

Abstract
A classification system for analytical methods was developed for the first time to determine the presence of aflatoxins B1, B2, G1 and G2 in traditional Chinese medicines (TCMs) based on different matrix types using ultra-performance liquid chromatography–tandem mass spectrometry. A useful characteristic of the approach was that the TCMs could be systematically divided into four categories (i.e., volatile oils, proteins, polysaccharides and fatty oils) depending on the matrix types. The approach concluded that different types of TCMs required different optimal sample preparation procedures. Based on the optimized analytical conditions, the limits of detection and quantification, average recoveries and linearity of four aflatoxins were determined and conformed to research limits. Of 22 TCMs samples, 14 samples were contaminated with at least one type aflatoxin at concentrations ranging from 0.2 to 7.5 μg/kg, and the average contents of aflatoxins were significantly different for the different matrix types. Moreover, we found a potential link between the contamination levels of aflatoxins and matrix types. TCMs containing fatty oils were the most susceptible to contamination by aflatoxins and followed by TCMs containing polysaccharides and proteins; TCMs containing abundant amounts of volatile oils were less prone to contamination.
Aflatoxins (AFs), namely aflatoxins B1 (AFB1), B2 (AFB2), G1 (AFG1) and G2 (AFG2), are secondary metabolites produced by fungal species, such as Aspergillus flavus, Aspergillus parasiticus and Aspergillus nomius1. AFs are carcinogenic, hepatotoxic, immunosuppressive, genotoxic, antinutritional, teratogenic and mutagenic to humans2,3,4 and AFB1 was defined as a Group 1A carcinogen by the International Agency for Research on Cancer (IARC)5. Due to the pernicious nature of AFs, many countries have established regulations to control the levels of AFs in food and agricultural products which are susceptible to fungal growth.
In China, traditional Chinese medicines (TCMs) with long histories of use are susceptible to mildew and fungus pollution and produce harmful mycotoxins during the production, processing, transportation and storage processes6. Therefore, China has formulated the following relevant standards: The limits for AFB1 and total AFs (sum of AFB1, AFG1, AFB2, and AFG2) in herbs and decoction pieces are 5 and 10 μg/kg, respectively (Chinese Pharmacopoeia, 2015). Other countries have established similar standards, the European Union in the Commission Regulation (EC) No. 1881/2006 has established the maximum residue limits (MRLs) of AFs: 2 μg/kg for AFB 4 μg/kg for the sum of the four AFs7. More than 1.5 billion people all over the world trust the efficacy and safety of TCMs8, and the daily consumption of TCMs is so huge. Hence our understanding of these materials should be strengthened to develop aflatoxin (AF) detection methods to ensure the safety of TCMs. Currently, detection methods exist for the monitoring of AF contamination in some TCMs9,10 such as licorice roots, fritillary bulbs, Fructus Bruceae, but comprehensive and systematic investigations on TCMs are lacking.
In recent years, many analytical techniques have been developed for the detection of AFs including thin layer chromatography (TLC)11, high performance liquid chromatography with fluorescence detector (HPLC-FD)12, iodine derivation after column(Chinese Pharmacopoeia 2015), enzyme-linked immunosorbent assays (ELISA)13,14 and high- (or ultra-) performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS or UPLC-MS/MS)15,16,17,18,19. UPLC-MS/MS methodologies with high resolution, high sensitivity and high selectivity have become a powerful tool for conducting research on complex chemical components20,21,22. The use of UPLC-MS/MS has been increasingly focusing on quantitative and qualitative analyses of traditional data obtained on AFs in TCMs.
Due to the complexity of TCMs, the matrix effect has become a main factor that has affected the accuracy of detecting AFs in TCMs9,23. Thus, the methods used for sample pre-treatment are very important for the accurate detection of AFs in TCMs. Sample pre-treatment mainly includes extraction and purification processes, and existing literature reports have shown that samples of different matrix types can be adopted using appropriate sample pre-treatment methods9,18,23,24. For instance, samples with fatty oils have high proportions of fatty oil contents; Huang B et al. adopted an extraction method using homogenization and a reliable solid phase extraction-based clean-up method to process such samples25. For cereal samples with high protein and polysaccharide (starch) contents, the extraction methods for AF samples frequently used ultrasonography12,26 and clean-up methods for AFs employed solid-phase extraction (SPE) methodologies27,28. The studies described above mostly utilized complex sample processing methods to accurately determine the presence of AFs in one or several samples. However, to accommodate the large number and variety of TCMs, much work is required to develop corresponding sample pre-treatment methods. If classified sample pre-treatment mode were to be established, the accuracy of the measurements and efficiency of work would improve.
Scholars have adopted classification methods18,29, and TCMs have always been divided into different medical parts, i.e., rhizomes, roots, seeds, flowers, grasses and leaves, which were extracted and purified using the same procedures depending on the medical parts. However, this classification method has some defects. For example, there may be a major difference in the matrix of the same medicinal parts of different medicinal materials. The same sample pre-treatment methods developed by such researchers were not suitable for extracting or detecting AFs in TCMs.
The aim of this work was to develop a novel classification of analytical method to detect aflatoxins B1, B2, G1 and G2 in widely applied TCMs based on different matrix types. Research efforts have focused on the influence of different sample pre-treatment methods on the samples and optimization of UPLC-MS/MS parameters. This research can offer a reference for systematically establishing analytical methods for the detection of AFs in TCMs. Meanwhile, data on the contamination levels of AFs and the contents of different matrix types were processed and analyzed using statistics software, and an inner relationship was found, which could be used to infer the susceptibility of fungus contamination based on the matrix types of TCMs and to provide a reference point for the safety of TCMs.
Results and Discussion
Analysis results of sample matrix types
The matrix types of all 22 TCMs were divided according to the contents of the basic components. The results are shown in Table 1. By comparing the content ratios of the four types of components in each sample, all 22 samples were divided into four matrix types, i.e., volatile oils, proteins, polysaccharides and fatty oils. In our work, the polysaccharide content was determined to be 2.2–31.8% in the 22 samples, among which the content of polysaccharides of five samples was larger than 20%; these five samples were eventually categorized into the polysaccharides group. Of the TCMs, 6 out of the 22 samples were classified as volatile oils, in which the range of the volatile oil content was between 1.6% and 2.9%. Similarly, fatty oil and protein contents ranged from 23.8% to 50.5% and from 15.9% to 21.3%, respectively, and these samples were categorized as the fatty oils and proteins, respectively.
Table 1. The content determination of volatile oils, fatty oils, polysaccharides and proteins in 22 TCMs, classification of samples matrix types, and the contamination levels of AFs in TCMs of different matrix types.
| Category | Samples | The content ratioa (%) |
The contentsa (μg/kg) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Volatile oil | Protein | Polysaccharide | Fatty oil | AFB1 | AFB2 | AFG1 | AFG2 | Totalb | ||
| Volatile oils | Rhizoma Alpiniae Officinarum | 1.6 | 1.4 | 17.5 | 1.7 | 0.4 | 0.1 | N.D | N.D | 0.5 |
| Fructus Anisi Stellati | 2.8 | 3.5 | 9.0 | 1.6 | N.D | N.D | N.D | N.D | — | |
| Fructus Citri Sarcodactylis | 2.2 | 6.4 | 13.4 | 3.0 | 0.2 | 0.2 | N.D | N.D | 0.4 | |
| Pericarpium Citri Reticulatae | 2.9 | 6.0 | 15.7 | 3.1 | N.D | N.D | N.D | N.D | — | |
| Fructus Tsaoko | 2.1 | 4.9 | 11.8 | 1.9 | 0.2 | N.D | N.D | N.D | 0.2 | |
| Flos Caryophylli | 2.3 | 5.1 | 15.6 | 3.7 | N.D | N.D | N.D | N.D | — | |
| Proteins | Semen Phaseoli | N.D | 21.3 | 12.8 | 1.5 | 2.9 | 0.4 | 0.2 | N.D | 3.5 |
| Semen Lablab Album | N.D | 17.0 | 9.0 | 0.8 | 0.6 | 0.2 | N.D | N.D | 0.8 | |
| Semen Coicis | N.D | 17.1 | 6.6 | 1.1 | N.D | N.D | N.D | N.D | — | |
| Semen Euryales | N.D | 15.9 | 2.2 | 2.2 | N.D | N.D | N.D | N.D | — | |
| Semen Nelumbinis | N.D | 19.7 | 5.6 | 1.8 | 0.3 | 0.1 | N.D | N.D | 0.4 | |
| Polysaccharides | Fructus Mume | N.D | 3.1 | 26.8 | 1.4 | 1.4 | 0.4 | 0.1 | N.D | 1.9 |
| Fructus Jujubae | N.D | 4.0 | 31.8 | 1.1 | 3.2 | 0.5 | 0.8 | N.D | 4.5 | |
| Fructus Hippophae | N.D | 10.4 | 21.8 | 1.8 | N.D | N.D | N.D | N.D | — | |
| Fructus Momordicae | N.D | 10.6 | 29.4 | 17.4 | 2.1 | 1.4 | 0.4 | N.D | 3.9 | |
| Fructus Rubi | N.D | 10.8 | 25.5 | 2.2 | N.D | 1.2 | N.D | N.D | 1.2 | |
| Fatty oils | Semen Pruni | N.D | 14.6 | 15.0 | 39.2 | 2.7 | 1.4 | 0.3 | N.D | 4.4 |
| Fructus Cannabis | N.D | 12.9 | 6.2 | 23.8 | N.D | N.D | N.D | N.D | — | |
| Semen Raphani | N.D | 14.0 | 15.6 | 37.7 | 3.8 | 1.2 | 0.1 | N.D | 5.1 | |
| Semen Armeniacae Amarum | N.D | 13.4 | 19.5 | 43.9 | 4.8 | 2.3 | 0.3 | 0.1 | 7.5 | |
| Fructus Perillae | N.D | 14.9 | 2.2 | 46.3 | N.D | N.D | N.D | N.D | — | |
| Semen Sesami Nigrum | N.D | 11.8 | 7.6 | 50.5 | 2.3 | 1.0 | 0.3 | 0.2 | 3.8 | |
N.D not detected.
aMean ± SD, n = 3.
bThe sum of AFB1, AFB2, AFG1 and AFG2.
Moisture contents of samples were tested based on the Chinese Pharmacopoeia (2015). The results were shown in Supplementary Table 1. For volatile oils, proteins, polysaccharides and fatty oils, the moisture content was 7.52–8.92%, 7.49–10.44%, 7.16–10.54%, and 5.37–7.13%, respectively. Moisture content results met the requirements of Chinese Pharmacopoeia.
Optimization of the extraction procedure
For the TCMs samples of four different matrix types, the effectiveness of various extraction methods was investigated. Four duplicate samples of the four types of matrices were extracted through shaking, homogenizing and ultrasonicating the samples. By comparing the extraction efficiencies of three methods, each sample of the four types of matrices required its own extraction methods (Fig. 1). Based on the results, ultrasonic extraction was selected as the best extraction method for the protein and volatile oil samples. Shaking extraction methods were determined to be the optimal methods for the samples of polysaccharides, and homogenization extraction was chosen for fatty oils. Because TCMs with high contents of fatty oils and polysaccharides were more viscous, an ultrasonography extraction method was prone to aggregating the extracts, and its use to extract AFs was not conducive to dissolution of the compounds.
Figure 1. Efficiency of extraction for AFs in TCMs of different matrix types using different extract methods.

In addition, to allow for higher extraction efficiencies, the extraction solvents and time were optimized. Five ratios of extraction solvents were investigated: 65%, 70%, 75%, 80%, and 85% aqueous methanol solutions were used for the samples of each type, and the samples were also subjected to different extraction times. The results of this optimization study are shown in Supplementary Fig. 1. For volatile oils, the samples were extracted in 75% aqueous methanol using ultrasonography for 45 min. The samples containing proteins were sonicated in 85% aqueous methanol for 45 min. For the samples with polysaccharides, they were extracted in 70% aqueous methanol for 3 h with shaking, and the samples of fatty oils were homogenized in 70% aqueous methanol for 4 min.
Because the samples had different matrices, each category of the samples required the use of a different extraction method. The obtained results were consistent with observations reported in previously published articles. A.S. Luna et al. conducted research on peanuts with more oil and used a homogenization extraction method to process the samples30. Wen J. et al. adopted an extraction procedure using ultrasonication to extract AFs from ginger and products related to volatile oils31. Kong W.J. et al. developed a method to analyse multi-class mycotoxins in Coix seeds32. However, in our work, shaking extraction was an optimal extraction method for samples containing polysaccharides.
Optimization of the clean-up procedure
To optimize extraction efficiencies and the recovery of materials, different methods were tested and compared. In our study, the use of Welchrom C18E columns and silica gel columns for the clean-up procedures after extraction was evaluated. The first two methods were compared to samples that were not subjected to purification methods, which showed that the recoveryno purification > the recoveryC18 columns > the recoverysilicagel columns (Supplementary Table 2). Because the fatty samples contained more nonpolar and weakly polar compounds which could pollute and damage the UPLC column and consequently shorten the service life of the column upon purification, the samples needed to be processed after being subjected to a clean-up procedure. In general, the three types of TCMs mentioned above were extracted without purification, which resulted in a higher recovery rate and lower loss rate. Samples of fatty oils were purified by C18-SPE columns to protect the columns against damage, and the obtained recovery was 70–110% using the clean-up method and matched the recovery amount of the standard.
Method validation
The ranges of linearity, the coefficients of determination and correlation, as well as the limits of detection (LOD) and quantification (LOQ) for each aflatoxin were determined. The working standard solutions of AFs were diluted immediately with methanol from the original stock solutions every weekday and which were used to make the mixed working standards. A set of four standard solutions containing different concentrations in the range of 0.0502–10.4 ng/mL for AFB1, 0.0350–7.0 ng/mL for AFB2, 0.0295–11.8 ng/mL for AFG1 and 0.0295–11.8 ng/mL for AFG2, which were prepared in methanol and were used for method calibration. These solutions were kept at −20 °C and were renewed weekly. The linearities obtained for all the analytes were good, and the correlation coefficients (R2) ranged from 0.9985 to 0.9996. LOD and LOQ values were 0.008–0.022 μg/kg and 0.011–0.029 μg/kg, respectively, which showed that the method developed, met the EU legislative requirements of 2 and 4 μg/kg for AFB1 and total AFs contents. The relative standard deviation (RSD) of precision at the middle concentration of the AFs mixture was 2.9–6.7% (n = 6). The data are shown in Table 2.
Table 2. ESI-MS/MS parameters, concentration ranges (ng/mL), limits of detection (LOD), limits of quantification (LOQ) and linearity values (R 2 ) for AFs.
| AFs | MW | Q1 (m/z) | Q3 (m/z) | CE (e/V) | DP (V) | range (ng/mL) | R2 | LOD (μg/ kg) | LOQ (μg/kg) | RSD (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| AFB1 | 312.3 | 313.3 | 285.3a | 30 | 178 | 0.0502–10.4 | 0.9987 | 0.008 | 0.011 | 2.9 |
| 313.3 | 241.0 | 47 | 166 | |||||||
| AFB2 | 314.3 | 315.3 | 287.1a | 33 | 161 | 0.0350–7.0 | 0.9992 | 0.015 | 0.023 | 3.5 |
| 315.3 | 259.0 | 38 | 159 | |||||||
| AFG1 | 328.3 | 329.2 | 311.2a | 30 | 143 | 0.0295–11.8 | 0.9985 | 0.022 | 0.029 | 4.6 |
| 329.2 | 243.1 | 34 | 158 | |||||||
| AFG2 | 330.3 | 331.2 | 217.0a | 46 | 131 | 0.0295–11.8 | 0.9991 | 0.020 | 0.027 | 3.4 |
| 331.2 | 245.3 | 38 | 114 |
aQuantitative ion.
Recovery estimations were carried out using the standard addition method, which comprised three spiked samples at different levels. Different types of TCMs were used for the recovery test to ensure that the method had broad applicability. Each sample was selected at random, and aliquots (n = 9) of the samples were spiked with the mixed standard solutions at a high concentration level (10.4 ng/mL for AFB1, 3.5 ng/mL for AFB2, 11.8 ng/mL for AFG1 and 5.9 ng/mL for AFG2), a medium concentration level (4.16 ng/mL for AFB1, 1.4 ng/mL for AFB2, 4.72 ng/mL for AFG1 and 2.36 ng/mL for AFG2) and a low concentration level (1.04 ng/mL for AFB1, 0.35 ng/mL for AFB2, 1.18 ng/mL for AFG1 and 0.59 ng/mL for AFG2). In general, a sample (2.0 g) was spiked with high, medium or low levels of the AF standards; and were treated and tested following the procedures outlined above. All recovery amounts ranged from 80.4% to 103.3% (Table 3). The spiked samples were extracted and analysed by UPLC-MS/MS, as previously described.
Table 3. Recovery results of AFB1, AFB2, AFG1 and AFG2 a (%).
| Category | Samples | Levels | AFB1 | AFB2 | AFG1 | AFG2 |
|---|---|---|---|---|---|---|
| Volatile oils | Rhizoma Alpiniae Officinarum | Low | 89.4 | 87.3 | 90.4 | 85.6 |
| Medium | 91.4 | 88.0 | 96.2 | 84.9 | ||
| High | 90.3 | 82.0 | 95.1 | 87.1 | ||
| Fructus Anisi Stellati | Low | 90.9 | 84.2 | 89.4 | 88.8 | |
| Medium | 94.4 | 96.7 | 100.5 | 85.2 | ||
| High | 85.4 | 84.9 | 86.1 | 89.9 | ||
| Fructus Citri Sarcodactylis | Low | 93.6 | 82.3 | 86.8 | 83.7 | |
| Medium | 87.0 | 92.3 | 100.1 | 96.2 | ||
| High | 83.7 | 88.2 | 85.8 | 84.0 | ||
| Pericarpium Citri Reticulatae | Low | 91.4 | 94.3 | 91.5 | 91.4 | |
| Medium | 90.9 | 89.6 | 84.8 | 91.3 | ||
| High | 82.6 | 85.9 | 97.6 | 86.5 | ||
| Fructus Tsaoko | Low | 95.8 | 92.3 | 82.4 | 81.2 | |
| Medium | 100.3 | 84.7 | 92.7 | 84.3 | ||
| High | 81.3 | 94.3 | 86.5 | 86.2 | ||
| Flos Caryophylli | Low | 96.4 | 90.0 | 88.4 | 93.9 | |
| Medium | 81.7 | 83.9 | 91.4 | 100.6 | ||
| High | 93.7 | 89.7 | 89.4 | 85.9 | ||
| Proteins | Semen Phaseoli | Low | 84.6 | 101.1 | 90.8 | 93.7 |
| Medium | 100.2 | 97.9 | 88.1 | 89.0 | ||
| High | 101.2 | 84.4 | 86.3 | 90.4 | ||
| Semen Lablab Album | Low | 83.4 | 84.7 | 81.2 | 83.4 | |
| Medium | 91.1 | 82.2 | 101.0 | 80.6 | ||
| High | 82.1 | 81.1 | 92.7 | 84.2 | ||
| Semen Coicis | Low | 87.9 | 91.6 | 80.4 | 96.1 | |
| Medium | 97.1 | 98.3 | 90.8 | 89.2 | ||
| High | 88.1 | 98.5 | 92.1 | 96.5 | ||
| Semen Euryales | Low | 82.8 | 95.3 | 100.5 | 99.4 | |
| Medium | 85.6 | 85.0 | 88.6 | 100.0 | ||
| High | 83.0 | 95.7 | 80.9 | 92.4 | ||
| Semen Nelumbinis | Low | 91.4 | 91.2 | 87.6 | 94.2 | |
| Medium | 84.9 | 89.2 | 92.8 | 85.4 | ||
| High | 89.7 | 100.1 | 86.4 | 84.7 | ||
| Polysaccharides | Fructus Mume | Low | 83.4 | 93.6 | 81.2 | 83.4 |
| Medium | 91.1 | 82.2 | 101.4 | 80.6 | ||
| High | 102.2 | 95.6 | 94.6 | 93.4 | ||
| Fructus Jujubae | Low | 91.4 | 93.8 | 100.1 | 99.7 | |
| Medium | 95.8 | 83.5 | 100.5 | 81.3 | ||
| High | 97.7 | 92.9 | 88.0 | 98.8 | ||
| Fructus Hippophae | Low | 88.3 | 83.3 | 100.6 | 91.3 | |
| Medium | 87.6 | 83.0 | 102.8 | 80.4 | ||
| High | 80.8 | 90.6 | 84.4 | 93.4 | ||
| Fructus Momordicae | Low | 96.7 | 101.2 | 90.5 | 101.0 | |
| Medium | 84.5 | 86.3 | 86.4 | 99.4 | ||
| High | 88.3 | 91.9 | 84.2 | 88.7 | ||
| Fructus Rubi | Low | 103.1 | 90.5 | 85.6 | 88.0 | |
| Medium | 97.5 | 87.4 | 98.4 | 90.3 | ||
| High | 93.4 | 88.0 | 91.9 | 81.2 | ||
| Fatty oils | Semen Pruni | Low | 91.1 | 101.5 | 90.2 | 84.6 |
| Medium | 83.6 | 92.5 | 81.7 | 82.0 | ||
| High | 94.0 | 89.3 | 92.4 | 84.5 | ||
| Fructus Cannabis | Low | 88.2 | 82.9 | 88.7 | 86.1 | |
| Medium | 91.4 | 98.3 | 98.4 | 81.9 | ||
| High | 90.7 | 82.4 | 94.0 | 82.1 | ||
| Semen Raphani | Low | 90.6 | 84.4 | 86.5 | 85.3 | |
| Medium | 88.4 | 97.9 | 100.2 | 90.7 | ||
| High | 86.5 | 81.8 | 82.6 | 86.9 | ||
| Semen Armeniacae Amarum | Low | 83.3 | 92.8 | 97.0 | 99.7 | |
| Medium | 83.9 | 86.3 | 93.4 | 99.4 | ||
| High | 88.3 | 91.9 | 84.2 | 88.7 | ||
| Fructus Perillae | Low | 102.1 | 90.5 | 85.6 | 88.0 | |
| Medium | 97.5 | 87.4 | 98.4 | 90.3 | ||
| High | 93.4 | 88.0 | 91.9 | 81.2 | ||
| Semen Sesami Nigrum | Low | 84.6 | 101.1 | 90.8 | 93.7 | |
| Medium | 100.2 | 97.9 | 88.1 | 89.0 | ||
| High | 101.2 | 84.4 | 103.3 | 90.4 |
aEach value represents the mean ± SD of at least three measurements.
For the four AFs the results indicated good accuracy of the method for the detection of aflatoxins B1, B2, G1, G2 in TCMs of different matrix types, and the recoveries were also in compliance with the requirements of the European Union (70–110%).
Method application
Following the optimization and validation of the analytical approach, it was successfully utilized to determine the contamination levels of four AFs in 22 classified TCMs. The levels of total and individual AFs are summarized in Table 1.Typical UPLC–MS/MS chromatograms of the four AFs in standard solutions (A) and in contaminated samples (B) are shown in Supplementary Fig. 2. Of the 22 samples, 14 samples were detected to be positive with four AFs at concentrations ranging from 0.2 to 7.5 μg/kg, and 13 samples were detected to be contaminated with AFB1. The incidence rate was as high as 63.6%, and four positive samples (18.2%) exceeded the maximum limit set by the European Union (4 μg/kg). With regards to individual AFs, the levels of AFB1, AFB2, AFG1, and AFG2 were detected in ranges of 0.2–4.8, 0.1–2.3, 0.1–0.8, 0.1–0.2 μg/kg, respectively. For the four types of TCMs (i.e., volatile oils, proteins, polysaccharides and fatty oils), the levels of AFB1 were 0.2–0.4, 0.3–2.9, 1.4–3.2, 2.3–4.8 μg/kg, respectively, and the levels of AFs were 0.2–0.5, 0.4–3.5, 1.2–4.5, 3.8–7.5 μg/kg, respectively. Based on these results, we inferred that contamination of AFB1 was the most serious in the 22 TCMs samples.
Correlation analysis
To further analyse the contamination levels of the 22 TCMs, we compared the contents of AFB1 and AFs in the samples of four matrix types. The effects of the matrix types on the contamination levels of AFs are thought to be due to their different abilities for breeding fungus. The average contamination levels of AFB1 and total AFs in the samples of four matrix types are shown in Fig. 2. The content of AFs in the samples of different matrix types was varied significantly. Results showed that TCMs with an abundant of fatty oils had the highest amounts of AFB1 and total AFs, while these contamination levels were very low for samples with an abundance of volatile oils. Furthermore, the internal relation between the contamination levels of AFs and matrix types was studied.
Figure 2. Average contamination levels of AFB1 and total AFs of samples from four matrix types.
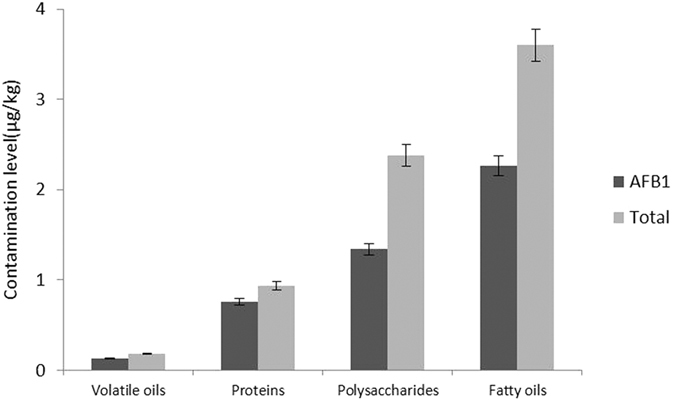
In our study, the results obtained by Pearson correlation analysis indicated that the contents of AFB1 and total AFs had varying degrees of influence on the different matrices. As shown in Table 4, the contents of AFB1 and AFs were negatively correlated with the contents of volatile oils, and the correlation coefficients (r) were −0.612 and −0.556 (P < 0.05). respectively. The content of fatty oil exhibited a positive correlation to the contamination levels of AFB1 (r = 0.661, P < 0.01) and AFs (r = 0.749, P < 0.01). The contents of AFB1 and AFs were not significantly positively correlated with the contents of polysaccharides and proteins.
Table 4. The correlation between the contents of volatile oils, fatty oils, polysaccharides, and proteins in AFB1 and total AFs.
| Component | Volatile oil | Protein | Polysaccharide | Fatty oil |
|---|---|---|---|---|
| AFB1 | r = −0.612* | r = 0.266 | r = 0.361 | r = 0.661** |
| AFs | r = −0.556* | r = 0.240 | r = 0.289 | r = 0.749** |
**extremely significant, P < 0.01;
*significant, P < 0.05.
Our results indicate that TCMs with fatty oils may easily multiply Aspergillus flavus and A. parasiticus, resulting in the production of secondary metabolites (AFs). Polysaccharides and proteins also provided nutritional ingredients for fungus and promote their growth; the contents of AFs were relatively high in both types of TCMs. TCMs with volatile oils, such as Fructus Tsaoko, Fructus Anisi Stellati and Flos Caryophylli contain the active chemical components, known as essential oils, which possessed antifungal effects that reduced or prevented fungal infection and subsequent AFs production. The essential oils can decrease the damaged effect of aflatoxins by two different ways. Firstly, DNA binding formation of aflatoxins is reduced by essential oils. Secondly, aflatoxins cause increase of reactive oxygen species and essential oils react with reactive oxygen species. Therefore, essential oils protect the cells from harmful impact of aflatoxins33,34. Similar results have been reported for studies conducted on Ocimum basilicum L.35, Radix Puerariae Lobatae and Semen Persicae samples18.
Conclusions
In this study, a classification method for the simultaneous detection of AFB1, AFB2, AFG1 and AFG2 in TCMs based on matrix types was established by UPLC-MS/MS for the first time, and the classification approach was successfully applied to analyse a total of 22 different matrix types of TCMs. This study provides a novel research approach for establishing the use of analytical methods to detect AFs in a large number of TCMs.
Furthermore, we found that there was significant relationship between matrix types and the contamination levels of AFs. The contents of fatty oils, polysaccharides and proteins to the contamination levels of AFB1 and AFs were positively correlated, whereas the contents of AFs were negatively correlated with the contents of volatile oils. Meanwhile, a possible association between the contamination levels of AFs and the different matrix types of TCMs was presented. The possibility for AFs contamination of medicinal materials containing fatty oils and polysaccharides was high, but the possibility of those containing volatile oils was low. These results indicate that the processing and storage methods used for medicinal materials are likely associated with the matrix types of their components, especially regarding the amounts of fatty oils of TCMs.
Methods
Materials and reagents
AF standards including AFB1, AFB2, AFG1 and AFG2 were purchased from Sigma-Aldrich (St. Louis, MO, USA). Solid powders of each aflatoxin standard were weighed accurately, and the standards were dissolved in methanol to prepare stock standard solutions and stored at −20 °C in a dark place. Distilled water was purified using a Milli-Q Gradient A 10 system (Millipore, Billerica, MA, USA). Acetonitrile, methanol and formic acid were of LC grade (Merck, Darmstadt, Germany). All the other solvents were of analytical grade. Welchrom C18E (500 mg/3 mL) columns were purchased from Welch (USA).
A total of 22 samples were randomly purchased from June to August 2014 from several local markets and drug stores in Chongqing China; the samples were authenticated by Professor Dan Zhang at Chongqing Medical University. All the samples were ground into powders, sieved through a 60-mesh filter and stored in sealed plastic bags below 4 °C for further analysis.
UPLC-MS/MS analysis
The UPLC chromatography system (Shimadzu Corp., Kyoto, Japan) was equipped with a solvent delivery pump (LC-30AD), an auto-sampler (SIL-30AC) and a column oven (CTO-20AC). The separations were performed on a Phenomenex Luna 3 μC18 (2) 100A column (50 × 2.00 mm) (Phenomenex, USA). Chromatographic analyses were carried out using a gradient elution, where eluent A was an aqueous solution of ammonium formate (5 mM) and eluent B consisting of acetonitrile. The analysis started with 30% of acetonitrile, which was held for 0.5 min, and was then changed to 80% acetonitrile at 4.5 min and held 1.5 min. Then, the eluent was changed to 30% acetonitrile at 6.1 min. The column was conditioned with 30% acetonitrile for 1.9 min before the next injection. The flow rate was set at 0.35 mL/min, and the injection volume was 3 μL. Moreover, the column temperature was maintained at 30 °C.
Electrospray mass spectrometry (ESI-MS) was carried out using an API 4000 triple-quadrupole instrument from Applied Biosystems (AB Sciex, Framingham, MA, USA), equipped with an electro-spray ionization (ESI) source. The mass spectrometer was operated in positive ESI modes with multiple reaction monitoring (MRM) at unit mass resolution. Data acquisition and processing of the ESI-MS were obtained using AnalystTF software (AB Sciex), and the accurate mass data for the molecular ions were processed by PeakViewTM 1.1.1 software (AB Sciex).The source/gas conditions were as follows: the curtain gases CAD and CUR were set at 4 and 25 psi, respectively. The ion source gas 1 (GS1) and ion source gas 2 (GS2) were set at 55 psi and 55 psi, respectively. The ionization source of the MS/MS detector had a capillary voltage of 5.5 kV, and the source temperature was set to 600 °C. The compound conditions were Entrance Potential (10.0) and Collision cell potential (12.0). The MRM transitions, applied cone voltages and collision energies are summarized in Table 2.
Analysis of samples matrix types
To determine the matrix composition of various medicinal materials, the contents of volatile oils, proteins, polysaccharides and fatty oils of 22 samples were determined. The contents of these materials were determined according to the Chinese Pharmacopoeia (2015), the Kjeldahl determination method36, the phenol-sulfuric acid method37 and the Soxhlet extraction method38, respectively. The content ratios were then calculated to classify the samples according to the matrix types.
Sample Preparation
Extraction
To optimize the extraction procedure of AFs in TCMs, the influence of different extraction methods and factor levels based on the classification results of different matrix was investigated. (1) Extraction methods: For the TCMs of four matrix types, duplicate samples of each type were extracted through shaking, homogenization and ultrasonication. (2) Extraction solution: Five different ratios of extraction solvents were investigated: 65%, 70%, 75%, 80%, and 85% aqueous methanol solutions were used for samples of each type. (3) Extraction time: Samples of four matrix types were extracted for four different periods of time. Four different extraction procedures were then used for samples of different matrix types, which are described below:
Volatile oils: A 2 g portion of ground sample was soaked in 10 mL of a methanol/water (75:25, v/v) solution for 1 h and was sonicated for 45 min. The sample was then centrifuged at 3000 rpm for 5 min, and1 mL of the supernatant was filtered through a 0.22 μm syringe filter prior to analysis.
Proteins: A 2 g portion of ground samples was soaked in 10 mL of a methanol/water (85:15, v/v) solution for 1 h and was sonicated for 45 min, The following procedure was the same as that used for the extraction procedures for volatile oils.
Polysaccharides: A 2 g portion of a ground sample was extracted in 10 mL of a methanol/water (70:30, v/v) solution for 3 h by shaking the sample. The sample was then centrifuged at 3000 rpm for 5 min, and 1 mL of the supernatant was filtered through a 0.22 μm syringe filter prior to analysis.
Fatty oils: A 2 g portion of a ground sample was homogenized in 10 mL of a methanol/water (70:30, v/v) solution for 4 min and was centrifuged at 3000 rpm for 5 min. Then, 2 mL of the supernatant was subjected to the for clean-up procedure.
Clean-up
To evaluate the efficiency of the clean-up procedure, results obtained using Welchrom C18E columns and silica gel columns were compared to samples that were not subjected to a purification method.
Samples of fatty oils were purified using the following procedure. A 2 mL aliquot of the final filtrate was passed through a Welchrom C18E column. The C18E column was pre-treated with 6 mL methanol before washing it with 6 mL distilled water. After the sample was loaded into the column, the column was first washed with 6 mL distilled water, and then the C18E column was rinsed with 4 mL methanol. The obtained elutes were completely evaporated under a steam of nitrogen gas at 30 °C, and the sample was re-dissolved in 1 mL methanol. The solution containing the AFs was vortexed for 30 s, and approximately 50 μL of the solution was filtered through a 0.22 μm filter. A 3 μL aliquot of the filtrate was injected into the UPLC-MS/MS system.
Method validation
Quantification of the AFs in TCMs followed testing for linearity, recovery, LOD and LOQ. To check the linearity of the method, calibration curves based on the peak area were constructed in the range of 0.0295–11.8 ng/mL. To interpolate the results, concentrations outside the calibration range were performed with proper dilutions. Quantification was performed by plotting concentration versus peak area, and the regression curve was evaluated by using variance (ANOVA) analysis.
The LODs were obtained using a signal-to-noise ratio of S/N = 3:1, and the LOQ was considered the lowest point of the calibration curve that was adopted when the concentration of a compound resulted in S/N = 10:1.
Recovery analysis was performed by testing replicate spiked samples at three different concentrations (low, medium and high levels). The recovery values were estimated by relating the concentration of the AFs found to the expected concentration.
Statistical treatment of data
To obtain further details of the differences, the UPLC-MS/MS datasets of the four groups were subjected to correlate analyses. The contents of AFB1, total AFs (AFB1, AFB2, AFG1 and AFG2) and the content ratios of the four types of matrices were expressed as mean ± standard deviation of three replicates. The significance of each group was checked by a one-way analysis of Variance (ANOVA) followed by a Pearson correlation. A bivariate correlate analysis was used to determine the relationship between AFB1 contents, total AF contents and the content ratios of the four different types of matrices. Correlate analysis was conducted using SPSS 19.0 statistical software (SPSS Inc., Chicago, IL, USA). The significant value was set at P < 0.05.
Additional Information
How to cite this article: Zhao, S.-P. et al. Analysis of aflatoxins in traditional Chinese medicines: Classification of analytical method on the basis of matrix variations. Sci. Rep. 6, 30822; doi: 10.1038/srep30822 (2016).
Supplementary Material
Acknowledgments
We thank Dr. Yanlei Guo (Institute of Life Science, Chongqing Medical University) for his help with UPLC-MS/MS analyses. This study was supported by the Provincial Key Subjects of the Chongqing Municipal Education Commission of China (Grant No. 40010200010042).
Footnotes
Author Contributions S.-P.Z., D.Z. and W.-G.C. designed the experiments. S.-P. Z., L.-H.T. and B.Y. performed the experiments. S.-P.Z. and D.Z. analysed the data. S.-P.Z. and D.Z. wrote the manuscript. W.-G.C., L.-H.T. and B.Y. contributed to and edited the manuscript. All the authors have reviewed and approved the final version of this manuscript.
References
- Scaglioni P. T., Becker-Algeri T., Drunkler D. & Badiale-Furlong E. Aflatoxin B(1) and M(1) in milk. Anal Chim Acta 829, 68–74, doi: 10.1016/j.aca.2014.04.036 (2014). [DOI] [PubMed] [Google Scholar]
- Partanen H. A. et al. Aflatoxin B1 transfer and metabolism in human placenta. Toxicol Sci 113, 216–225, doi: 10.1093/toxsci/kfp257 (2010). [DOI] [PubMed] [Google Scholar]
- Sweeney M. J. & Dobson A. D. W. Molecular biology of mycotoxin biosynthesis. FEMS Microbiol Lett 175, 149–163 (1999). [DOI] [PubMed] [Google Scholar]
- Hussein H. S. & Brasel J. M. Toxicity, metabolism, and impact of mycotoxins on humans. Toxicology 167, 101–134 (2001). [DOI] [PubMed] [Google Scholar]
- International Agency for Research on Cancer(IARC). Some Naturally Occurring Substances: Food Items and Constituents, Heterocyclic Aromatic Amines and Mycotoxins, Monographs on the Evaluation of Carcinogenic Risks to Humans. World Health Organization, Lyon 82, 171–300 (2002). [Google Scholar]
- Qi M. & Zhen A. Pondering over the Applicat ion ofGene - chip Technology to the Research in TCM Symptoms. Chinese Archives of Traditional Chinese Medcine 24, 2025–2026, doi: 10.13193/j.archtcm.2006.11.59.zhangzhl.025 (2006). [DOI] [Google Scholar]
- Commission Regulation (EC). No 1881/2006 of 19 December 2006 setting maximum levels for certain contaminants in foodstuffs. Official Journal of the European Communities L364, 5–24 (2006). [Google Scholar]
- Hosbach I., Neeb G., Hager S., Kirchhoff S. & Kirschbaum B. In defence of traditional Chinese herbal medicine. Anaesthesia 58, 280–300 (2003). [DOI] [PubMed] [Google Scholar]
- Cao J. et al. Simultaneous determination of aflatoxins B(1), B(2), G(1), G(2) in Fructus Bruceae by high-performance liquid chromatography with online postcolumn photochemical derivatization. J Sep Sci 37, 2771–2778, doi: 10.1002/jssc.201400501 (2014). [DOI] [PubMed] [Google Scholar]
- Wang L. et al. Simultaneous determination of aflatoxin B1 and ochratoxin A in licorice roots and fritillary bulbs by solid-phase extraction coupled with high-performance liquid chromatography–tandem mass spectrometry. Food Chem 138, 1048–1054, 10.1016/j.foodchem.2012.11.066 (2013). [DOI] [PubMed] [Google Scholar]
- Hitokoto H., Morozumi S., Wauke T., Sakai S. & Kurata H. Fungal contamination and mycotoxin detection of powdered herbal drugs. Appl Environ Microbiol 36, 252–256 (1978). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinto M., Spadaccino G., Palermo C. & Centonze D. Determination of aflatoxins in cereal flours by solid-phase microextraction coupled with liquid chromatography and post-column photochemical derivatization-fluorescence detection. J Chromatogr A 1216, 8636–8641, doi: 10.1016/j.chroma.2009.10.031 (2009). [DOI] [PubMed] [Google Scholar]
- Ardic M., Karakaya Y., Atasever M. & Durmaz H. Determination of aflatoxin B(1) levels in deep-red ground pepper (isot) using immunoaffinity column combined with ELISA. Food Chem Toxicol 46, 1596–1599, doi: 10.1016/j.fct.2007.12.025 (2008). [DOI] [PubMed] [Google Scholar]
- Saha D., Acharya D., Roy D., Shrestha D. & Dhar T. K. Simultaneous enzyme immunoassay for the screening of aflatoxin B1 and ochratoxin A in chili samples. Analy Chim Acta 584, 343–349, doi: 10.1016/j.aca.2006.11.042 (2007). [DOI] [PubMed] [Google Scholar]
- Matumba L. et al. A survey of the incidence and level of aflatoxin contamination in a range of locally and imported processed foods on Malawian retail market. Food Control 39, 87–91, doi: 10.1016/j.foodcont.2013.09.068 (2014). [DOI] [Google Scholar]
- Gerding J., Cramer B. & Humpf H. U. Determination of mycotoxin exposure in Germany using an LC-MS/MS multibiomarker approach. Mol Nutr Food Res 58, 2358–2368, doi: 10.1002/mnfr.201400406 (2014). [DOI] [PubMed] [Google Scholar]
- Solfrizzo M., Gambacorta L., Lattanzio V. M., Powers S. & Visconti A. Simultaneous LC-MS/MS determination of aflatoxin M1, ochratoxin A, deoxynivalenol, de-epoxydeoxynivalenol, alpha and beta-zearalenols and fumonisin B1 in urine as a multi-biomarker method to assess exposure to mycotoxins. Anal Bioanal Chem 401, 2831–2841, doi: 10.1007/s00216-011-5354-z (2011). [DOI] [PubMed] [Google Scholar]
- Han Z. et al. An ultra-high-performance liquid chromatography-tandem mass spectrometry method for simultaneous determination of aflatoxins B1, B2, G1, G2, M1 and M2 in traditional Chinese medicines. Anal Chim Acta 664, 165–171, doi: 10.1016/j.aca.2010.02.009 (2010). [DOI] [PubMed] [Google Scholar]
- Liu L., Jin H., Sun L., Ma S. & Lin R. Determination of aflatoxins in medicinal herbs by high-performance liquid chromatography-tandem mass spectrometry. Phytochem Anal 23, 469–476, doi: 10.1002/pca.2343 (2012). [DOI] [PubMed] [Google Scholar]
- Jager A. V., Tonin F. G., Souto P. C., Privatti R. T. & Oliveira C. A. Determination of urinary biomarkers for assessment of short-term human exposure to aflatoxins in Sao Paulo, Brazil. Toxins 6, 1996–2007, doi: 10.3390/toxins6071996 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura M. et al. Ultra-performance liquid chromatography/tandem mass spectrometry for the simultaneous analysis of aflatoxins B1, G1, B2, G2 and ochratoxin A in beer. Rapid Commun Mass Spectrom 20, 3199–3204, doi: 10.1002/rcm.2723 (2006). [DOI] [PubMed] [Google Scholar]
- Soleimany F., Jinap S., Faridah A. & Khatib A. A UPLC–MS/MS for simultaneous determination of aflatoxins, ochratoxin A, zearalenone, DON, fumonisins, T-2 toxin and HT-2 toxin, in cereals. Food Control 25, 647–653, doi: 10.1016/j.foodcont.2011.11.012 (2012). [DOI] [Google Scholar]
- Chen S. & Zhang H. Development of a microwave-assisted-extraction-based method for the determination of aflatoxins B1, G1, B2, and G2 in grains and grain products. Anal Bioanal Chem 405, 1623–1630, doi: 10.1007/s00216-012-6564-8 (2013). [DOI] [PubMed] [Google Scholar]
- Zheng R. S., Xu H., Wang W. L., Zhan R. T. & Chen W. W. Determination of aflatoxin B1, B2, G1, G2 in armeniacae semen amarum by high-performance liquid chromatography-tandem mass spectrometry. Zhongguo Zhong Yao Za Zhi 38, 3534–3538 (2013). [PubMed] [Google Scholar]
- Huang B., Han Z., Cai Z., Wu Y. & Ren Y. Simultaneous determination of aflatoxins B1, B2, G1, G2, M1 and M2 in peanuts and their derivative products by ultra-high-performance liquid chromatography-tandem mass spectrometry. Anal Chim Acta 662, 62–68, doi: 10.1016/j.aca.2010.01.002 (2010). [DOI] [PubMed] [Google Scholar]
- Liu X. et al. Development of hyperbranched polymers with non-covalent interactions for extraction and determination of aflatoxins in cereal samples. Anal Chim Acta 797, 40–49, doi: 10.1016/j.aca.2013.08.020 (2013). [DOI] [PubMed] [Google Scholar]
- Asghar M. A. et al. Development and validation of a high-performance liquid chromatography method with post-column derivatization for the detection of aflatoxins in cereals and grains. Toxicol Ind Health, doi: 10.1177/0748233714547732 (2014). [DOI] [PubMed] [Google Scholar]
- Liu X. Y. et al. Development of hyperbranched polymers with non-covalent interactions for extraction and determination of aflatoxins in cereal samples. Anal Chim Acta 797, 40–49, doi: 10.1016/j.aca.2013.08.020 (2013). [DOI] [PubMed] [Google Scholar]
- Zheng R., Xu H., Wang W., Zhan R. & Chen W. Simultaneous determination of aflatoxin B1, B2, G1, G2, ochratoxin A, and sterigmatocystin in traditional Chinese medicines by LC–MS–MS. Anal Bioanal Chem 406, 3031–3039, doi: 10.1007/s00216-014-7750-7 (2014). [DOI] [PubMed] [Google Scholar]
- Luna A. S. et al. Simultaneous determination of aflatoxins B2 and G2 in peanuts using spectrofluorescence coupled with parallel factor analysis. Anal Chim Acta 778, 9–14, doi: 10.1016/j.aca.2013.03.038 (2013). [DOI] [PubMed] [Google Scholar]
- Wen J., Kong W., Wang J. & Yang M. Simultaneous determination of four aflatoxins and ochratoxin A in ginger and related products by HPLC with fluorescence detection after immunoaffinity column clean-up and postcolumn photochemical derivatization. J Sep Sci 36, 1–20, doi: 10.1002/jssc.201300885 (2013). [DOI] [PubMed] [Google Scholar]
- Kong W. J. et al. Development of a sensitive and reliable high performance liquid chromatography method with fluorescence detection for high-throughput analysis of multi-class mycotoxins in Coix seed. Anal Chim Acta 799, 68–76, doi: 10.1016/j.aca.2013.08.042 (2013). [DOI] [PubMed] [Google Scholar]
- Borges L. L., Conceicao E. C. & Silveira D. Active compounds and medicinal properties of Myrciaria genus. Food Chem 153, 224–233, doi: 10.1016/j.foodchem.2013.12.064 (2014). [DOI] [PubMed] [Google Scholar]
- Bakkali F., Averbeck S., Averbeck D. & Idaomar M. Biological effects of essential oils–a review. Food Chem Toxicol 46, 446–475, doi: 10.1016/j.fct.2007.09.106 (2008). [DOI] [PubMed] [Google Scholar]
- Abou El-Soud N. H., Deabes M., Abou El-Kassem L. & Khalil M. Chemical Composition and Antifungal Activity of Ocimum basilicum L. Essential Oil. OAMJMS 3, 374, doi: 10.3889/oamjms.2015.082 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. M. et al. The comparison with Kjeldah and Coomassie Brilliant Blue method on testing protein content of the polysaccharide from Sipunculus nudus. Chin J Exp Med Formul 19, 96–98, doi: 10.11653 /syfj2013190096 (2013). [Google Scholar]
- Masuko T. et al. Carbohydrate analysis by a phenol-sulfuric acid method in microplate format. Anal Biochem 339, 69–72, doi: 10.1016/j.ab.2004.12.001 (2005). [DOI] [PubMed] [Google Scholar]
- Wu F. M., Li M. & Wang D. Q. Content determination of fat oil and protein in Cannabis sativa. Res Prac Chin Med 25, 68–70 (2011). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.