

The 2.4 Å resolution crystal structure of human mitochondrial TRAP1NM is reported, which together with epitope-mapping results supports a role for the M-domain in mediating the specific interaction with mitochondrial Hsp70 that is required for TRAP1-dependent protein folding.
Keywords: Hsp90 paralog, TRAP1, molecular chaperones, mitochondrial matrix
Abstract
TRAP1 is an organelle-specific Hsp90 paralog that is essential for neoplastic growth. As a member of the Hsp90 family, TRAP1 is presumed to be a general chaperone facilitating the late-stage folding of Hsp90 client proteins in the mitochondrial matrix. Interestingly, TRAP1 cannot replace cytosolic Hsp90 in protein folding, and none of the known Hsp90 co-chaperones are found in mitochondria. Thus, the three-dimensional structure of TRAP1 must feature regulatory elements that are essential to the ATPase activity and chaperone function of TRAP1. Here, the crystal structure of a human TRAP1NM dimer is presented, featuring an intact N-domain and M-domain structure, bound to adenosine 5′-β,γ-imidotriphosphate (ADPNP). The crystal structure together with epitope-mapping results shows that the TRAP1 M-domain loop 1 contacts the neighboring subunit and forms a previously unobserved third dimer interface that mediates the specific interaction with mitochondrial Hsp70.
1. Introduction
Tumor necrosis factor receptor-associated protein 1 (TRAP1) is a mitochondria-specific Hsp90 homolog that is found in metazoans and some protozoans but is absent in yeast. It is widely presumed that TRAP1 is a general chaperone that facilitates the late-stage folding and maturation of Hsp90 client proteins in the mitochondrial matrix, although the physiological function of TRAP1 remains poorly understood (Rasola et al., 2014 ▸). Interestingly, it has been reported that TRAP1 is widely expressed in many tumors (Kang et al., 2007 ▸; Leav et al., 2010 ▸; Sciacovelli et al., 2013 ▸) but not in mitochondria of most normal tissues (Kang et al., 2007 ▸) or highly proliferating, nontransformed cells (Sciacovelli et al., 2013 ▸), underscoring the potential of TRAP1 as an anticancer drug target (Lee et al., 2015 ▸).
At the molecular level, TRAP1 is a multi-domain protein consisting of an N-terminal ATP-binding domain (N-domain), a middle domain (M-domain) and a C-terminal dimerization domain (C-domain). However, TRAP1 lacks both the charged linker and the C-terminal MEEVD motif of eukaryotic cytosolic Hsp90. Instead, TRAP1 is preceded by a mitochondria-targeting signal peptide that is cleaved off during import (Felts et al., 2000 ▸). Crystal structures of the mature form of zebrafish TRAP1 with ADPNP or in complex with different transition-state mimics have been reported (Lavery et al., 2014 ▸). All of these structures show an N-terminally intertwined, closed dimer that resembles the X-ray structure of full-length yeast Hsp90–ADPNP but without the bound p23/Sba1 co-chaperone (Ali et al., 2006 ▸), confirming that p23 binding is not required for dimer closure. More recently, a 3.3 Å resolution crystal structure of the human TRAP1 NM-domain (TRAP1NM) bound to ADPNP has also been reported (Lee et al., 2015 ▸). Although the overall structure and domain organization of human and zebrafish TRAP1NM are very similar, the three-dimensional structure of the TRAP1 M-domain is either incomplete or partially disordered in the previously reported crystal structures (Lavery et al., 2014 ▸; Lee et al., 2015 ▸), leaving the functional importance of the conserved M-domain loops unclear.
Here, we present the 2.4 Å resolution crystal structure of a closed-state human TRAP1NM–ADPNP dimer determined from a new crystal form. Although the overall structure is very similar to the previously reported crystal structure of human TRAP1NM (Lee et al., 2015 ▸), the present structure is complete, of higher resolution and better refined. Notably, we find that the conserved M-domain loop 1 contacts the neighboring subunit, enabling intersubunit signalling in the closed-state TRAP1 dimer conformation. Finally, we demonstrate using peptide-array technology that the M-domain loop 1 presents a protein–protein binding site that facilitates specific interaction with mitochondrial Hsp70, which is essential for TRAP1-dependent protein folding.
2. Materials and methods
2.1. Macromolecule production
Human TRAP1NM (residues 60–554) was cloned by PCR from a human TRAP1 cDNA clone into the pProEx HTb bacterial expression vector. The resulting plasmid (pTRAP1NM) was transformed into chemically competent Escherichia coli BL21-Codon Plus (DE3)-RIL cells (Stratagene) and grown at 310 K to an OD600 of ∼0.6 in lysogeny broth (LB) supplemented with 100 µg ml−1 ampicillin and 34 µg ml−1 chloramphenicol. The cells were induced with 0.4 mM isopropyl β-d-1-thiogalactopyranoside to overexpress TRAP1NM with a TEV protease-cleavable N-terminal His6 tag and growth was continued at 289 K for a further 16 h before harvesting.
The cell pellet was resuspended in buffer A (40 mM Tris–HCl pH 7.5, 300 mM KCl, 6 mM β-mercaptoethanol) and lysed using an M-110Y cell disrupter (Microfluidics). The cleared lysate was loaded onto a pre-equilibrated Ni Sepharose High Performance column (GE Healthcare) and was washed with buffer A containing 30 mM imidazole. Bound His6-(TEV)-TRAP1NM was eluted using a linear gradient from 30 to 500 mM imidazole in buffer A. Peak fractions were pooled, mixed with His6-TEV protease and dialyzed overnight against 25 mM Tris–HCl pH 8.0, 150 mM NaCl, 6 mM β-mercaptoethanol. The liberated His6 tag and His6-TEV protease were removed by passing the sample over a 5 ml Ni Sepharose High Performance column in negative binding mode, with TRAP1NM preceded by a Gly-Ala-Met-Gly-Ser leader peptide in the flowthrough. Ammonium sulfate was added to the flowthrough to a final concentration of 0.5 M, followed by loading the sample onto a pre-equilibrated Toyopearl Butyl-650S (Tosoh Bioscience) column and washing the column with 50 mM Tris–HCl pH 8.0, 6 mM β-mercaptoethanol, 0.5 M ammonium sulfate. Bound TRAP1NM was eluted with the same buffer using a linear gradient of 0.5–0.0 M ammonium sulfate and dialyzed against 25 mM Tris–HCl pH 8.0, 1 mM tris(2-carboxyethyl)phosphine (TCEP). The protein concentration was estimated by the method of Gill & von Hippel (1989 ▸), using a calculated molar extinction coefficient of 51 800 M −1 cm−1 for human TRAP1NM.
The overexpression and purification of human TRAP1, of the mitochondrial Hsp70 system consisting of human mortalin, yeast Mdj1 and yeast Mge1 (MJE), of human His6-Hsp70ΔC and of the E. coli Hsp70 system consisting of DnaK, DnaJ and GrpE (KJE) have been described previously (Sung et al., 2016 ▸; Lee et al., 2013 ▸; Sielaff & Tsai, 2010 ▸).
2.2. Crystallization
TRAP1NM was crystallized by the vapor-diffusion method at 287 K using VDX plates. TRAP1NM protein (0.44 mM) in 25 mM Tris–HCl pH 8.0, 1 mM TCEP was mixed with 5 mM ADPNP and 10 mM MgCl2 and incubated for 30 min on ice. Hanging drops were set up by mixing the protein sample with an equal volume (1.5 µl) of reservoir solution consisting of 1.8 M ammonium sulfate, 0.1 M MES pH 6.3, 1% dioxane. Crystals reached maximum dimensions of 600 × 500 × 200 µm after three weeks, were harvested in reservoir solution supplemented with 25% glycerol and were flash-cooled in liquid nitrogen.
2.3. Data collection and processing
A complete data set was collected from a single crystal on the SBC 19-ID beamline of the Advanced Photon Source, Argonne, Illinois, USA (Table 1 ▸). All data were processed using the HKL-3000 software suite (Minor et al., 2006 ▸).
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Wavelength (Å) | 0.97929 |
| Temperature (K) | 93 |
| Detector | ADSC Q315r |
| Space group | P212121 |
| a, b, c (Å) | 93.437, 104.946, 156.909 |
| α, β, γ (°) | 90.0, 90.0, 90.0 |
| Resolution range (Å) | 49.76–2.43 (2.47–2.43) |
| Total No. of reflections | 110286 |
| No. of unique reflections | 58241 |
| Completeness (%) | 99.0 (90.7) |
| Multiplicity | 6.8 (5.1) |
| 〈I/σ(I)〉 | 10.60 (1.49) |
| R merge † | 0.038 |
| Overall B factor from Wilson plot (Å2) | 59.5 |
| Structure refinement | |
| Resolution range (Å) | 49.76–2.43 (2.49–2.43) |
| Completeness (%) | 98.6 (87.0) |
| No. of reflections, working set | 56114 |
| No. of reflections, test set | 1998 |
| Final R cryst ‡ | 0.1951 (0.3339) |
| Final R free ‡ | 0.2379 (0.3871) |
| No. of non-H atoms | |
| Protein | 7648 |
| Ion | 2 |
| Ligand | 106 |
| Water | 135 |
| Total | 7891 |
| R.m.s. deviations | |
| Bonds (Å) | 0.007 |
| Angles (°) | 1.035 |
| Average B factors (Å2) | |
| Protein | 80.9 |
| Ion | 60.5 |
| Ligand | 74.1 |
| Water | 72.4 |
| All atoms | 80.0 |
| Ramachandran plot | |
| Most favored (%) | 97.2 |
| Allowed (%) | 2.8 |
| Outliers (%) | 0.0 |
R
merge = 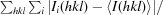
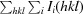 , where 〈I(hkl)〉 is the mean of i observations I
i(hkl) of reflection hkl.
, where 〈I(hkl)〉 is the mean of i observations I
i(hkl) of reflection hkl.
R
cryst and R
free = 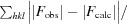
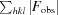 , where F
obs and F
calc are the observed and calculated structure factors, respectively, calculated for recorded data (R
cryst) and for 3.4% of the data omitted in refinement (R
free).
, where F
obs and F
calc are the observed and calculated structure factors, respectively, calculated for recorded data (R
cryst) and for 3.4% of the data omitted in refinement (R
free).
2.4. Structure solution and refinement
The crystal structure of TRAP1NM–ADPNP was determined by molecular replacement using Phaser (McCoy, 2007 ▸), with the crystal structure of zebrafish TRAP1 (residues 85–566; PDB entry 4ipe, chain A; Lavery et al., 2014 ▸) as the search model. 3.4% of the data were excluded from refinement for cross-validation purposes. Structure refinement using PHENIX (Adams et al., 2002 ▸) and REFMAC5 (Murshudov et al., 2011 ▸) was interspersed with several rounds of manual model building in Coot (Emsley & Cowtan, 2004 ▸). Water molecules were fitted automatically. The refined structure has excellent stereochemical properties, with none of the residues in generously allowed or disallowed regions of the Ramachandran plot (Table 1 ▸).
2.5. Biolayer interferometry
Biolayer interferometry was used to examine TRAP1–mortalin interaction and was performed with an Octet RED96 instrument (ForteBio). Biotinylation of TRAP1 and BSA, respectively, was carried out using the EZ-link NHC-LC-LC-biotin labeling kit (Thermo Scientific) in 25 mM potassium phosphate pH 6.5, 100 mM KCl, 1 mM dithiothreitol (DTT) for 24 h at 277 K to preferentially label the N-terminal α-amino group (Sélo et al., 1996 ▸). Biotinylated protein was immobilized onto streptavidin-coated biosensors (ForteBio) at a concentration of 0.5 µM in binding buffer (25 mM Tris–HCl pH 8.0, 100 mM KCl, 1 mg ml−1 BSA, 1 mM DTT) followed by blocking of the biosensors with 0.5 µM biotinylated BSA in binding buffer. TRAP1–mortalin and BSA–mortalin binding curves were obtained at 303 K at different mortalin concentrations in binding buffer plus buffer blanks.
2.6. Peptide-array synthesis and analysis
Miniaturized peptide libraries immobilized on a cellulose support membrane were synthesized by standard Fmoc chemistry using an ASP222 autospot robot (Intavis AG), essentially as described previously (Rees et al., 2006 ▸; Sielaff et al., 2011 ▸; Lee et al., 2013 ▸). For probing with human mortalin, a peptide array consisting of 197 overlapping 12-mer peptides was generated by walking through the human TRAP1 sequence (UniProtKB Q12931), advancing three to four amino acids at each position, in addition to several binding control peptides. The membrane was blocked with 1× SuperBlock (Pierce) in TBS buffer and was probed at 295 K for 1 h with 0.5 µM purified human His6-mortalin and 3 mM ATP in TBS buffer containing 10% SuperBlock and 5% sucrose. The membrane was washed three times for 10 min in TBS buffer with 50 µM ATP. Protein binding was detected by probing the membrane directly with an anti-His6 monoclonal antibody conjugated to horseradish peroxidase (mAB-HRP; BD Bioscience).
For probing with human cytosolic Hsp70, a second human TRAP1 peptide array consisting of 161 overlapping 12-mer peptides was generated together with binding control peptides and probed as described above but with 1 µM His6-Hsp70ΔC. Peptides that were recognized by both His6-mortalin and His6-Hsp70ΔC were eliminated to derive mortalin-specific binding motifs.
2.7. TRAP1 chaperone assay
Chaperone activity was measured by monitoring the recovery of heat-denatured firefly luciferase (FFL; Promega) using a coupled-chaperone assay (Sung et al., 2016 ▸) consisting of human cytosolic Hsp90 (Hsp90), mitochondrial Hsp90 (TRAP1) or bacterial Hsp90 (HtpG) and untreated rabbit reticulocyte lysate (RRL; Promega), the mitochondrial Hsp70 system (MJE) or the bacterial Hsp70 system (KJE). Briefly, 160 nM FFL was mixed with 20 µM TRAP1, Hsp90 or HtpG in the presence of 5 mM ATP in 30 mM Tris–HCl pH 7.5, 2 mM DTT and heat-denatured for 5 min at 318 K. Samples were cooled on ice for 5 min and diluted tenfold in refolding buffer (25 mM HEPES pH 7.5, 50 mM KCl, 5 mM ATP, 5 mM MgCl2, 2 mM DTT) supplemented with 50% RRL, 4 µM MJE or 4 µM KJE. The recovery of FFL activity was measured at 303 K after 120 min using an LS55 fluorescence spectrophotometer (Perkin Elmer).
3. Results
3.1. Overall structure of the TRAP1NM–ADPNP dimer
To provide an accurate understanding of the mitochondrial proteostasis network, in order that this information might be exploited to develop new anticancer drugs, we wished to determine the crystal structure of human TRAP1 in both unliganded and nucleotide-bound states. Because our attempts to crystallize the mature protein were unsuccessful, we adopted a divide-and-conquer approach to determine crystal structures of human TRAP1 fragments that are more amenable to high-resolution structural studies.
Here, we present the 2.4 Å resolution crystal structure of a human TRAP1NM–ADPNP complex in an orthorhombic P212121 crystal form. The structure was determined by molecular replacement, which yielded a clear solution with one TRAP1NM dimer in the crystallographic asymmetric unit. As observed in previous crystal structures of TRAP1NM–ADPNP complexes (Lavery et al., 2014 ▸; Lee et al., 2015 ▸), TRAP1NM–ADPNP crystallized as an N-terminally intertwined, closed-state dimer with each subunit bound to ADPNP (Fig. 1 ▸ a). The overall structure of the TRAP1NM–ADPNP dimer resembles those of other GHL (Gyrase, Hsp90, MutL) ATPases in the ATP-bound state (Wigley et al., 1991 ▸; Ban et al., 1999 ▸; Ali et al., 2006 ▸; Lavery et al., 2014 ▸; Lee et al., 2015 ▸) and occludes 3867 Å2 of solvent-accessible area as calculated with PISA (Krissinel & Henrick, 2007 ▸). The two TRAP1NM monomers of the dimer superimpose with an r.m.s.d. of only 0.60 Å2 over 424 out of 482 Cα atoms. The 2F o − F c map is of excellent quality and enabled tracing of all but the first ten residues of TRAP1NM (Fig. 2 ▸). Notably, the final structure includes both M-domain loops 1 and 2. Loop 1 was disordered in previous structures (Lavery et al., 2014 ▸; Lee et al., 2015 ▸) and therefore could not be modeled. We note that the region comprising residues 353–360 of molecule A has high B factors (206.4 Å2 on average) compared with the average for the protein (80.9 Å2), indicating intrinsic flexibility. In contrast, the electron density for molecule B is well defined (average B factor of 95.8 Å2) and enabled tracing of the complete M-domain loops. The final structure was refined to an R cryst of 19.5% (R free of 23.8%) with excellent stereochemistry (Table 1 ▸). Thus, the present structure of human TRAP1NM is of higher resolution, is more complete and is better refined than the previously reported structure of human TRAP1NM (Lee et al., 2015 ▸).
Figure 1.

Crystal structure of human TRAP1NM bound to ADPNP. (a) Ribbon diagram depicting the TRAP1NM dimer with ADPNP shown as a CPK model. Each subunit is colored differently. Key structural elements are labeled. (b) Enlarged view of the N-terminal ATP-binding pocket of one TRAP1NM subunit with bound ADPNP (stick model). Ordered water molecules are shown as red spheres and the bound Mg2+ ion as a yellow sphere. (c) Enlarged view of the bound ADPNP molecule with the ATP-lid colored red.
Figure 2.

Stereoview of a section of the simulated-annealed composite OMIT map contoured at the 1.5σ level. The figure shows the ATP-binding pocket of TRAP1NM with bound ADPNP. The bound Mg2+ ion is shown as a green sphere.
3.2. Structure of the N-terminal ATP-binding domain of TRAP1
The N-domain shares the Bergerat fold common to Hsp90 chaperones and consists of a two-layer α/β sandwich composed of an eight-stranded mixed β-sheet and a segregated layer of five α-helices on one side (Fig. 3 ▸ and Supplementary Fig. S1). The Bergerat fold is shared with DNA gyrase B (Wigley et al., 1991 ▸; Tsai et al., 1996 ▸), MutL (Ban et al., 1999 ▸) and the catalytic domains of sensor kinases such as PhoQ (Guarnieri et al., 2008 ▸), and is characterized by several conserved motifs (Figs. 1 ▸ b, 1 ▸ c and 3 ▸). Motif I (Glu115) serves as the catalytic glutamate that activates a water molecule for in-line attack on the γ-phosphate of ATP. The bound nucleotide has a compact conformation, with the triphosphate moiety being ‘curled’ as a result of the α,β,γ-tridentate coordination to Mg2+, which is further coordinated by the side chain of Asn119 and a water molecule (Fig. 1 ▸ b). The sole base-specific interaction is formed between the carboxyl side chain of the evolutionarily conserved Asp158 (motif II) and the N6 amino group of the adenine ring, which together with Gly162 confers specificity for adenine over guanine nucleotides. The ATP-lid (residues 177–202) is folded over the nucleotide-binding pocket and features a C-terminal glycine-rich region (motif III) which clamps the γ-phosphate of the bound ADPNP (Fig. 1 ▸ c). Finally, Thr251 (motif IV), which is adjacent to motif II, makes an additional, water-mediated contact with the N1 site of adenine. Strikingly, only few direct contacts between the ATP-lid and the bound nucleotide are observed, which include interactions between the side chain of Ser178 and the β-phosphate and between the main-chain amides of Gly179 and Ser180 and the 3′-hydroxyl of the ribose sugar. The lack of specific interactions may suggest that lid closure is largely driven by steric interference resulting from dimer closure as opposed to nucleotide binding.
Figure 3.

Topology diagram of TRAP1NM. Secondary-structure elements with residue numbers are shown as blue cylinders (α-helices) or orange arrows (β-strands). Motifs that define the Bergerat fold are indicated by green symbols and roman numerals.
The most notable structural feature is the N-strap that extends the β-strand swap of GHL ATPases and straddles the N-domain of the neighboring subunit (Figs. 1 ▸ a and 3 ▸). Interestingly, deletion of the N-strap (TRAP1Δ84) resulted in a ∼25-fold increase in ATPase activity (Partridge et al., 2014 ▸; Sung et al., 2016 ▸), suggesting that the N-strap negatively regulates the ATPase activity of TRAP1.
3.3. Structure of the TRAP1 M-domain
Similar to cytosolic Hsp90, the TRAP1 M-domain can be divided into three segments (Fig. 3 ▸ and Supplementary Fig. S1). The first segment consists of an α/β sandwich composed of a mixed, five-stranded β-sheet flanked by α-helices and interspersed with two intervening loop regions, termed loop 1 and loop 2 (Figs. 1 ▸ a and 3 ▸). The second segment consists of three short α-helices and leads to a C-terminal α/β segment unique to Hsp90 chaperones (Meyer et al., 2003 ▸).
The conserved Arg402 residue on loop 2 functions as the ATP sensor (motif V), which senses the presence of the γ-phosphate of the cis-bound nucleotide (Figs. 1 ▸ b and 1 ▸ c), as previously shown for other Hsp90 chaperones (Cunningham et al., 2012 ▸). In addition, loop 2 residues (Asn399 and Gln406) form hydrogen-bond interactions with the main chain of an intervening loop that spans helices 1 and 2 in the N-domain of the neighboring subunit (residues 103–106), which stabilize the closed-state dimer conformation. However, as in all Hsp90 structures, no direct contacts between the cis subunit and the nucleotide bound to the trans subunit are observed (Jeng et al., 2015 ▸).
Unexpectedly, we find that loop 1, which was disordered in previously reported TRAP1 structures, contacts the M-domain from the neighboring subunit, burying an additional 690 Å2 of solvent-occluded surface area and providing a surface for other proteins to bind. Taken together, our findings suggest that the M-domain contributes to dimer formation, supporting a role for loop 1 of the M-domain in intersubunit communication and perhaps in protein–protein interaction.
3.4. M-domain loop 1 mediates the specific interaction with mitochondrial Hsp70
Using heat-aggregated FFL as a model substrate, we found that active folding by Hsp90 chaperones requires functional cooperation between Hsp90 and the cognate Hsp70 system (Fig. 4 ▸). No substrate recovery was observed either with TRAP1 alone (TRAP1/−) or when TRAP1 was omitted (−/MJE) (Fig. 4 ▸, right). Similar results were also obtained with both eukaryotic and prokaryotic cytosolic Hsp90 (Fig. 4 ▸, left and middle). Interestingly, we previously showed that the mitochondrial MJE system could not be replaced by rabbit reticulocyte lysate (RRL), which provides a rich source of cytosolic chaperones but is devoid of mitochondrial chaperones (Sung et al., 2016 ▸). Together, these findings support a direct, physical interaction between human TRAP1 and one or more components of the mitochondrial Hsp70 system, with human TRAP1 and mortalin (mtHsp70) forming a binary complex, as confirmed by biolayer interferometry (Fig. 5 ▸).
Figure 4.

Hsp90 chaperones (Hsp90, HtpG and TRAP1) require the cognate Hsp70 system (RRL, KJE and MJE) for active protein folding. Recovery of heat-denatured FFL is shown in the absence of chaperones (−/−), with Hsp90 chaperones only, with the Hsp70 system only and with Hsp90 together with the cognate Hsp70 system. Recovered FFL activities are expressed relative to the corresponding bi-chaperone system. Averages of three independent measurement ± SD are shown.
Figure 5.

TRAP1 and mortalin interact directly as determined by biolayer interferometry. The binding of mortalin to immobilized biotinylated TRAP1 (blue) and biotinylated BSA (gray), respectively, was measured by light distance shift (nm). Binding curves for different mortalin concentrations (30 and 60 µM) are shown in different hues.
To map the mortalin-binding sites in TRAP1, we synthesized miniaturized arrays of overlapping 12-mer peptides by walking through the human TRAP1 amino-acid sequence. One array was probed with highly purified His-tagged mortalin and the second array with His-tagged cytosolic Hsp70. Peptides recognized by both mortalin and cytosolic Hsp70 were eliminated to identify mortalin-specific binding motifs (Supplementary Fig. S2). This resulted in one peptide motif (motif L) with at least two consecutive spots on the membrane (Figs. 6 ▸ a and 6 ▸ b) comprising residues 351–362 (Ser-Met-Phe-Asp-Val-Ser-Arg-Glu-Leu-Gly-Ser-Ser), which map to M-domain loop 1 (Fig. 6 ▸ c). A second peptide motif (motif G) was identified (Figs. 6 ▸ a and 6 ▸ b), revealing an overlapping but non-identical binding motif for mortalin and cytosolic Hsp70 (Supplementary Fig. S2) and comprising N-domain residues 261–272 (Cys-Lys-Glu-Phe-Ser-Ser-Glu-Ala-Arg-Val-Arg-Asp). Although the two mortalin-specific binding peptides are found in different TRAP1 domains, it is immediately evident from mapping the interacting peptides onto a three-dimensional model of TRAP1 that the two motifs are closely spaced and define a surface for mortalin to bind (Fig. 6 ▸ c). Taken together, our findings indicate an asymmetric mortalin-binding site involving one TRAP1 subunit on one surface of the dimer and potentially a second mortalin-binding site on the opposite dimer surface via the other TRAP1 subunit.
Figure 6.

Identifying the mortalin-binding sites of TRAP1 using peptide-array technology. (a) TRAP1 peptide array probed with His6-mortalin. After eliminating binding peptides that were also observed with His6-Hsp70ΔC, overlapping peptides containing at least two consecutive spots are boxed and colored according to domain location, with TRAP1N in green and TRAP1M in magenta. (b) Sequence of mortalin-specific binding peptides. The dominant binding signal observed for each motif is boxed. (c) The location of binding motifs mapped onto a composite three-dimensional model of TRAP1 consisting of human TRAP1NM and zebrafish TRAP1C (PDB entry 4ipe) shown in different hues.
4. Discussion
The present work provides the first complete description of the human TRAP1NM structure at high resolution. Most importantly, we show that the M-domain contributes towards dimerization by mediating intersubunit contacts in the closed-state conformation when ATP is bound, revealing a novel role of the M-domain loop 1 in mediating a direct physical interaction with mitochondrial Hsp70.
Although all ATP-bound Hsp90 dimers show the structure of an N-terminally intertwined, closed-state dimer, it is surprising that no contacts between the cis subunit and the nucleotide bound to the neighboring subunit of the TRAP1 dimer are observed. The latter argues against a mechanism in which ATP binding in trans drives dimer closure. We previously showed that the N-strap undergoes a structural transition from α-helix (in the unliganded state) to β-strand when ATP is bound (Sung et al., 2016 ▸). Thus, it is tempting to speculate that dimer closure is largely the result of changes in local structure upon nucleotide binding in cis, which globally affects the TRAP1 dimer conformation.
In addition to the N-strap, TRAP1 features additional regulatory elements that are shared with other members of the Hsp90 chaperone family. This includes the ATP-lid (Sung et al., 2016 ▸) and loop 2 featuring the ATP sensor (Arg402), both of which are evolutionarily conserved across Hsp90 chaperones. Arg402 senses the presence of the γ-phosphate of the cis-bound adenine nucleotide and helps to stabilize a hydrolysis-competent Hsp90NM conformation (Cunningham et al., 2012 ▸). Although reminiscent of the arginine-finger residue in AAA+ ATPases, which reaches across subunits to complete the ATP-binding pocket in trans (Biter et al., 2012 ▸), the ATP sensor of Hsp90 chaperones is structurally analogous to the sensor-2 motif of AAA+ chaperones by sensing the nucleotide bound to the cis subunit (Hattendorf & Lindquist, 2002 ▸).
The M-domain loop 1 has not been observed in previously reported TRAP1 structures. Loop 1 extends away from the rest of the molecule and contacts the M-domain of the neighboring subunit, forming a previously unknown third dimer interface that mediates the specific interaction with mitochondrial Hsp70. In light of the high structural and functional conservation of Hsp90 chaperones, we speculate that the formation of an Hsp90–Hsp70 complex is a common feature of different Hsp90 and Hsp70 isoforms. Since Hsp90 clients are critically dependent on the functional and perhaps the physical interaction of Hsp70 and Hsp90 chaperones, preventing the formation of a TRAP1–mortalin complex may provide a new avenue for drug development.
Supplementary Material
PDB reference: human mitochondrial TRAP1NM, 5hph
Supplementary Figures S1 and S2.. DOI: 10.1107/S2059798316009906/qh5042sup1.pdf
Acknowledgments
We thank S. Felts and D. Toft for the human TRAP1 cDNA clone, E. Craig for the E. coli HtpG construct and J. Tsai for editing this manuscript. This work was supported by grants R01-GM111084, R01-GM104980 and R01-GM115501 from the National Institutes of Health and Q-1530 from the Welch Foundation. The use of the SBC beamlines at the Advanced Photon Source was supported by the US Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357.
References
- Adams, P. D., Grosse-Kunstleve, R. W., Hung, L.-W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K. & Terwilliger, T. C. (2002). Acta Cryst. D58, 1948–1954. [DOI] [PubMed]
- Ali, M. M., Roe, S. M., Vaughan, C. K., Meyer, P., Panaretou, B., Piper, P. W., Prodromou, C. & Pearl, L. H. (2006). Nature (London), 440, 1013–1017. [DOI] [PMC free article] [PubMed]
- Ban, C., Junop, M. & Yang, W. (1999). Cell, 97, 85–97. [DOI] [PubMed]
- Biter, A. B., Lee, J., Sung, N., Tsai, F. T. F. & Lee, S. (2012). J. Struct. Biol. 179, 172–180. [DOI] [PMC free article] [PubMed]
- Cunningham, C. N., Southworth, D. R., Krukenberg, K. A. & Agard, D. A. (2012). Protein Sci. 21, 1162–1171. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Felts, S. J., Owen, B. A., Nguyen, P., Trepel, J., Donner, D. B. & Toft, D. O. (2000). J. Biol. Chem. 275, 3305–3312. [DOI] [PubMed]
- Gill, S. C. & von Hippel, P. H. (1989). Anal. Biochem. 182, 319–326. [DOI] [PubMed]
- Guarnieri, M. T., Zhang, L., Shen, J. & Zhao, R. (2008). J. Mol. Biol. 379, 82–93. [DOI] [PubMed]
- Hattendorf, D. A. & Lindquist, S. (2002). Proc. Natl Acad. Sci. USA, 99, 2732–2737. [DOI] [PMC free article] [PubMed]
- Jeng, W., Lee, S., Sung, N., Lee, J. & Tsai, F. T. F. (2015). F1000Research, 4, 1448. [DOI] [PMC free article] [PubMed]
- Kang, B. H., Plescia, J., Dohi, T., Rosa, J., Doxsey, S. J. & Altieri, D. C. (2007). Cell, 131, 257–270. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol. 372, 774–797. [DOI] [PubMed]
- Lavery, L. A., Partridge, J. R., Ramelot, T. A., Elnatan, D., Kennedy, M. A. & Agard, D. A. (2014). Mol. Cell, 53, 330–343. [DOI] [PMC free article] [PubMed]
- Leav, I., Plescia, J., Goel, H. L., Li, J., Jiang, Z., Cohen, R. J., Languino, L. R. & Altieri, D. C. (2010). Am. J. Pathol. 176, 393–401. [DOI] [PMC free article] [PubMed]
- Lee, J., Kim, J.-H., Biter, A. B., Sielaff, B., Lee, S. & Tsai, F. T. F. (2013). Proc. Natl Acad. Sci. USA, 110, 8513–8518. [DOI] [PMC free article] [PubMed]
- Lee, C., Park, H.-K., Jeong, H., Lim, J., Lee, A.-J., Cheon, K. Y., Kim, C.-S., Thomas, A. P., Bae, B., Kim, N. D., Kim, S. H., Suh, P.-G., Ryu, J.-H. & Kang, B. H. (2015). J. Am. Chem. Soc. 137, 4358–4367. [DOI] [PubMed]
- McCoy, A. J. (2007). Acta Cryst. D63, 32–41. [DOI] [PMC free article] [PubMed]
- Meyer, P., Prodromou, C., Hu, B., Vaughan, C., Roe, S. M., Panaretou, B., Piper, P. W. & Pearl, L. H. (2003). Mol. Cell, 11, 647–658. [DOI] [PubMed]
- Minor, W., Cymborowski, M., Otwinowski, Z. & Chruszcz, M. (2006). Acta Cryst. D62, 859–866. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Partridge, J. R., Lavery, L. A., Elnatan, D., Naber, N., Cooke, R. & Agard, D. A. (2014). Elife, 3, e03487. [DOI] [PMC free article] [PubMed]
- Rasola, A., Neckers, L. & Picard, D. (2014). Trends Cell Biol. 24, 455–463. [DOI] [PMC free article] [PubMed]
- Rees, I., Lee, S., Kim, H. & Tsai, F. T. F. (2006). Biochim. Biophys. Acta, 1764, 1073–1079. [DOI] [PubMed]
- Sciacovelli, M., Guzzo, G., Morello, V., Frezza, C., Zheng, L., Nannini, N., Calabrese, F., Laudiero, G., Esposito, F., Landriscina, M., Defilippi, P., Bernardi, P. & Rasola, A. (2013). Cell Metab. 17, 988–999. [DOI] [PMC free article] [PubMed]
- Sélo, I., Négroni, L., Créminon, C., Grassi, J. & Wal, J. M. (1996). J. Immunol. Methods, 199, 127–138. [DOI] [PubMed]
- Sielaff, B., Lee, K. S. & Tsai, F. T. F. (2011). J. Mol. Biol. 405, 831–839. [DOI] [PMC free article] [PubMed]
- Sielaff, B. & Tsai, F. T. F. (2010). J. Mol. Biol. 402, 30–37. [DOI] [PMC free article] [PubMed]
- Sung, N., Lee, J., Kim, J.-H., Chang, C., Joachimiak, A., Lee, S. & Tsai, F. T. F. (2016). Proc. Natl Acad. Sci. USA, 113, 2952–2957. [DOI] [PMC free article] [PubMed]
- Tsai, F. T. F., Subramanya, H. S., Brannigan, J. A., Wilkinson, A. J. & Wigley, D. B. (1996). Acta Cryst. D52, 1216–1218. [DOI] [PubMed]
- Wigley, D. B., Davies, G. J., Dodson, E. J., Maxwell, A. & Dodson, G. (1991). Nature (London) 351, 624–629. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: human mitochondrial TRAP1NM, 5hph
Supplementary Figures S1 and S2.. DOI: 10.1107/S2059798316009906/qh5042sup1.pdf