

Abstract
X-linked adrenoleukodystrophy (X-ALD) is a peroxisomal disorder caused by mutations in the ABCD1 gene, leading to a defect in the peroxisomal adrenoleukodystrophy protein (ALDP), which inhibits the β-oxidation of very long chain fatty acids (VLCFAs). It is a complex disease where the same mutation in the peroxisomal ABCD1 can lead to clinically diverse phenotypes ranging from the fatal disorder of cerebral ALD (cALD) to mild adult disorder of adrenomyeloneuropathy (AMN). This suggests a role of epigenetic factors/modifier genes in disease progression of X-ALD which is not understood at present. To examine the possible role of microRNA (miRNA) in X-ALD disease mechanisms for differences in cALD and AMN phenotype, we profiled 1008 known miRNA in cALD, AMN, and normal human skin fibroblasts using miScript miRNA PCR array (Qiagen) and selected miRNAs which had differential expression in cALD and AMN fibroblasts. Eleven miRNA which were differentially regulated in cALD and AMN fibroblasts were identified. miR-196a showed a significant differential expression between cALD and AMN and is further characterized for target gene regulation. The predicted role of miR-196a in inhibition of inflammatory signaling factors (IKKα and IKKβ) and ELOVL1 expression suggests the pathological role of altered expression of miR-196a. This study indicates that miR-196a participated in differential regulation of ELOVL1 and inflammatory response between cALD as compared to AMN and may be a possible biomarker to differentiate between cALD and AMN.
Keywords: miRNA, X-ALD, Phenotype, Differential expression, Inflammation
Introduction
X-linked adrenoleukodystrophy (X-ALD) is the most common peroxisomal disorder, with an incidence of approximately 1:17,000. It is a complex disease where the same mutation in peroxisomal ABCD1 (also known as adrenoleukodystrophy protein or ALDP) can lead to clinically diverse phenotypes, the fatal disorder of cerebral ALD (cALD) and adult disorder of adrenomyeloneuropathy (AMN). The cALD is characterized by progressive cerebral demyelination with a strong inflammatory response in the white matter leading to neurodegeneration and death often before the patient reaches adolescence [1–3]. AMN affects adults (second to fourth decade) and is characterized by axonopathy (resembling spastic paraparesis or spastic paraplegia) without significant neuroinflammation and neurodegeneration. However, approximately 35 % of AMN patients subsequently develop CNS involvement with inflammatory demyelination and these patients share the same poor prognosis as children with cALD [1, 4]. VLCFA-induced inflammatory disease in cALD vs. the milder phenotypes in patients with AMN resulting from the same mutation in ABCD1 suggests a role of epigenetic factors/modifier genes in X-ALD disease progression. However, the proposed role of epigenetic factors/modifier genes that modulate the clinical outcome of the disease is not understood at present [2, 4].
The biochemical signature of X-ALD and AMN is the pathognomonic accumulation of saturated very long chain fatty acids (VLCFAs; >C22:0). VLCFAs accumulate in all tissues as various lipid classes; however, the degree of accumulation is higher in the cholesterol ester and sphingolipid fractions of brain white matter and adrenal cortex [5]. This pathognomonic accumulation of VLCFAs is also used as diagnostic test for X-ALD; however, it does not distinguish between X-ALD phenotypes [2, 3]. The ALD gene (ABCD1), identified by positional cloning [6], encodes ALDP which is a member of peroxisomal ATP-binding cassette (ABCD) transmembrane transporter proteins [7, 8]. The loss of ABCD1 and thus its function results in defective β-oxidation of VLCFA [9] and supplementation of ALDP gene corrects the metabolic defects in ALD fibroblasts indicating a role of ALDP loss in VLCFA accumulation [10]. In addition, three other half transporters have been identified: ABCD1-related protein (ABCD2), peroxisomal membrane protein (PMP70 or ABCD3), and PMP70-related protein (ABCD4) [11, 12]. Homo or hetero dimers of these proteins function as ATP-binding cassette peroxisomal membrane transporters [13] but their precise functions are not understood at present. In addition, increased synthesis of VLCFA is also reported in X-ALD cells [14, 15]. Thus, the elevation of VLCFAs in X-ALD is the consequence of both reduced VLCFA peroxisomal β-oxidation [9] and increased activity of FA elongases (ELOVLs) for VLCFA synthesis [15, 16]. However, relative contribution of these two mechanisms to overall cellular load of VLCFA is not understood at present.
The pathobiology of X-ALD is complex as the same defect can lead to clinically diverse phenotype where patients with CNS inflammatory disease of cALD die in early childhood while patients with peripheral CNS disease without inflammatory disorder go on to live to the age of 50 or 60 years. Recently, Singh and Pujol proposed a three-hit hypothesis as a framework to better understand the molecular mechanisms associated with the disease pathogenesis of X-ALD [4]; gene mutation induced pathognomonic accumulation of VLCFA as the “first hit”; VLCFA-induced epigenetic/stochastic mechanism leading to oxidative disease as the “second hit”; and induction of inflammatory disease as the “third hit” thus creating a vicious cycle resulting in cell loss and progressive inflammatory demyelinating disease.
MicroRNAs (miRNAs) are small, about 21–25 nucleotides in length, single stranded, non-coding RNAs that bind to the 3′ untranslated region of target messenger RNA (mRNA) and negatively regulate the expression of genes involved in important cellular processes depending on the degree of complementarity by either degradation or translational repression of a target mRNA [17]. Recent studies describe the potential role of miRNA in epigenetic mechanisms in post-transcriptional regulations. In recent years, miRNA have been recognized to play an important role in the pathogenesis of various diseases including neurodegenerative disorders [18–25]. While both cALD and AMN disease pathology starts with mutation or deletion of ALD gene, the observed differential accumulation of VLCFA in different cell types causes different degrees of oxidative and inflammatory pathology affecting different parts of the CNS and possibly different phenotypes [26]. This study describes the complexity of X-ALD disease pathology.
To examine the possible role of miRNA in X-ALD disease mechanisms for differences in cALD and AMN phenotype, we profiled 1008 known miRNA in cALD, AMN, and normal human skin fibroblasts using miScript miRNA PCR array (Qiagen) and selected miRNAs based on their differential expressions in cALD and AMN fibroblasts. We identified 11 miRNA which were differentially regulated in cALD and AMN fibroblasts (i.e., miR-103b, miR-106b, miR-135a, miR-142-3p, miR-142-5p, miR-152, mir-196a, mir-504, miR-708, miR-10b*, and miR-19a*). Based on the high degree of differential expression of miR-196a between cALD and AMN, it is further characterized for target gene regulation and disease pathogenesis.
Material and Methods
Reagents
Dulbecco's modified Eagle's medium (DMEM) and Hanks' balanced salt solution (HBSS) were purchased from Invitrogen Life Technologies; fetal bovine serum (FBS) was purchased from BioAbChem Inc. (Ladson, SC). Antibodies P-IkBα, P-NFkB, and Ikkα were purchased from Cell Signaling Technology Inc. (MA, USA). RNA extraction kit, mRNA profiling Array, and miScript Primer Assays were purchased from Qiagen.
Cell Culture-Fibroblasts
Human skin fibroblasts derived from control (normal; GM03348, GM08398, and GM00495), X-ALD (GM04932, GM04934, GM04904, GM04496, and GM04933), and AMN (GM07530 and GM07531) patients were obtained from the National Institute of General Medical Sciences (NIGMS) Human Genetic Mutant Cell Repository at the Coriell Institute for Medical Research (ccr.coriell.org/). The fibroblasts were cultured in DMEM supplemented with 10 % FBS and antibiotic/antimycotic solution in a humidified incubator (37 °C and 5 % CO2). X-ALD fibroblast cell line GM04932 was derived from a patient with positive family history of X-ALD (10-year-old male with elevated C26:0 fatty acids). GM04934 was from a 7-year-old boy with elevated C26:0 fatty acids levels. GM04904 was from a 11-year-old male with elevated C26:0/C22:0 ratio in fibroblasts; adrenal insufficiency; progressive white matter disease; and progressively deteriorating intellectual function with a family history of X-ALD. GM04496 was from a 6-year-old male with elevated levels of C26 fatty acids in fibroblasts and pathological autopsy in both the adrenal and central nervous system. GM04933 was from a 8-year-old male with adrenal insufficiency; elevated C26:C22 fatty acid ratio in fibroblasts and plasma; and with affected uncles and cousins. AMN fibroblast GM07530 was from a 26-year-old male characterized as AMN with adrenal insufficiency; hyperreflexia; progressive spastic paraparesis; occasional nausea and vomiting; increased frequency of urination; and elevated levels of C26 fatty acids in plasma and fibroblasts with negative family history. GM07531 was from a 19-year-old male characterized as AMN with adrenal insufficiency since age 11 years; unsteady gait since age 13 years; slight weakness and hyperreflexia since age 19 years; and elevated level of C26 fatty acid in plasma and fibroblasts (ccr.coriell.org/). Normal (control) fibroblasts GM03348 were from an apparently healthy 10-year-old male. GM08398 was from an apparently healthy 8-year-pold male and GM00495 was from an apparently healthy 29-year-old male.
Stable Lentiviral Vector-Mediated Gene Silencing of Abcd1
A set of three human (SK-009605-00-10) and rat (SK-098142-00-10) specific SMART vector 2.0 lentiviral shRNA particles (108 TU/ml) for Abcd1 were purchased from Thermo Fisher Scientific Dharmacon (CO, USA). The vector had an hCMV promoter, a TurboGFP reporter gene, and a puromycin selection gene. Human U87 astrocytes (ATCC, VA, USA) and rat B12 oligodendrocytic cells (Sigma-Aldrich, MO, USA) were cultured in DMEM with 10% FBS in the presence of antibiotics. Viral particles (Abcd1) were added with a multiplicity of infection (MOI) of 2.5 and 3.0, respectively, for U87 astrocytes and B12 oligodendrocytes. TurboGFP expression was analyzed using microscopy, and GFP-positive cells were selected using puromycin after 72 h (3.0 mg/ml for U87 astrocytes and 2.0 mg/ml for B12 oligodendrocytes). The puromycin selection was done until 100% cells were GFP positive and selected cells were maintained in culture media containing puromycin at 0.5 mg/ml until further use [27]
Human Brain Samples
Frozen (−80 °C) postmortem brain tissues from cerebral cALD patients (6–9 years), AMN patients (35–40 year), and age matched healthy male control subjects were obtained from the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore, MD, USA. The tissue sections were obtained from equivalent cerebrum-white matter area of cALD, AMN, and control subjects. The post-mortem interval for these samples was between 5 and 10 h. Samples from three cerebral cALD, AMN, and healthy male subjects were used. Out of the three AMN samples used for the study, two samples (B1 and B2) were pure AMN samples but B3 sample was from the patient that subsequently developed cerebral involvement.
miRNA Extraction and complementary DNA (cDNA) Synthesis
Total RNA including miRNA was extracted from cell lines and tissues using a miRNeasy mini kit (Qiagen) per the manufacturer's protocol. RNA concentrations were determined with NanoDrop (ThermoScientific). Total RNA that contains miRNA (600 ng) was used for cDNA synthesis using miScript II RT Kit (Qiagen) using miScript Reverse Transcriptase Mix, 10× miScript Nucleics Mix, and 5× miScript HiSpec Buffer. The mixture is incubated for 60 min at 37 °C and for 5 min at 95 °C to inactivate miScript Reverse transcriptase mix and placed on ice. This cDNA is diluted in RNase free water (20 μl of cDNA obtained above mixed with 180 μl of water).
miRNA Profiling
miRNA profiling was performed using Human miRNome miRNA PCR array (96 well format from Qiagen-MIHS-216Z). The array profiles the expression of the 1008 most abundantly expressed and best characterized miRNA sequences in the human miRNA genome (miRNome) as annotated in miRBase Release 16 (www.mirbase.org). A set of controls are included on each plate which enables data analysis using ΔΔCT method of relative quantification, assessment of reverse transcription performance, and assessment of PCR performance. The miScript miRNA PCR array enables SYBR Green-based real-time PCR analysis using Biorad CFX96 real-time PCR system as follows: 95 °C for 15 min; 40 cycles of 94 °C for 15 s; 55 °C for 30 s; and 70 °C for 30 s. The relative expression was calculated using the ΔΔCT method (relative gene expression= 2-(ΔCT sample-ΔCT control)] and is presented in fold increase relative to control.
miScript Primer Assays and Western Blotting
To validate the levels of miRNA selected from miRNA profile in wide range of samples, we used Qiagen miScript Primer Assays and SYBR Green PCR kit. Values were normalized to SNORD61 and data analyzed using ΔΔCT method of relative quantification and presented as fold change. Protein extraction and western blot analysis were performed as described earlier [28] using antibodies raised against P-p65, p-IκB, and β-actin (Cell Signaling, MA, USA).
Transfection of miR-196a Mimic
ABCD1-silenced U87 Astrocytes and B12 Oligodendrocytes were transfected with miScript miRNA mimic for miR-196a (Qiagen) using HiPerFect transfection reagent (Qiagen) at a final concentration of 8 nM. Transfection efficiency was assessed by miScript PCR assay.
Target Gene Prediction
Prediction of putative miRNA targets was performed by using the online softwares, TargetScan (http://www.targetscan.org/), PicTar (http://pictar.mdc-berlin.de/), miRDB (http://mirdb. org/miRDB/), and NPInter (http://www.bioinfo.org/NPInter/).
Results
miRNA Profiling
To identify the miRNAs which are differentially expressed in cALD and AMN, we profiled a miRNA library consisting of 1008 miRNA in human cALD, AMN, and control fibroblasts cell lines each in triplicate. About 80 % of miRNA profiled were detectable in all cell lines with a raw CT-value< 35. The Web-based miScript miRNA PCR array data analysis tool was used to analyze the real-time PCR data. This tool calculates the fold change using ΔΔCT method of relative quantification. We selected those miRNAs which were at least twofold up- or downregulated as compared to control fibroblasts. From these miRNAs, we identified miRNAs which were differentially regulated between cALD and AMN fibroblasts. Among 1008 miRNAs, 11 miRNAs were significantly differentially regulated between cALD and AMN. These are miR-103b, miR-106b, miR-135a, miR-142-3p, miR-142-5p, miR-152, miR-196a, miR-504, miR-708, miR-10b*, and miR-19a* (Fig. 1). Among these differentially regulated miRNAs, miR-196a is of special interest as it shows the highest fold change (~82-fold downregulated in cALD as compared to control and ~4-fold upregulated in AMN as compared to control) and is evaluated further.
Fig. 1.

Differentially expressed miRNA in cALD (grey) and AMN (white) fibroblasts from miRNome miRNA PCR array. The expression of 1008 most abundantly expressed and best characterized miRNA sequences in the human miRNA genome was profiled using Human miRNome miRNA PCR array (Qiagen-MIHS-216Z) in triplicate. The relative expression was calculated using the ΔΔCT method and is presented as fold change relative to control. Among 1008 miRNAs, 11 miRNAs were significantly differentially regulated between cALD and AMN as shown in the figure. miR-196a was most significantly differentially regulated
Validation of miR-196a Expression by Real-Time PCR
To validate the miRNA profiling data, we performed real-time RT-qPCR assay for miR-196a in cALD and AMN fibroblast cell lines, cALD and AMN human brain tissue samples, and ABCD1-silenced Astrocytes (U87) and oligodendrocyte (B12) cell lines. As shown in Fig. 2a, miR-196a was down-regulated in all the five cALD fibroblasts cell lines and upregulated in two AMN fibroblast cell lines studied. miR-196a was also downregulated in human brain tissues from three cALD patients and upregulated in two out of three AMN brain samples (Fig. 2b). Third AMN brain sample # UMB1836 showing downregulation of miR-196a was from AMN patient that in later life had cerebral involvement similar to one observed in cALD, a secondary cALD disease indicating the relationship between neuroinflammatory disease and downregulation of miR-196a. miR-196a expression was also evaluated in brain cell lines (B12 oligodendrocytes and U87 astrocytes) stably silenced for ABCD1 using lentiviral shRNA as described previously [27, 29]. The transduction efficiency was shown by mRNA levels of ABCD1 in U87 astrocytes and B12 oligodendrocytes (Fig. 3a, b). Silencing of ABCD1 increased the level of VLCFA by 3.1- and 3.15-fold in ABCD1-silenced U87 astrocytes and B12 oligodendrocytes when compared with their respective controls [27]. Thus, both cell models recapitulate events of the cALD pathology. Figure 3c, d shows that silencing of ABCD1 downregulates miR-196a in both ABCD1-silenced U87 astrocyte and B12 oligodendrocyte cell lines. These studies document that miR-196a express differentially in cALD and AMN samples.
Fig. 2.

Validation of miR-196a expression in human skin fibroblasts (a) and human brain samples (b) by qRT-PCR. miR-196a expression was validated in human skin fibroblasts and human brain samples (from equivalent brain regions) using Qiagen miScript primer assays by qRT-PCR. The values were normalized to SNORD61 and presented as fold change. As represented by figure, miR-196a expression was downregulated in cALD fibroblasts and brain samples and upregulated in AMN fibroblasts and brain samples. B3 is brain sample from AMN that subsequently developed CNS disease. Downregulation of miR-196a expression in brain sample B3 indicates of cerebral involvement. Thirty-five percent of AMN patients develop cerebral involvement
Fig. 3.

miR-196a expression in ABCD1-silenced U87 astrocytes and B12 oligodendrocytes. Human U87 astrocytes cells and rat B12 oligodendrocyte cells were transduced with ABCD1 Lentiviral (ABCD1-Lenti) shRNA particles followed by selection with puromycin. There was significant decrease in ABCD1 expression as analysed by qRT-PCR (a and b). **P < 0.05 vs. WT. Similar to cALD cells/brain, miR-196a expression was downregulated in both ABCD1-silenced U87 and B12 (c and d). An unpaired student t test was used to determine the statistical significance of the data from three independent experiments
Identification of Possible Target Genes of miR-196a
To elucidate the molecular mechanism by which miR-196a is involved in cALD and AMN pathogenesis, its potential mRNA targets were analyzed using bioinformatics programs: TargetScan, PicTar, miRDB, miRecords, and NPInter. Among the targets predicted by these softwares, we selected the targets which are thought to be involved in disease pathogenesis. The selected targets are MAP4K3, MAP3K2, IKKα (IKBKA), IKKβ (IKBKB), and ELOVL1 (Table 1). In X-ALD, VLCFA are elevated in plasma and tissues [30]. In addition to the role of ABCD1 dysfunction, the biosynthesis of VLCFA by ELOVL1 (elongase) also reported to contribute to VLCFA load in X-ALD [31] indicating that ELOVL1 may be one of the important targets of miR-196a. Moreover, based on the neuroinflammatory responses in cALD, other potential targets of miR-196a are involved in inflammatory signaling in cALD as compared to AMN. We further analyzed the target relationships of other differentially regulated miRNAs from the miRNA profile and found that miRNAs such as miR-106b, miR-135a, miR-504, and miR-196a are involved in VLCFA synthesis (i.e., ELOVL1 and ELOVL6), metabolism (ABCD2 and ABCD3), and inflammatory signaling (IKKα, IKKβ, MAP3K2, NFκB1, and NFκB2).
Table 1.
Selected targets of miR-196a
| MAP3K2 | Mitogen-activated protein kinase kinase kinase 2 |
| MAP4K3 | Mitogen-activated protein kinase kinase kinase kinase 3 |
| IKKα | Inhibitor of nuclear factor kappa-B kinase subunit alpha (IKBKA) |
| IKKβ | Inhibitor of nuclear factor kappa-B kinase subunit beta (IKBKB) |
| ELOVL1 | Elongation of very chain fatty acids protein |
SAHA Upregulates Expression of miR-196a in ABCD1 Knockdown Cells
Suberoylanilide hydroxamic acid (SAHA) is a potent and selective class I and II HDAC inhibitor [32]. Previously, it was shown to normalize the levels of VLCFA in ABCD1-silenced U87 and B12 cells by correction of fatty acid β-oxidation via induction of ABCD1-related proteins ABCD2/ABCD3 [27]. Treatment with SAHA was also reported to decrease the expression of ELOVL1 and ELOVL3 in ABCD1-silenced U87 and B12 cells [27]. miR-196a expression was downregulated in ABCD1-silenced U87 cells (U87 Lenti) but treatment of these U87 Lenti cells with SAHA reversed (upregulated) the expression of miR-196a (Fig. 4). SAHA treatment was also shown to reduce the expression of ELOVL1 [27] which is one of the target of miR-196a. These observations further support the relationship between ALD gene, VLCFA, and expressions of miR-196a and ELOVL1.
Fig. 4.
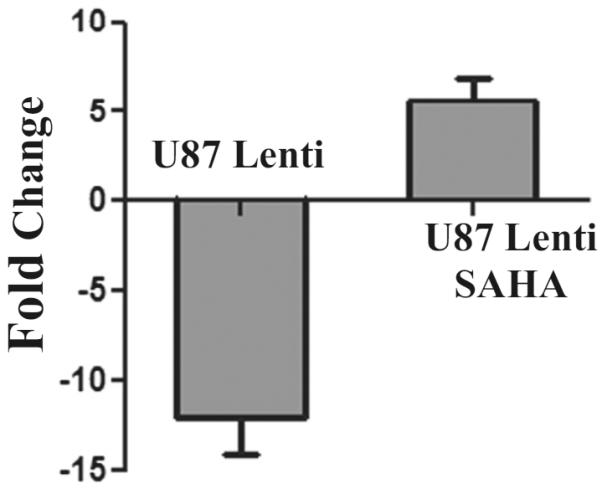
SAHA upregulates the expression of miR-196a in ABCD1-silenced U87 astrocytes (U87 lenti). Previously, we reported that the treatment of ABCD1-silenced cells with SAHA corrects the metabolic defect of X-ALD (28), therefore, we tested the effect of SAHA on expression of miR-196a in ABCD1 silenced cells. U87 Lenti cells were treated with 5 μM SAHA for 72 h and evaluated for miR-196a expression by qRT-PCR. miR-196a was downregulated in U87 Lenti cells but its expression was upregulated on treatment with SAHA indicating the role of epigenetic regulation of miR-196a in X-ALD
Expression of Target Genes in cALD Brains as Compared to AMN
U87 cells were selected for further studies because in X-ALD, accumulation of VLCFA C26:0 has been documented to cause metabolic alterations leading to membrane perturbation, redox imbalance, and changes in membrane lipid composition, as well as the induction of inflammatory mediators in cultured astrocytes [28, 33]. Since silencing of ABCD1 gene sensitizes astrocytes for inflammation and we were interested to study the inflammatory cells, we choose U87 for experimentation. For the study of neuroinflammatory disease of X-ALD, ABCD1-silenced U87 astrocyte cell lines and equivalent brain regions of cALD and AMN were studied for the expression of miR-196a and inflammatory gene signaling. The qRT-PCR analysis showed that the predicted target genes of miR-196a including MAP4K3, MAP3K2, IKKα (IKBKA), IKKβ (IKBKB), and ELOVL1 were markedly increased in cALD human brain tissues as compared to control as well as AMN brains. Therefore, it is of interest to evaluate whether decreased expression of miR-196a in cALD is responsible for increased expression of these target genes in cALD brain and thus a pathological mechanism for enhanced inflammatory reactions in CNS of cALD (Fig. 5). The NF-κB/Rel transcription factors are present in the cytosol in an inactive state complexed with the inhibitory IκB proteins. Activation occurs via phosphorylation of IκBα followed by its proteasome-mediated degradation and nuclear translocation of active NF-κB [34, 35]. The activation of NF-κB pathway is shown by increased expression of phosphorylated IκBα and p65 in cALD brain tissues as compared to AMN (Fig. 6). These observations document that downregulation of miR-196a parallel increased expression of inflammatory signaling pathway in cALD.
Fig. 5.

Analysis of mRNA expression of miR-196a target genes. The target genes selected for miR-196a were validated in human cALD and AMN brain samples taken from equivalent brain regions by qRT-PCR relative to RPLP0 levels and represented as percent of control. These target genes (MAP4K3, MAP3K2, IKKα, IKKβ, and ELOVL1) were upregulated in brain samples of cALD patients as compared to control and AMN patients. **P < 0.005 vs. control. There was no significant difference between control and AMN groups. An unpaired student t test was used to determine the statistical significance of the data from three independent experiments
Fig. 6.
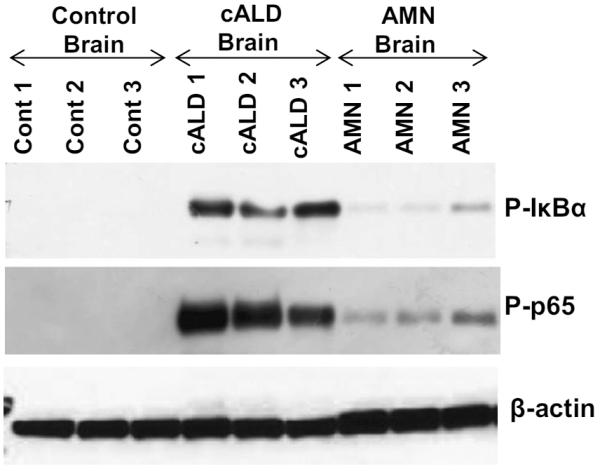
Western blot analysis of target genes of miR-196a. Lysates from equivalent brain regions of healthy, cALD and AMN patients were analyzed by western blotting. Representative immunoblots depicts increased NFκB activation (phosphorylation of IκBα and NFκB p65) in cALD group as compared to control and AMN groups as levels of PIκBα, P-p65, and β-actin in three different brain samples
Effect of miR-196a Mimic on the Expression of Target Genes
To further establish the relationship between miR-196a and its target genes and to determine the biological function of miR-196a, we used miR-196a mimic in ABCD1-silenced U87 astrocytes, B12 oligodendrocytes, and cALD fibroblast cells. miRNA mimics are small, chemically modified double-stranded RNAs that mimic endogenous miRNAs following transfection into cells, and enable its functional analysis by upregulation of miRNA activity. miScript miR-196a mimic (Qiagen) was used to overexpress miR-196a in ABCD1-silenced oligodendrocytes, astrocytes, and cALD fibroblasts. The transfection efficiency was determined by miScript qRT-PCR assay (Fig. 7a). These overexpressed cells were then analyzed for the expression of miR-196a targets, IKKα, IKKβ, and ELOVL1. Figure 7b, c shows that silencing of ALD gene results in upregulation of inflammatory genes (IKKα and IKKβ) in U87 astrocytes cells and expression of these target genes was normalized/decreased following the transfection of miR-196a mimic. ELOVL1 with chain length specificity toward VLCFA [36] contributes to VLCFA load in X-ALD and is also one of the targets of miR-196a. The expression of ELOVL1 was measured following transfection of miR-196a mimic. There was no significant difference in levels of mRNA for ELOVL1 between U87 control and ABCD1-silenced astrocyte cells as well as between control and cALD fibroblasts as reported earlier in our studies [37] but there was significant decrease in the expression of ELOVL1 following transfection of miR-196a mimics in cALD fibroblasts and ABCD1-silenced U87 cells (Fig. 8a, c). In B12 oligodendrocyte cell line, the expression of ELOVL1 was significantly increased following ABCD1 silencing (B12 Lenti) as compared to B12 control and transfection of miR-196a mimic in B12 Lenti cells decreased the expression of ELOVL1 (Fig. 8b). These studies document that expression levels of miR-196a regulate the expression of its target gene ELOVL1 for VLCFA load and NFkB inflammatory signaling pathways in X-ALD (Fig. 9).
Fig. 7.
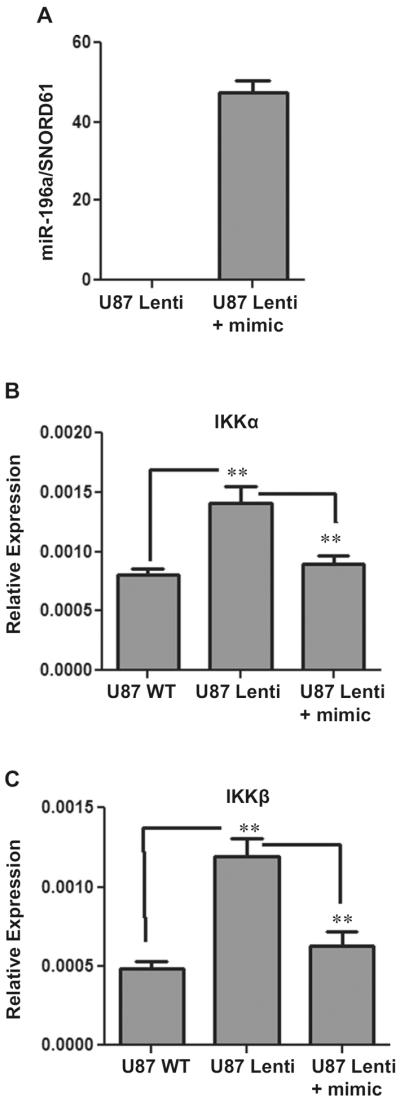
Roles of miR-196a in target gene expression. To establish the regulation of target genes by miR-196a, miR-196a mimic was transfected in ABCD1 knockdown U87 astrocytes cells (U87 Lenti) and transfection efficiency evaluated by miScript qRT-PCR. SNORD61 was used as an internal control and represented as miR-196a/SNORD61 (a). The expression of target genes IKKα and IKKβ was analysed after the transfection of miR-196a mimic and normalized to GAPDH. The expression of these genes was significantly increased in ABCD1 silenced cells as compared to wild type but significantly decreased after transfection of mimic in U87 Lenti cells (b and c). **P < 0.05. An unpaired student t test was used to determine the statistical significance of the data from three independent experiments
Fig. 8.

Role of miR-196a in target gene (ELOVL1) expression. miR-196a mimic was transfected in cALD fibroblasts (GMO4934), ABCD1-silenced U87 astrocytes cells (U87 Lenti), and ABCD1-silenced B12 oligodendrocytes cells (B12 Lenti) and analysed for ELOVL1 expression. There was no significant difference between normal (GMO3348) and cALD (GMO4934) fibroblasts and astrocytes (U87 WT and U87 Lenti cells) but there was significant difference between oligodendrocytes (B12 WT and B12 Lenti cells) in ELOVL1 expression. The expression of ELOVL1 was decreased after transfection of miR-196a mimic in all the three cell types. The expression of ELOVL1 was normalized to GAPDH. *P<0.01; **P<0.05; NS non significant. An unpaired student t test was used to determine the statistical significance of the data from three independent experiments
Fig. 9.

Schematic diagram showing the participation of genetic and epigenetic contributions to VLCFA accumulations
Discussion
MicroRNAs regulate multiple pathways during physiological and pathological conditions. Emerging evidence suggests that miRNAs are essential for the normal tissue development where specific miRNAs are reported to control the specific tissue development at particular stages of development as well as development of disease pathologies [38]. Altered expression or function of particular miRNAs has been identified in various CNS pathological conditions, including neuroinflammation, neurodegeneration, and autoimmune diseases [39]. This study identifies the role of miR-196a in differential accumulation of VLCFA in different cell types via the regulation of enzyme system for VLCFA synthesis (ELOVL1) and thus VLCFA load. Moreover, miR-196a also participates in differential expression of inflammatory genes in cALD vs. AMN disorders of X-ALD.
X-ALD is a complex disease where the same mutation in the ALD gene (ABCD1) can lead to clinically very distinct phenotypes [40]. Recently, Sutovsky et al. 2014 reported that two unrelated patients with same ABCD1 gene mutation where the first patient, a 47-year-old man had frontal lobe involvement, and in the second patient, a 16-year-old boy with Addison's disease since age of 9 years developed cognitive deficit and parieto-occipital involvement in the course of 1 year [41]. Thus, the same mutation can lead to the development of different manifestations of disease. These observations suggest that, in addition to VLCFA defect caused by ABCD1 deletion, epigenetic factors may contribute to these different disease phenotypes. In X-ALD, excessive accumulation of VLCFA starts in utero, with the subsequent neuroinflammatory disease (cALD) in early childhood (4–8 years of age), leading to loss of oligodendrocytes and myelin, and eventually to death. What triggers the transition from metabolic to neuroinflammatory disease is still unknown. Two clinical variants (cALD and AMN), caused by the same ABCD1 mutation, have different degrees of inflammation that affect different parts of the nervous system [42]. In cALD (the childhood/cerebral form of X-ALD), the neuroinflammatory disease affects the white matter and related cells in the CNS, whereas in AMN, the adult form of X-ALD, axonal tracks of the spinal cord are affected [43]. In addition, the neuropathology of cALD is associated with vascular immune cell infiltration and related pathologies [44, 45]. First, the accumulation of VLCFA differs in different organs with major accumulations in brain, adrenal cortex, and testis in X-ALD. Also, excessive accumulation of VLCFA was reported to differ in different cell types of the same organ such as brain [26, 33]. Recently, we reported that oligodendrocytes and astrocytes derived from induced pluripotent cells (IPSC) generated from cALD and AMN fibroblasts accumulate VLCFA; however, the levels of saturated VLCFAwere higher in cALD cells than in AMN cells. Second, cALD oligodendrocytes have higher VLCFA load compared to AMN oligodendrocytes. Third, ELOVL1 gene for synthesis of VLCFA was induced to a higher degree in cALD oligodendrocytes as compared to AMN oligodendrocytes and ELOVL1 mRNA expression appears to parallel the levels of VLCFA accumulation. Furthermore, cALD astrocytes expressed higher levels of proinflammatory cytokines than AMN astrocytes and control astrocytes with or without stimulation with lipopolysaccharides. These studies document that cell-specific induction of ELOVL1 as a result of ABCD1 deletion contributes to the differential load of VLCFA in different cell types and induction of inflammatory cytokines in response to VLCFA load in cALD vs. AMN [26, 29]
The observed differential load of VLCFA between cALD vs. AMN cells [26, 33]) suggests for a role of modifier genes or environmental/epigenetic stochastic factors in the clinical outcome of the disease. The epigenetic mechanisms, including DNA methylation and histone modification, not only regulate the expression of protein-encoding genes but also the expression of miRNAs from noncoding genes [46]. To understand the role of epigenetic modification under X-ALD conditions in the expression of miRNAs, we performed miRNA profiling with human skin fibroblasts from normal, cALD and AMN patients using human miRNome miScript miRNA PCR Array (Qiagen). Among 1008 miRNA profiled in triplicate, 11 miRNAs were found to be differentially regulated in cALD and AMN fibroblasts. These differentially regulated miRNAs are miR-103b, miR-106b, miR-135a, miR-142-3p, miR-142-5p, miR-152, miR-196a, miR-504, miR-708, miR-10b*, and miR-19a* (Fig. 1). Further, to explore the mechanisms by which these miRNAs contribute to X-ALD pathogenesis, we analyzed relationships of these differentially expressed miRNAs to target genes participating in X-ALD pathology using bioinformatics programs. Analysis of these databases suggests that these differentially expressed miRNAs may be involved in VLCFA synthesis (i.e., ELOVL1 and ELOVL6), catabolism (i.e., ABCD2 and ABCD3), and inflammatory signaling (NFκB).
Among these differentially expressed miRNAs between cALD and AMN, miR-196a is of specific interest because of its marked decreased expression in cALD (~82-fold as compared to control) and increased expression in AMN (~4-fold as compared to control). This pattern of expression was confirmed by qRT-PCR in fibroblast cell lines as well as in the brains of cALD and AMN patients (Fig. 2). Moreover, ABCD1-silenced astrocytes expressing proinflammatory cytokines and oligodendrocytes expressing proapototic molecules with cALD phenotype [27] had downregulated miR-196a expression as compared to unsilenced cells (Fig. 3) thus documenting a direct relationship between ABCD1 deletion induced VLCFA derangements and expression of miR-196a. The molecular basis for the differential expression of miR-196a between cALD and AMN is not understood at present.
Since one miRNA is known to regulate multiple gene products [47], the expression of miR-196a is also implicated in other disease conditions [48–51]. miR-196a was significantly downregulated in serum of patients with chronic hepatitis C (CHC), and thus, circulating miR-196a was identified as a specific and non-invasive candidate biomarker for the diagnosis of CHC [51]. Overexpression of miR-196a was reported to protect against liver injury in chronic hepatitis C virus infection [52]. Similarly, the serum levels of miR-196a were also significantly downregulated in localized scleroderma patients (LSc) and thus potentially a diagnostic marker for LSC [53]. miR-196a was shown to be upregulated in the mouse model of Spinal and bulbar muscular atrophy (SBMA) [24] and Huntington disease (HD) models and overexpression of miR-196a was shown to provide protection against the respective disease process [48–50]. The expression of miR-196a in cALD is downregulated and overexpression of its mimic corrected the ABCD1 deletion induced abnormalities in cALD cells, thus establishing cause and effect relationship between disease processes and expression of miR-196a in cALD.
Silencing of ABCD1 in astrocytes and oligodendrocytes increased the VLCFA load and administration of HDAC inhibitor corrected the VLCFA load as well as the expression of proinflammatory cytokines in ABCD1-silenced astrocytes and proapoptotic proteins in ABCD1-silenced oligodendrocytes [37]. The observed downregulation of the expression of miR-196a following silencing of ABCD1 in astrocytes and SAHA-mediated reversal/upregulation of miR-196a in ABCD1-silenced U87 cells (Fig. 4) indicate a role for miR-196a in regulation of cALD disease processes. The expressions of predicted inflammatory target genes of miR-196a including MAP4K3, MAP3K2, IKKα and IKKβ, and ELOVL1 expression were markedly increased in brain samples from cALD patients as compared to equivalent brain samples from control and AMN patients (Fig. 5). Accordingly, NFκB activation (phosphorylation of IκB and p65) was increased in the brains of cALD as compared to control and AMN groups (Fig. 6) which explain the increase in inflammation in cALD as compared to AMN. Further, miR-196a mimic transfection in ABCD1-silenced cells decreased the expression of IKKα and IKKβ in these cells (Fig. 7). These findings indicate that the observed decreased expression of miR-196a in ABCD1-silenced cells could be one of the pathological factors responsible for inflammatory reaction in the central nervous system of cALD. Interestingly, transfection of skin fibroblasts from cALD patients or ABCD1-silenced U87MG (astrocytic) cells or B12 (oligodendrocytes) cells with miR-196a mimic decreased the expression of its target gene, ELOVL1 (Fig. 8). Although ELOVL1 expression in skin fibroblasts of cALD patients and U87 astrocyte cells silenced for ABCD1 gene was not altered significantly as compared to control groups, but its expression was significantly increased in B12 oligodendrocyte cells. This increase was abrogated by transfection with miR-196a mimic documenting the role of miR-196a expression in altered ELOVL1 expression and thus VLCFA accumulation in cALD disease progression (Fig. 8).
In conclusion, studies using fibroblasts and brain samples from cALD and AMN patients and ABCD1-silenced oligodendrocytes and astrocytes document for the first time the differential expression of miR-196a in cALD and AMN. Although altered miR-196a expression is described in other disease conditions but the expression of miR-196a along with the quantification of VLCFA may potentially serve as a prenatal/postnatal diagnostic marker in differentiating between the fatal disease of cALD from adult disease of AMN. The differential expression of miR-196a and VLCFA metabolism as expression of ELOVL1 and inflammatory responses as activation of NF-κB and expression of proinflammatory cytokines documents a relationship to disease pathologies of cALD and AMN. Secondly, downregulation of miR-196a following silencing of ABCD1 (ALD gene) establishes a cause and effect relationship between ABCD1 loss associated cellular derangements and downregulation of miR-196a. The observed correction of miR-196a expression by SAHA, an agent previously reported to correct VLCFA load [42] and miR-196a mimic-induced correction of VLCFA metabolism as the expression of ELOVL1 and the inflammatory signaling pathway as NFκB indicate that targeting of these epigenetic mechanisms may potentially prove to be of therapeutic value in cALD.
Acknowledgments
The authors thank Drs Baarine Mauhammad, Je-Seong Won, and Mushfiquddin Khan for helpful discussion. We thank Dr Avtar K. Singh for brain sectioning. We also thank Dr Nishant Saxena and Ms Danielle Lowe for the help in figure formatting and Ms Joyce Bryan for procurement of chemicals. These studies were supported by grants (NS22576 and NS37766) from the National Institutes of Health, Bethesda, MD. This work was also supported by the National Institutes of Health Grants C06 RR018823 and C06 RR015455 from the Extramural Research Facilities Program of the National Center for Research Resources.
Footnotes
Compliance with ethical standards
Conflict of Interest The authors declare that they have no competing interests.
References
- 1.Berger J, Gartner J. X-linked adrenoleukodystrophy: clinical, biochemical and pathogenetic aspects. Biochim Biophys Acta. 2006;1763(12):1721–1732. doi: 10.1016/j.bbamcr.2006.07.010. doi:10.1016/j.bbamcr.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 2.Bezman L, Moser AB, Raymond GV, Rinaldo P, Watkins PA, Smith KD, Kass NE, Moser HW. Adrenoleukodystrophy: incidence, new mutation rate, and results of extended family screening. Ann Neurol. 2001;49(4):512–517. [PubMed] [Google Scholar]
- 3.Dubois-Dalcq M, Feigenbaum V, Aubourg P. The neurobiology of X-linked adrenoleukodystrophy: a demyelinating peroxisomal disorder. Trends Neurosci. 1999;22(1):4–12. doi: 10.1016/s0166-2236(98)01319-8. [DOI] [PubMed] [Google Scholar]
- 4.Singh I, Pujol A. Pathomechanisms underlying X-adrenoleukodystrophy: a three-hit hypothesis. Brain Pathol. 2010;20(4):838–844. doi: 10.1111/j.1750-3639.2010.00392.x. doi:10.1111/j.1750-3639.2010.00392.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moser HW, Loes DJ, Melhem ER, Raymond GV, Bezman L, Cox CS, Lu SE. X-linked adrenoleukodystrophy: overview and prognosis as a function of age and brain magnetic resonance imaging abnormality: a study involving 372 patients. Neuropediatrics. 2000;31(5):227–239. doi: 10.1055/s-2000-9236. [DOI] [PubMed] [Google Scholar]
- 6.Moser HW. Lorenzo oil therapy for adrenoleukodystrophy: a prematurely amplified hope. Ann Neurol. 1993;34(2):121–122. doi: 10.1002/ana.410340202. doi:10.1002/ana.410340202. [DOI] [PubMed] [Google Scholar]
- 7.Contreras M, Sengupta TK, Sheikh F, Aubourg P, Singh I. Topology of ATP-binding domain of adrenoleukodystrophy gene product in peroxisomes. Arch Biochem Biophys. 1996;334(2):369–379. doi: 10.1006/abbi.1996.0467. doi:10.1006/abbi.1996.0467. [DOI] [PubMed] [Google Scholar]
- 8.Contreras M, Mosser J, Mandel JL, Aubourg P, Singh I. The protein coded by the X-adrenoleukodystrophy gene is a peroxisomal integral membrane protein. FEBS Lett. 1994;344(2–3):211–215. doi: 10.1016/0014-5793(94)00400-5. [DOI] [PubMed] [Google Scholar]
- 9.Singh I, Moser AE, Goldfischer S, Moser HW. Lignoceric acid is oxidized in the peroxisome: implications for the Zellweger cerebro-hepato-renal syndrome and adrenoleukodystrophy. Proc Natl Acad Sci U S A. 1984;81(13):4203–4207. doi: 10.1073/pnas.81.13.4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shinnoh N, Yamada T, Yoshimura T, Furuya H, Yoshida Y, Suzuki Y, Shimozawa N, Orii T, Kobayashi T. Adrenoleukodystrophy: the restoration of peroxisomal β-Oxidaton by transfection of normal cDNA. Biochem Biophys Res Commun. 1995;210(3):830–836. doi: 10.1006/bbrc.1995.1733. doi:10.1006/bbrc.1995.1733. [DOI] [PubMed] [Google Scholar]
- 11.Lombard-Platet G, Savary S, Sarde CO, Mandel JL, Chimini G. A close relative of the adrenoleukodystrophy (ALD) gene codes for a peroxisomal protein with a specific expression pattern. Proc Natl Acad Sci U S A. 1996;93(3):1265–1269. doi: 10.1073/pnas.93.3.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holzinger A, Kammerer S, Berger J, Roscher AA. cDNA cloning and mRNA expression of the human adrenoleukodystrophy related protein (ALDRP), a peroxisomal ABC transporter. Biochem Biophys Res Commun. 1997;239(1):261–264. doi: 10.1006/bbrc.1997.7391. doi:10.1006/bbrc.1997.7391. [DOI] [PubMed] [Google Scholar]
- 13.Liu LX, Janvier K, Berteaux-Lecellier V, Cartier N, Benarous R, Aubourg P. Homo- and heterodimerization of peroxisomal ATP-binding cassette half-transporters. J Biol Chem. 1999;274(46):32738–32743. doi: 10.1074/jbc.274.46.32738. [DOI] [PubMed] [Google Scholar]
- 14.Tsuji S, Sano T, Ariga T, Miyatake T. Increased synthesis of hexacosanoic acid (C23:0) by cultured skin fibroblasts from patients with adrenoleukodystrophy (ALD) and adrenomyeloneuropathy (AMN) J Biochem. 1981;90(4):1233–1236. doi: 10.1093/oxfordjournals.jbchem.a133578. [DOI] [PubMed] [Google Scholar]
- 15.Kemp S, Valianpour F, Denis S, Ofman R, Sanders RJ, Mooyer P, Barth PG, Wanders RJ. Elongation of very long-chain fatty acids is enhanced in X-linked adrenoleukodystrophy. Mol Genet Metab. 2005;84(2):144–151. doi: 10.1016/j.ymgme.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 16.Koike R, Tsuji S, Ohno T, Suzuki Y, Orii T, Miyatake T. Physiological significance of fatty acid elongation system in adrenoleukodystrophy. J Neurol Sci. 1991;103(2):188–194. doi: 10.1016/0022-510x(91)90163-2. [DOI] [PubMed] [Google Scholar]
- 17.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 18.Kim J, Inoue K, Ishii J, Vanti WB, Voronov SV, Murchison E, Hannon G, Abeliovich A. A MicroRNA feedback circuit in midbrain dopamine neurons. Science. 2007;317(5842):1220–1224. doi: 10.1126/science.1140481. doi: 10.1126/science.1140481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karres JS, Hilgers V, Carrera I, Treisman J, Cohen SM. The conserved microRNA miR-8 tunes atrophin levels to prevent neurodegeneration in Drosophila. Cell. 2007;131(1):136–145. doi: 10.1016/j.cell.2007.09.020. doi:10.1016/j.cell.2007.09.020. [DOI] [PubMed] [Google Scholar]
- 20.Lee Y, Samaco RC, Gatchel JR, Thaller C, Orr HT, Zoghbi HY. miR-19, miR-101 and miR-130 co-regulate ATXN1 levels to potentially modulate SCA1 pathogenesis. Nat Neurosci. 2008;11(10):1137–1139. doi: 10.1038/nn.2183. doi:10.1038/nn.2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams AH, Valdez G, Moresi V, Qi X, McAnally J, Elliott JL, Bassel-Duby R, Sanes JR, Olson EN. MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science. 2009;326(5959):1549–1554. doi: 10.1126/science.1181046. doi:10.1126/science.1181046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bilen J, Liu N, Burnett BG, Pittman RN, Bonini NM. MicroRNA pathways modulate polyglutamine-induced neurode-generation. Mol Cell. 2006;24(1):157–163. doi: 10.1016/j.molcel.2006.07.030. doi:10.1016/j.molcel.2006.07.030. [DOI] [PubMed] [Google Scholar]
- 23.Schaefer A, O'Carroll D, Tan CL, Hillman D, Sugimori M, Llinas R, Greengard P. Cerebellar neurodegeneration in the absence of microRNAs. J Exp Med. 2007;204(7):1553–1558. doi: 10.1084/jem.20070823. doi:10.1084/jem.20070823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miyazaki Y, Adachi H, Katsuno M, Minamiyama M, Jiang YM, Huang Z, Doi H, Matsumoto S, Kondo N, Iida M, Tohnai G, Tanaka F, Muramatsu S, Sobue G. Viral delivery of miR-196a ameliorates the SBMA phenotype via the silencing of CELF2. Nat Med. 2012;18(7):1136–1141. doi: 10.1038/nm.2791. doi:10.1038/nm.2791. [DOI] [PubMed] [Google Scholar]
- 25.Lu M, Zhang Q, Deng M, Miao J, Guo Y, Gao W, Cui Q. An analysis of human microRNA and disease associations. PLoS One. 2008;3(10):e3420. doi: 10.1371/journal.pone.0003420. doi:10.1371/journal.pone.0003420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baarine M, Khan M, Singh A, Singh I. Functional characterization of IPSC-derived brain cells as a model for X-linked adrenoleukodystrophy. PLoS One. 2015;10(11):e0143238. doi: 10.1371/journal.pone.0143238. doi:10.1371/journal.pone.0143238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh J, Khan M, Pujol A, Baarine M, Singh I. Histone deacetylase inhibitor upregulates peroxisomalfatty acid oxidation and inhibits apoptotic cell death inabcd1-deficient glial cells. PLoS One. 2013;8(7):e70712. doi: 10.1371/journal.pone.0070712. doi:10.1371/journal.pone.0070712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh J, Khan M, Singh I. Silencing of Abcd1 and Abcd2 genes sensitizes astrocytes for inflammation: implication for X-adrenoleukodystrophy. J Lipid Res. 2009;50(1):135–147. doi: 10.1194/jlr.M800321-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baarine M, Beeson C, Singh A, Singh I. ABCD1 deletion-induced mitochondrial dysfunction is corrected by SAHA: implication for adrenoleukodystrophy. J Neurochem. 2014 doi: 10.1111/jnc.12992. doi:10.1111/jnc.12992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moser AB, Kreiter N, Bezman L, Lu S, Raymond GV, Naidu S, Moser HW. Plasma very long chain fatty acids in 3,000 peroxisome disease patients and 29,000 controls. Ann Neurol. 1999;45(1):100–110. doi: 10.1002/1531-8249(199901)45:1<100::aid-art16>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 31.Ofman R, Dijkstra IM, van Roermund CW, Burger N, Turkenburg M, van Cruchten A, van Engen CE, Wanders RJ, Kemp S. The role of ELOVL1 in very long-chain fatty acid homeostasis and X-linked adrenoleukodystrophy. EMBO Mol Med. 2010;2(3):90–97. doi: 10.1002/emmm.201000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gottlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG, Heinzel T. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001;20(24):6969–6978. doi: 10.1093/emboj/20.24.6969. doi:10.1093/emboj/20.24.6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khan M, Singh J, Gilg A, Uto T, Singh I. Very long-chain fatty acid accumulation causes lipotoxic response via 5-lipoxygenase in cerebral adrenoleukodystrophy. J Lipid Res. 2010;51(7):1685–1695. doi: 10.1194/jlr.M002329. doi:10.1194/jlr.M002329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science. 1995;267(5203):1485–1488. doi: 10.1126/science.7878466. [DOI] [PubMed] [Google Scholar]
- 35.Finco T, Beg A, Baldwin AJ. Inducible phosphorylation of I kappa B alpha is not sufficient for its dissociation from NF-kappa B and is inhibited by protease inhibitors. Proc Natl Acad Sci U S A. 1994;91(25):11884–11888. doi: 10.1073/pnas.91.25.11884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tvrdik P, Westerberg R, Silve S, Asadi A, Jakobsson A, Cannon B, Loison G, Jacobsson A. Role of a new mammalian gene family in the biosynthesis of very long chain fatty acids and sphingolipids. J Cell Biol. 2000;149(3):707–717. doi: 10.1083/jcb.149.3.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singh J, Khan M, Singh I. HDAC inhibitor SAHA normalizes the levels of VLCFAs in human skin fibroblasts from X-ALD patients and downregulates the expression of proinflammatory cytokines in Abcd1/2-silenced mouse astrocytes. J Lipid Res. 2011;52(11):2056–2069. doi: 10.1194/jlr.M017491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shafi G, Aliya N, Munshi A. MicroRNA signatures in neurological disorders. Can J Neurol Sci. 2010;37(2):177–185. doi: 10.1017/s0317167100009902. [DOI] [PubMed] [Google Scholar]
- 39.Su W, Aloi MS, Garden GA. MicroRNAs mediating CNS inflammation: small regulators with powerful potential. Brain Behav Immun. 2016;52:1–8. doi: 10.1016/j.bbi.2015.07.003. doi:10.1016/j.bbi.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moser HW, Loes DJ, Melhem ER, Raymond GV, Bezman L, Cox CS, Lu SE. X-Linked adrenoleukodystrophy: overview and prognosis as a function of age and brain magnetic resonance imaging abnormality. A study involving 372 patients. Neuropediatrics. 2000;31(5):227–39. doi: 10.1055/s-2000-9236. doi:10.1055/s-2000-9236. [DOI] [PubMed] [Google Scholar]
- 41.Sutovsky S, Kolnikova M, Petrovic R, Kollar B, Siarnik P, Chandoga J, Fischerova M, Turcani P. Differing clinical presentations of two unrelated cases of X-linked adrenoleukodystrophy with identical mutation Y296C in the ABCD1 gene. Neuro Endocrinol Lett. 2014;35(5):411–416. [PubMed] [Google Scholar]
- 42.Moser HW. Therapy of X-linked adrenoleukodystrophy. NeuroRx. 2006;3(2):246–253. doi: 10.1016/j.nurx.2006.01.004. doi:10.1016/j.nurx.2006.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Powers JM, Moser HW. Peroxisomal disorders: genotype, phenotype, major neuropathologic lesions, and pathogenesis. Brain Pathol. 1998;8(1):101–120. doi: 10.1111/j.1750-3639.1998.tb00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gilg AG, Singh AK, Singh I. Inducible nitric oxide synthase in the central nervous system of patients with X-adrenoleukodystrophy. J Neuropathol Exp Neurol. 2000;59(12):1063–1069. doi: 10.1093/jnen/59.12.1063. [DOI] [PubMed] [Google Scholar]
- 45.Powers JM, Pei Z, Heinzer AK, Deering R, Moser AB, Moser HW, Watkins PA, Smith KD. Adreno-leukodystrophy: oxidative stress of mice and men. J Neuropathol Exp Neurol. 2005;64(12):1067–1079. doi: 10.1097/01.jnen.0000190064.28559.a4. [DOI] [PubMed] [Google Scholar]
- 46.Sato F, Tsuchiya S, Meltzer SJ, Shimizu K. MicroRNAs and epigenetics. FEBS J. 2011;278(10):1598–1609. doi: 10.1111/j.1742-4658.2011.08089.x. doi:10.1111/j.1742-4658.2011.08089.x. [DOI] [PubMed] [Google Scholar]
- 47.Thomson DW, Bracken CP, Goodall GJ. Experimental strategies for microRNA target identification. Nucleic Acids Res. 2011;39(16):6845–6853. doi: 10.1093/nar/gkr330. doi:10.1093/nar/gkr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Packer A, Xing Y, Harper S, Jones L, Davidson B. The bifunctional microRNA miR-9/miR-9* regulates REST and CoREST and is downregulated in Huntington's disease. J Neurosci. 2008;28(53):14341–14346. doi: 10.1523/JNEUROSCI.2390-08.2008. doi:10.1523/JNEUROSCI.2390-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gaughwin P, Ciesla M, Lahiri N, Tabrizi S, Brundin P, Björkqvist M. Hsa-miR-34b is a plasma-stable microRNA that is elevated in pre-manifest Huntington's disease. Hum Mol Genet. 2011;20(11):2225–2237. doi: 10.1093/hmg/ddr111. doi:10.1093/hmg/ddr111. [DOI] [PubMed] [Google Scholar]
- 50.Cheng PH, Li CL, Chang YF, Tsai SJ, Lai YY, Chan AW, Chen CM, Yang SH. miR-196a ameliorates phenotypes of Huntington disease in cell, transgenic mouse, and induced pluripotent stem cell models. Am J Hum Genet. 2013;93(2):306–312. doi: 10.1016/j.ajhg.2013.05.025. doi:10.1016/j.ajhg.2013.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu B, Xiang Y, Zhang H. Circulating microRNA-196a as a candidate diagnostic biomarker for chronic hepatitis C. Mol Med Rep. 2015;12(1):105–110. doi: 10.3892/mmr.2015.3386. doi:10.3892/mmr.2015.3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hou W, Tian Q, Zheng J, Bonkovsky HL. MicroRNA-196 represses Bach1 protein and hepatitis C virus gene expression in human hepatoma cells expressing hepatitis C viral proteins. Hepatology. 2010;51(5):1494–1504. doi: 10.1002/hep.23401. doi:10.1002/hep.23401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Makino T, Jinnin M, Etoh M, Yamane K, Kajihara I, Makino K, Ichihara A, Igata T, Sakai K, Fukushima S, Ihn H. Down-regulation of microRNA-196a in the sera and involved skin of localized scleroderma patients. Eur J Dermatol: EJD. 2014;24(4):470–476. doi: 10.1684/ejd.2014.2384. doi:10.1684/ejd.2014.2384. [DOI] [PubMed] [Google Scholar]