

The structure of the N-terminal half of the human RAD52 protein containing three alanine substitutions was solved at 2.4 Å resolution. The structure revealed a previously unobserved interaction between RAD52 rings in which the C-terminal end of one of the monomers in the RAD52 ring associates with the positively charged groove of a symmetry-related RAD52 ring in the crystal lattice.
Keywords: ssDNA-binding protein, single-strand annealing proteins, homologous recombinational repair, surface-entropy reduction, higher order interaction, RAD52
Abstract
The Rad52 protein is a eukaryotic single-strand DNA-annealing protein that is involved in the homologous recombinational repair of DNA double-strand breaks. The isolated N-terminal half of the human RAD52 protein (RAD521–212) forms an undecameric ring structure with a surface that is mostly positively charged. In the present study, it was found that RAD521–212 containing alanine mutations of the charged surface residues (Lys102, Lys133 and Glu202) is highly amenable to crystallization. The structure of the mutant RAD521–212 was solved at 2.4 Å resolution. The structure revealed an association between the symmetry-related RAD521–212 rings, in which a partially unfolded, C-terminal region of RAD52 extended into the DNA-binding groove of the neighbouring ring in the crystal. The alanine mutations probably reduced the surface entropy of the RAD521–212 ring and stabilized the ring–ring association observed in the crystal.
1. Introduction
Homology-based repair is an important mechanism for the accurate repair of DNA double-strand breaks. A key reaction in this repair process is DNA annealing, in which base-pair formation is promoted between the complementary single-stranded DNA regions (Morrical, 2015 ▸). This reaction is promoted by single-strand annealing proteins (SSAPs), which form a superfamily. SSAPs are widely distributed across viral, prokaryotic and eukaryotic species, which suggests the importance of DNA annealing in the DNA-repair process (Iyer et al., 2002 ▸).
Much of our knowledge on the promotion of DNA annealing by SSAPs was gained from studies of the yeast Rad52 and human RAD52 proteins. Yeast Rad52 and human RAD52 form oligomeric rings, a property that appears to be conserved among SSAPs. The crystal structures of the N-terminal half of the human RAD52 protein revealed a positively charged groove that runs around the ring structure (Kagawa et al., 2002 ▸; Singleton et al., 2002 ▸). Mutagenesis and footprinting studies suggested that the single-stranded DNA (ssDNA) binds inside the positively charged groove and wraps around the ring structure (Parsons et al., 2000 ▸; Kagawa et al., 2002 ▸; Lloyd et al., 2005 ▸). A more extensive mutagenesis study suggested the presence of a second DNA-binding site outside the groove (Kagawa et al., 2008 ▸). Thus, RAD52 has two possible DNA-binding sites. However, the precise mechanism by which RAD52 binds to ssDNA and promotes DNA annealing is still unclear. In order to elucidate the molecular mechanism of DNA annealing promoted by RAD52, a more detailed view of the RAD52–ssDNA interaction is required.
Although the structure of the DNA-free form of RAD52 has been determined, a co-crystal structure with DNA has yet to be solved. A possible reason for the difficulty in determining a co-crystal structure could be the inability of RAD52 to form a stable crystal lattice because of its highly positively charged surface that extends beyond the putative DNA-binding groove. In general, charged amino-acid residues on the surface of a protein increase the surface entropy and may inhibit crystallization (Mateja et al., 2002 ▸; Longenecker et al., 2001 ▸; McElroy et al., 1992 ▸). Proteins that are difficult to crystallize for this reason can be mutagenized to modify their surface entropy, which may promote favourable crystal contacts.
In the present study, we performed an X-ray crystallographic study of the N-terminal half of the human RAD52 protein (RAD521–212) containing alanine substitutions at amino-acid positions 102, 133 and 202. In the unsubstituted protein these positions harbour lysine (Lys102 and Lys133) and glutamic acid (Glu202) residues. The side chains of these amino-acid residues are exposed to the solvent and are completely free of interactions in the previously solved structures (PDB entries 1kn0 and 1h2i; Kagawa et al., 2002 ▸; Singleton et al., 2002 ▸). The mutant RAD521–212 readily crystallized, and its structure was determined at 2.4 Å resolution. The structure revealed a ring–ring interaction in the crystal, in which the C-terminal region containing the partially unfolded α-helix from one of the monomers in the RAD52 ring associates with the positively charged groove of the neighbouring ring.
2. Materials and methods
2.1. Protein expression and purification
The human RAD521–212 K102A/K133A/E202A protein was expressed as an N-terminally hexahistidine-tagged protein using the pET-15b vector (Merck Millipore). The human RAD521–212 K102A/K133A/E202A gene was inserted between the NdeI and BamHI sites of the pET-15b vector. The K102A, K133A and E202A mutations were incorporated using the QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies). RAD52 was expressed in Escherichia coli strain JM109(DE3) bearing a plasmid (pArg3Arg4, a gift from K. Sakamoto, RIKEN Center for Life Science Technologies, Yokohama, Japan) for the expression of low-abundance arginine tRNAs. To express RAD52, freshly grown colonies were directly transferred to 800 ml LB medium and grown at 30°C until the culture reached an optical density (A 600) of 0.6. RAD52 expression was then induced by adding β-d-1-thiogalactopyranoside to a final concentration of 0.5 mM. After an overnight culture, the cells were collected by centrifugation and resuspended in approximately 35 ml buffer A (50 mM Tris–HCl pH 7.8, 0.3 M KCl, 10% glycerol) containing 10 mM imidazole. The cells were lysed by sonication and the insoluble material was cleared from the resulting cell lysate by centrifugation at 35 200g for 30 min. The supernatant was mixed with 3 ml of nickel–nitrilotriacetic acid agarose beads (Qiagen) and was incubated at 4°C with gentle mixing for 1 h. The mixture was poured into an Econo-Column (Bio-Rad) and the unbound cell lysate was drained out. The agarose beads packed inside the column were washed with 90 ml buffer A containing 50 mM imidazole. The protein was eluted with a 90 ml linear gradient of 50–400 mM imidazole in buffer A. To remove the hexahistidine tag, the eluted protein was mixed with 5 units of thrombin protease (Wako Pure Chemical Industries) per milligram of RAD52 and was immediately dialyzed against buffer B (20 mM HEPES–KOH pH 7.5, 0.2 M KCl, 0.5 mM EDTA, 5% glycerol, 2 mM 2-mercaptoethanol). After several exchanges of buffer B, the dialyzed protein was collected and applied onto a 2.1 ml SP Sepharose (GE Healthcare) column equilibrated with buffer B. RAD52 was collected from the flowthrough fraction and applied onto a 2.1 ml Q Sepharose (GE Healthcare) column equilibrated with buffer B. RAD52 was collected from the flowthrough fraction and purified on a HiLoad 26/60 Superdex 200 (GE Healthcare) column equilibrated with buffer B. The purified RAD52 was concentrated to approximately 24 mg ml−1 using a Vivaspin 20 (50K MWCO) concentrator (Sartorius). The concentration of RAD52 was determined from the absorbance at 280 nm, using an extinction coefficient of 20 400 M −1 cm−1. The extinction coefficient was calculated with the ProtParam tool on the ExPASy website (http://web.expasy.org/protparam/).
2.2. Crystallization and diffraction data collection
The purified RAD521–212 K102A/K133A/E202A protein (24 mg ml−1) retained DNA-binding activity (data not shown) and thus was mixed with a polydeoxythymine 40-mer prior to crystallization. Crystals of RAD521–212 K102A/K133A/E202A were grown at 293 K using the hanging-drop vapour-diffusion method by mixing 1 µl protein solution with 1 µl reservoir solution, which consisted of a fourfold H2O-diluted solution from PEG/Ion or PEG/Ion 2 (Hampton Research). The reservoir contained 500 µl of the crystallization solution. The crystals typically grew to their full size within 1–3 d. The crystals were cryoprotected in a reservoir solution supplemented with 30%(v/v) ethylene glycol and flash-cooled in a stream of nitrogen gas (∼100 K). X-ray diffraction data were collected using an Eiger X 4M or a Pilatus 2M-F detector on BL-1A or a Pilatus3 S 6M detector on BL-17A at the Photon Factory, Tsukuba, Japan. The data sets were processed and scaled using XDS (Kabsch, 2010 ▸).
2.3. Structural determination and refinement
The structure of RAD521–212 K102A/K133A/E202A, which crystallized in multiple space groups, was solved by the molecular-replacement method using MOLREP (Vagin & Teplyakov, 2010 ▸). The coordinates of the RAD521–212 structure (PDB entry 1kn0), which lacks amino-acid substitutions, were used as the search model. To confirm that the molecular-replacement solution was correct, rigid-body refinement followed by energy minimization and B-factor refinement of the molecular-replacement solution were performed using CNS (Brünger et al., 1998 ▸; Brunger, 2007 ▸), and the resulting R free and R work values, along with the initially calculated 2F o − F c map, were inspected. The electron density for the ssDNA was absent from all of the structures. For the highest resolution data, the structural model was refined to completion using PHENIX (Adams et al., 2010 ▸) instead of CNS. Iterative rounds of manual model correction using Coot (Emsley et al., 2010 ▸) and refinement with PHENIX using individual isotropic atomic displacement parameters were performed until there were no Ramachandran outliers. A noncrystallographic symmetry restraint was applied to the 11 monomers within the RAD52 ring throughout the refinement. All structure figures were created using PyMOL (http://www.pymol.org).
3. Results and discussion
3.1. Crystallization of RAD521–212 containing surface mutations
The previously determined crystal structures of the isolated N-terminal half of the human RAD52 protein revealed that the surface of the ring structure is mostly positively charged (Kagawa et al., 2002 ▸; Singleton et al., 2002 ▸). As it is generally known that substituting charged surface residues with alanines reduces the surface entropy, and that the substitutions can make the protein more amenable to crystallization (Mateja et al., 2002 ▸; Longenecker et al., 2001 ▸; McElroy et al., 1992 ▸), we considered whether the introduction of alanine mutations at such residues could further promote the crystallization of RAD521–212. This could be particularly useful when attempting to co-crystallize RAD52 with DNA or other interacting proteins, provided that the mutation site is not the direct binding site for these factors. In the previously determined RAD521–212 structure, the side chains of Lys102, Lys133, Lys190 and Glu202 are completely exposed to the solvent and do not interact with other amino-acid residues. Lys102 and Lys133 constitute a binding site for double-stranded DNA (Kagawa et al., 2008 ▸), and thus the RAD52 mutant containing alanine substitutions of these residues may have limited usefulness for co-crystallization with double-stranded DNA. In contrast, Lys190 and Glu202 are located outside the highly conserved region (Fig. 1 ▸ f) and have no known functions.
Figure 1.

Crystal structure of the human RAD521–212 K102A/K133A/E202A protein. The mutant RAD521–212 structure is depicted as a ribbon representation, and its side (a), bottom (b) and top (c) views are shown. Amino-acid positions 102, 133 and 202 (coloured red, blue and purple, respectively) are shown as spheres. (d) A surface representation of the mutant RAD521–212 structure, viewed from the side [the same view as in (a)]. The surface is coloured according to the local electrostatic potential, from −5.0k B T (red) to +5.0k B T (blue). (e) A surface representation of nonmutated RAD521–212, coloured similarly to the mutant RAD521–212 shown in (d). (f) Amino-acid sequence comparison of human, mouse, chicken and yeast (Saccharomyces cerevisiae) RAD52 proteins. Amino-acid positions 102, 133 and 202 (coloured red, blue and purple, respectively) are indicated by circles. The amino-acid residues of human RAD52 that are important for DNA binding and self-association are indicated by black and grey triangles, respectively.
To examine the effects of substituting these residues with alanine on the crystallization, we constructed several mutant RAD521–212 proteins, each containing a different combination of mutations, and screened the crystallization conditions for each mutant. We found that RAD521–212 with K102A, K133A and E202A mutations yielded crystals that diffracted X-rays to the highest resolution (Fig. 1 ▸ a). The structure of RAD521–212 K102A/K133A/E202A was solved using the molecular-replacement method (Table 1 ▸). The diffraction data analysis revealed that the RAD521–212 K102A/K133A/E202A crystal belonged to the primitive monoclinic space group P21, with one undecameric RAD52 ring in the asymmetric unit. Notably, the solvent content of the crystal was 49.9%, which is similar to those of previously determined RAD52 structures (51.2% for PDB entry 1kn0 and 55.1% for PDB entry 1h2i).
Table 1. Statistics for crystallographic structural determination.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Beamline | BL-17A, Photon Factory |
| Wavelength (Å) | 0.9800 |
| Space group | P21 |
| Unit-cell parameters (Å, °) | a = 111.0, b = 98.5, c = 116.4, β = 92.0 |
| Resolution range (Å) | 49.26–2.40 (2.55–2.40) |
| Unique reflections | 190708 |
| Multiplicity | 3.5 (3.5) |
| Completeness (%) | 99.6 (99.0) |
| R merge † (%) | 9.5 (63.3) |
| R meas (%) | 11.3 (74.7) |
| 〈I/σ(I)〉 | 9.2 (1.9) |
| CC1/2 (%) | 99.5 (74.5) |
| Refinement | |
| Resolution (Å) | 49.3–2.4 |
| R factor/free R factor | 22.4/27.3 |
| B factor (Å2) | 58.6 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.008 |
| Bond angles (°) | 1.02 |
| Ramachandran plot (%) | |
| Favoured regions | 97.8 |
| Allowed regions | 2.2 |
| Outliers | 0 |
R
merge = 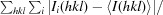
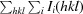 , where Ii(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations I of reflection hkl.
, where Ii(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations I of reflection hkl.
3.2. Structure of RAD521–212 K102/K133A/E202A
A superimposition of the RAD521–212 K102A/K133A/E202A structure with the previously determined RAD521–212 structure (PDB entry 1kn0) revealed that the root-mean-square difference between the two structures is 0.408 Å (over 12 553 atoms), indicating that the lysine-to-alanine substitutions did not affect the overall structure. Furthermore, a visual inspection of the mutation sites did not reveal any significant local structural differences between the two proteins. In contrast, the surface potential of the RAD521–212 K102A/K133A/E202A structure was quite different compared with that of nonmutated RAD521–212 (Figs. 1 ▸ d and 1 ▸ e). This significant change in the surface potential is likely to be owing to the fact that the mutation sites are located close to each other, near the central channel of the ring (Figs. 1 ▸ b and 1 ▸ c).
In the crystal, the three mutated amino-acid positions were located near each other at the crystal-packing interface (Figs. 2 ▸ a and 2 ▸ b). This ring–ring association suggested that this crystal packing would be destabilized if one of the residues remained as the wild-type amino-acid residue.
Figure 2.

Interaction between symmetry-related mutant RAD521–212 rings. (a) Symmetry-related RAD52 rings are shown in blue, green and magenta. The boxed region of the mutant RAD521–212 ring structure is engaged in the crystal packing. The C-terminal region beyond the partially unfolded α-helix from one of the monomers, which interacts with the neighbouring RAD52 ring, is encircled by a dashed red oval. (b) A close-up view of the boxed region in (a). Ala102, Ala133 and Ala202 are located close to each other at the crystal-packing interface.
3.3. Association between symmetry-related RAD521–212 K102/K133A/E202A rings
In the present structure, we unexpectedly found a partially unfolded, C-terminal α-helix in one of the monomers in the undecameric ring (Fig. 3 ▸ a). The remaining C-terminal α-helices in the other monomers in the ring structure were folded and their structures were quite similar to those in the previously determined RAD52 structures (Fig. 3 ▸ c). These C-terminal α-helices form approximately 2.2 turns and are positioned between the N-terminal α-helices (Figs. 3 ▸ a and 3 ▸ b). In contrast, the unfolded α-helix makes approximately 1.7 turns, and the C-terminal region beyond the helix points away from the central channel. Interestingly, the electron density of the C-terminal region was visible to the C-terminus (Fig. 3 ▸ d) and this region extended into the positively charged groove of the neighbouring undecameric ring in the crystal (Figs. 2 ▸ a and 3 ▸ d). Notably, Arg209 in the extended region was positioned between two RAD52 monomers and appeared to form hydrogen bonds to Glu145 from one monomer and to Glu140 from the other monomer of the neighbouring RAD52 ring (Fig. 3 ▸ e). We note that the mutant RAD52 construct used in the present study lacks the C-terminal 206 amino-acid residues (213–418); it has too many missing residues for it to be accomodated in the positively charged groove. The human RAD52 protein has been demonstrated to form higher order complexes containing multiple multimeric rings (Ranatunga et al., 2001 ▸). The deletion of the C-terminal half of the protein impairs the formation of higher order complexes, indicating that the C-terminal half is responsible for the association between rings. Further investigations are required to determine whether the ring–ring interactions observed in the present study are biologically relevant.
Figure 3.

The C-terminal structure of the mutant RAD521–212. (a) The location of the partially unfolded C-terminal α-helix. The RAD52 monomer containing the unfolded α-helix is coloured magenta. (b) A close-up view of the unfolded α-helix. The C-terminal helices are positioned between the N-terminal helices, such that the N- and C-terminal helices alternate at the surface of the ring near the central channel. The unfolded α-helix is about half a turn shorter than the nearby C-terminal helices. (c) Schematic diagram of the secondary structures of RAD521–212 K102A/K133A/E202A that were visible in the crystal structure. The top diagram (green) corresponds to the RAD52 monomers with a folded C-terminal α-helix. The bottom diagram (magenta) corresponds to the RAD52 monomer containing the unfolded C-terminal α-helix. The RAD52 monomer containing an unfolded α-helix was visible to the C-terminus, whereas the other RAD52 monomers were only visible to amino-acid position 208. (d) The C-terminal region from the mutated RAD521–212 ring (green) is depicted as a stick representation and its 2F o − F c electron-density map (contoured at 1.0σ) is shown. (e) Hydrogen bonding by Arg209 in the C-terminal region. Hydrogen bonds are depicted as dashed lines.
Supplementary Material
PDB reference: RAD521–212 K102A/K133A/E202A mutant, 5jrb
Acknowledgments
The synchrotron-radiation experiments were performed on the BL-1A and BL-17A beamlines at the Photon Factory (Proposal Nos. 2014G174 and 2014G556). We are grateful to the beamline scientists for their assistance with data collection. This work was supported in part by JSPS KAKENHI Grant Nos. 24570138 (to WK) and 25250023 (to HK). This work was also supported in part by MEXT KAKENHI Grant Nos. 26116521 (to WK) and 25116002 (to HK). HK was also supported by the Waseda Research Institute for Science and Engineering.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Brunger, A. T. (2007). Nature Protoc. 2, 2728–2733. [DOI] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Iyer, L. M., Koonin, E. V. & Aravind, L. (2002). BMC Genomics, 3, 8. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kagawa, W., Kagawa, A., Saito, K., Ikawa, S., Shibata, T., Kurumizaka, H. & Yokoyama, S. (2008). J. Biol. Chem. 283, 24264–24273. [DOI] [PMC free article] [PubMed]
- Kagawa, W., Kurumizaka, H., Ishitani, R., Fukai, S., Nureki, O., Shibata, T. & Yokoyama, S. (2002). Mol. Cell, 10, 359–371. [DOI] [PubMed]
- Lloyd, J. A., McGrew, D. A. & Knight, K. L. (2005). J. Mol. Biol. 345, 239–249. [DOI] [PubMed]
- Longenecker, K. L., Garrard, S. M., Sheffield, P. J. & Derewenda, Z. S. (2001). Acta Cryst. D57, 679–688. [DOI] [PubMed]
- Mateja, A., Devedjiev, Y., Krowarsch, D., Longenecker, K., Dauter, Z., Otlewski, J. & Derewenda, Z. S. (2002). Acta Cryst. D58, 1983–1991. [DOI] [PubMed]
- McElroy, H. E., Sisson, G. W., Schoettlin, W. E., Aust, R. M. & Villafranca, J. E. (1992). J. Cryst. Growth, 122, 265–272.
- Morrical, S. W. (2015). Cold Spring Harb. Perspect. Biol. 7, a016444. [DOI] [PMC free article] [PubMed]
- Parsons, C. A., Baumann, P., Van Dyck, E. & West, S. C. (2000). EMBO J. 19, 4175–4181. [DOI] [PMC free article] [PubMed]
- Ranatunga, W., Jackson, D., Lloyd, J. A., Forget, A. L., Knight, K. L. & Borgstahl, G. E. O. (2001). J. Biol. Chem. 276, 15876–15880. [DOI] [PubMed]
- Singleton, M. R., Wentzell, L. M., Liu, Y., West, S. C. & Wigley, D. B. (2002). Proc. Natl Acad. Sci. USA, 99, 13492–13497. [DOI] [PMC free article] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: RAD521–212 K102A/K133A/E202A mutant, 5jrb