. performed qRT-PCR experiments. S.B. and R.G. performed drug synergy studies using PDX-derived cell lines. L.S.F. and C.S. carried out kinase inhibitor screen. Y.S.L. performed and analyzed RNAseq. W.E.C., M.A., and N.L. provided CTx-0249885 for MIBs studies. J.S.D., J.C., J.R.P. and D.C.C. contributed to experimental design.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn SH, Kim M, Buratowski S. Phosphorylation of serine 2 within the RNA polymerase II c-terminal domain couples transcription and 3′ end processing. Mol Cell. 2004;13:67–76. doi: 10.1016/s1097-2765(03)00492-1. [DOI] [PubMed] [Google Scholar]
- Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotech. 2011;29:1039–1045. doi: 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baratta MG, Schinzel AC, Zwang Y, Bandopadhayay P, Bowman-Colin C, Kutt J, Curtis J, Piao H, Wong LC, Kung AL, et al. An in-tumor genetic screen reveals that the BET bromodomain protein, BRD4, is a potential therapeutic target in ovarian carcinoma. Proc Natl Acad Sci USA. 2015;112:232–237. doi: 10.1073/pnas.1422165112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartholomeeusen K, Xiang Y, Fujinaga K, Peterlin BM. Bromodomain and extra-terminal (BET) bromodomain inhibition activate transcription via transient release of positive transcription elongation factor b (P-TEFb) from 7SK small nuclear ribonucleoprotein. J Biol Chem. 2012;287:36609–36616. doi: 10.1074/jbc.M112.410746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbous J, Chalbos D, Hipskind R, Jariel-Encontre I, Piechaczyk M. Ubiquitin-independent proteasomal degradation of Fra-1 is antagonized by Erk1/2 pathway-mediated phosphorylation of a unique C-terminal destabilizer. Mol Cell Biol. 2007;27:3936–3950. doi: 10.1128/MCB.01776-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK, Baselga J, Rosen N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19:58–71. doi: 10.1016/j.ccr.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG, et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell. 2013;24:777–790. doi: 10.1016/j.ccr.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper MJ, Cox NJ, Zimmerman EI, Dewar BJ, Duncan JS, Whittle MC, Nguyen TA, Jones LS, Ghose Roy S, Smalley DM, et al. Application of multiplexed kinase inhibitor beads to study kinome adaptations in drug-resistant leukemia. PLoS One. 2013;8:e66755. doi: 10.1371/journal.pone.0066755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, Johnson NL, Granger DA, Jordan NV, Darr DB, et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell. 2012;149:307–321. doi: 10.1016/j.cell.2012.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov. 2014;13:337–356. doi: 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- Fong CY, Gilan O, Lam EYN, Rubin AF, Ftouni S, Tyler D, Stanley K, Sinha D, Yeh P, Morison J, et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature. 2015;525:538–542. doi: 10.1038/nature14888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanrahan AJ, Schultz N, Westfal ML, Sakr RA, Giri DD, Scarperi S, Janikariman M, Olvera N, Stevens EV, She QB, et al. Genomic complexity and AKT dependence in serous ovarian cancer. Cancer Discov. 2012;2:56–67. doi: 10.1158/2159-8290.CD-11-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar K, Raza SS, Knab LM, Chow CR, Kwok B, Bentrem DJ, Popovic R, Ebine K, Licht JD, Munshi HG. GLI2-dependent c-MYC upregulation mediates resistance of pancreatic cancer cells to the BET bromodomain inhibitor JQ1. Sci Rep. 2015;5:9489. doi: 10.1038/srep09489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokawa M, Kornbluth S. Caspases and kinases in a death grip. Cell. 2009;138:838–854. doi: 10.1016/j.cell.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockwood WW, Zejnullahu K, Bradner JE, Varmus H. Sensitivity of human lung adenocarcinoma cell lines to targeted inhibition of BET epigenetic signaling proteins. Proc Natl Acad Sci USA. 2012;109:19408–19413. doi: 10.1073/pnas.1216363109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte R, Sayad A, Brown KR, Sanchez-Garcia F, Reimand J, Haider M, Virtanen C, Bradner JE, Bader GD, Mills GB, et al. Functional genomic landscape of human breast cancer drivers, vulnerabilities, and resistance. Cell. 2016;164:293–309. doi: 10.1016/j.cell.2015.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L, Sims RJ. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci USA. 2011;108:16669–16674. doi: 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott CJ, Kopp N, Bird L, Paranal RM, Qi J, Bowman T, Rodig SJ, Kung AL, Bradner JE, Weinstock DM. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood. 2012;120:2843–2852. doi: 10.1182/blood-2012-02-413021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, Nekritz EA, Zeid R, Gustafson WC, Greninger P, et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 2013;3:308–323. doi: 10.1158/2159-8290.CD-12-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathert P, Roth M, Neumann T, Muerdter F, Roe JS, Muhar M, Deswal S, Cerny-Reiterer S, Peter B, Jude J, et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature. 2015;525:543–547. doi: 10.1038/nature14898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000;14:2501–2514. doi: 10.1101/gad.836800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segura MF, Fontanals-Cirera B, Gaziel-Sovran A, Guijarro MV, Hanniford D, Zhang G, González-Gomez P, Morante M, Jubierre L, Zhang W, et al. BRD4 sustains melanoma proliferation and represents a new target for epigenetic therapy. Cancer Res. 2013;73:6264–6276. doi: 10.1158/0008-5472.CAN-13-0122-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell. 2014;54:728–736. doi: 10.1016/j.molcel.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura T, Chen Z, Soucheray M, Carretero J, Kikuchi E, Tchaicha JH, Gao Y, Cheng KA, Cohoon TJ, Qi J, et al. Efficacy of BET bromodomain inhibition in Kras-mutant non-small cell lung cancer. Clin Cancer Res. 2013;19:6183–6192. doi: 10.1158/1078-0432.CCR-12-3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu S, Lin CY, He HH, Witwicki RM, Tabassum DP, Roberts JM, Janiszewska M, Jin Huh S, Liang Y, Ryan J, et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature. 2016;529:413–417. doi: 10.1038/nature16508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sos ML, Levin RS, Gordan JD, Oses-Prieto JA, Webber JT, Salt M, Hann B, Burlingame AL, McCormick F, Bandyopadhyay S, et al. Oncogene mimicry as a mechanism of primary resistance to BRAF inhibitors. Cell Rep. 2014;8:1037–1048. doi: 10.1016/j.celrep.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratikopoulos EE, Dendy M, Szabolcs M, Khaykin AJ, Lefebvre C, Zhou MM, Parsons R. Kinase and BET inhibitors together clamp inhibition of PI3K signaling and overcome resistance to therapy. Cancer Cell. 2015;27:837–851. doi: 10.1016/j.ccell.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuhlmiller TJ, Miller SM, Zawistowski JS, Nakamura K, Beltran AS, Duncan JS, Angus SP, Collins KAL, Granger DA, Reuther RA, et al. Inhibition of lapatinib-induced kinome reprogramming in ERBB2-positive breast cancer by targeting BET family bromodomains. Cell Rep. 2015;11:390–404. doi: 10.1016/j.celrep.2015.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Hobor S, Bertotti A, Zecchin D, Huang S, Galimi F, Cottino F, Prahallad A, Grernrum W, Tzani A, et al. Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep. 2014;7:86–93. doi: 10.1016/j.celrep.2014.02.045. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Ma P, Jing Y, Yan Y, Cai MC, Zhang M, Zhang S, Peng H, Ji ZL, Di W, et al. BET bromodomain inhibition as a therapeutic strategy in ovarian cancer by downregulating FoxM1. Theranostics. 2016;6:219–230. doi: 10.7150/thno.13178. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.