

Abstract
Catalytic allylboron additions to aldimines are presented for which small amounts of Zn(OMe)2 serve as co-catalyst to accelerate allyl exchange and 1,3-borotropic shift processes. Low yielding and moderately α-, and diastereoselective reactions are thus turned into highly efficient, γ-, diastereo- and enantioselective transformations that exhibit considerable scope.
Keywords: boron, borotropic shift, catalysis, diastereoselective synthesis, enantioselective catalysis, homoallylic amines
Graphical abstract

Otherwise α-selective crotyl additions to aldimines are rendered highly γ-selective by adding small amounts of Zn(OMe)2 into the reaction mixture. The Lewis acidic co-catalyst is believed to accelerate borotropic shifts that are responsible for the selectivity reversal, and cause faster exchange processes that generate the catalytic active chiral allylboron species.
Catalytic enantioselective addition of unsaturated organoboron compounds to imines is a convenient way of synthesizing homoallylic amines.[1] When the allylboron reagent is terminally substituted (R1 ≠ H, Scheme 1a), product identity and its isomeric purity hinges on how many 1,3-boryl shifts occur before a C–C bond is formed (Scheme 1a): α addition is observed if the C–B bond relocates once from the Cα to the Cγ carbon, and none or two relays means net γ addition. Rational strategies for reliable control of boryl isomerizations are therefore much needed.
Scheme 1.

The role of boryl relays and shifts and the challenge of controlling them in the design of a catalytic process.
Catalysts assembled in situ from (pin)B-derived (pin, pinacolato) reagents and an enantiomerically pure aminophenol (e.g., 1a, Scheme 1b) facilitate additions of allylboronates to imines.[2] These transformations proceed with high α selectivity (Scheme 1b)[3] because of a pair of γ-selective events: one delivers organoboron intermediate Z-i and the other the homoallylic amide product. However, a preferred approach would involve converting a more easily accessible achiral organoboron reagent to the same type of product by a γ-, diastereo- and enantioselective route (Scheme 1c).[4, 5] One way of achieving this would entail inclusion of a 1,3–boron shift in a re-designed pathway. Herein, we describe the implementation of this objective.
Our plan was to induce a borotropic shift (iv→v→Z-i, Scheme 2) that would take place between the γ-selective processes (ii→iii→iv and Z-i→vi→ii). The driving force would be lowering of steric strain caused by the tertiary boron-substituted carbon center in iv and formation of a more substituted alkene. The question then was: How might conditions be altered so that iv isomerizes to Z-i via v faster than it reacts with a phosphinoylimine?
Scheme 2.

A 1,3-borotropic shift (iv→v→Z-i) might give rise to preferential formation of γ-addition products.
1,3-Borotropic shifts have been examined extensively,[6] and several observations have been attributed to them.[7] Hancock has shown[6a–b] that the facility of these isomerizations depends on electronic attributes of the boron center. For instance, there is <2% rearrangement with the diaminoboryl system A at 170 °C after 24 hours (Scheme 3) but the (amino)alkyl derivative B isomerizes at 150 °C in six hours, and the boryl shift in dialkylboryl compound D takes place at −78 °C (6 h). Cross-over experiments indicate that rearrangements are intramolecular.[6a–b] Congruent with Hancock's conclusions is the proposal by Aggarwal[7o] that an (alkoxy)alkylboron species (C) rearranges at lower temperature (−78→22 °C, 14 h); the energy barrier calculated by us for this transformation supports the suggested scenario (Scheme 3).[8] In the systems more directly relevant to the studies below (E and F, Scheme 3), the ammonium salt derivative (cf. F) should rearrange more readily. Still, the high α selectivities in the original studies[2] (cf. Scheme 1b) mean that the boron atom within a chiral complex (e.g., iv, Scheme 2) is not Lewis acidic enough for swift borotropic shifts.
Scheme 3.

Previous observations and calculations performed in this study (with ωB97XD/Def2TZVPP functional, solvation in toluene) point to a significant substituent influence on the rates of borotropic shifts (the more π-donating, the slower the rearrangement). [a] For the experimental observation: alkyl = nBu; R = C(Me)2C(Me)2OLi. For calculation: alkyl = Et; R = Me.
The idea of deploying a Lewis acidic co-catalyst was based on the following reasoning: Firstly, as illustrated in I (Figure 1), by coordination to the aryloxy arm of the chiral complex, the boron center's Lewis acidity and thus the rate of isomerization could be enhanced (i.e., iv→v→Z-i, Scheme 2). Secondly, association of the amide carbonyl with a Lewis acid (II, Figure 1) could disrupt its coordination with the boron atom (III), facilitating allyl exchange (L = OMe) and/or 1,3-borotropic shift (L = allylic group). DFT calculations[8] intimated that complex III (without the Lewis acid) is favored, although reaction most probably proceeds via II (and/or the complex without a bound Lewis acid).
Figure 1.

Possible ways through which a Lewis acid may facilitate borotropic shift.
Our initial foray uncovered another problem (Scheme 4): under the previous conditions,[2] reaction of E-crotyl–B(pin) (E-3) with imine 2a resulted in only 35% conversion after 24 h [γ:α = 25:75; 88:12 diastereomeric ratio (d.r.) for the γ product]. We surmised that the low rate might be because the methyl substituent [vs. a terminal olefin in Scheme 1b or with allyl–B(pin)] causes the chiral allylboron intermediate to be generated inefficiently for steric factors (i.e., ii→iii→iv, Scheme 2). We wondered if coordination of the same Lewis acidic co-catalyst to the B(pin) moiety[9] might strengthen chelation of its boron center with the chiral complex's methoxy unit to accelerate allyl transfer as well (Scheme 4).
Scheme 4.

Possible role of a Lewis acidic co-catalyst in facilitating the initial allyl transfer.
Several Lewis acidic metal salts were screened,[8] resulting in the discovery that with 2.5 mol % Zn(OtBu)2 (Scheme 5a) not only may efficiency be improved substantially (>98% vs. 35% conv. with NaOtBu; Scheme 4), the γ-addition product was generated predominantly (γ:α = 25:75) in 89:11 d.r. and 95:5 enantiomeric ratio (e.r.). The t-butyl substituent meant that a diminutive co-catalyst could be better. Indeed, with the smaller Zn(OMe)2, γ:α ratio (90:10) and diastereo- and enantioselectivity were higher still (94:6 d.r. and 96.5:3.5 e.r.).[10] The transformation with the Z organoboron reagent proceeded to 97% conversion in three hours (vs. 24 h with E-3), furnishing the same diastereomer as with the E isomer in >98% γ selectivity, 93:7 d.r. and 96:4 e.r. (Scheme 5b). Control experiments indicate that the presence of Z-3 does not cause any isomerization.
Scheme 5.

The influence of Zn(OMe)2 and crotyl–B(pin) stereochemistry on efficiency and selectivity.
The data in Scheme 6 offer further insight.[11] Reaction with deuterium-labeled allyl–B(pin) 6 led to high α selectivity when NaOtBu was present (96:4 d2-7a:d2-7b; Scheme 6a) but with Zn(OMe)2 the γ product was formed preferentially (g:a = 70:30). The impact of the co-catalyst thus extends to the unsubstituted organoboron system. The γ:α ratio is lower (vs. with E- or Z-3, Scheme 5) likely because the less hindered chiral allylboron intermediate (cf. iv, G = H) reacts faster with the imine, competing more easily with a borotropic shift. There is no steric or electronic impetus for isomerization (see above). Furthermore, with rac-8, the product from α-addition was formed exclusively in 62:38 d.r. (Scheme 6b), indicating that conversion to E- and Z-i is probably followed by an eventual γ addition to the imine (2a).[12] The predominant formation of the anti diastereomer in the reaction with E- or Z-crotyl–B(pin) (E- or Z-3; Scheme 5) must therefore originate from a kinetic preference for isomerization to afford the chiral Z-allylboron intermediate (Z-i, Scheme 2).
Scheme 6.
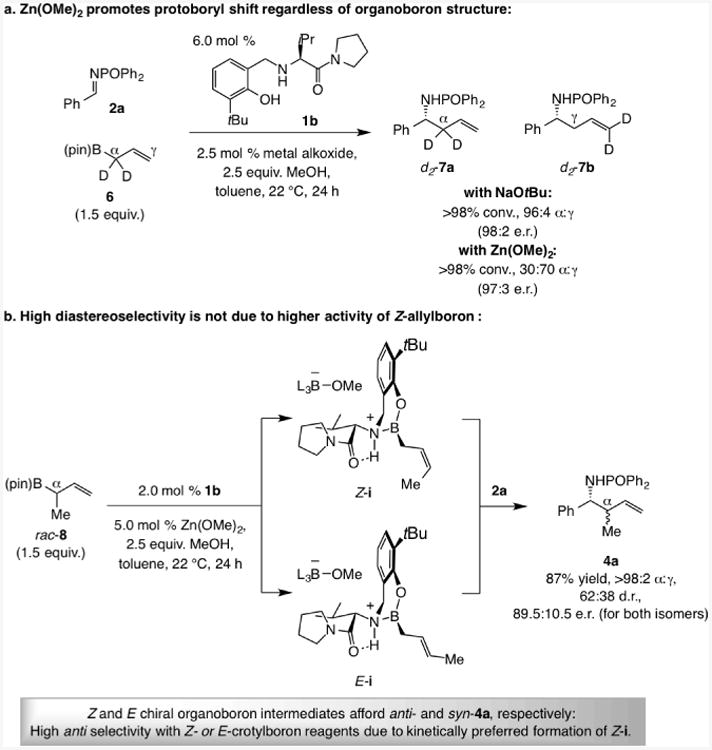
Experiments indicating that (a) Zn(OMe)2 promotes borotropic shift and (b) formation of anti diastereomer with E and Z organoboron reagent is due to a kinetic preference for the formation of the chiral Z-crotylboron intermediate.
The routes in Scheme 7 account for the kinetically controlled stereoselective Lewis acid promoted 1,3-borotropic shifts. With the higher energy Z reagent, complex iv is produced faster than isomeric viii, generated from the corresponding E isomer. In either instance, regardless of the stereochemistry of the initial allylboronate, isomerization proceeds via transition states wherein the substituent (G) is oriented away from the chiral complex (cf. vii and ix, Scheme 7). Allylboron intermediate Z-i[13] and the anti diastereomer of the γ-addition product are hence favored. That is, diastereoselectivities are high not because the Z and E isomers rapidly interconvert and the former reacts faster (i.e., Curtin–Hammett kinetics are not operative). Moreover, regardless of crotyl–B(pin) stereochemistry, product identity does not originate from the E and Z chiral allylboron isomers reacting via different transition structures to give the same stereoisomer.[4f]
Scheme 7.

Rationale for preferential generation of Z-crotylboryl intermediate (Z-i), irrespective of the reagent stereochemistry.
The catalytic protocol has considerable scope (Chart 1).[11] Reactions were often complete with 2.0 mol % aminophenol 1b and 5.0 mol % Zn(OMe)2 within three hours at ambient temperature. Aryl-substituted imines with varying electronic attributes were converted to the desired products efficiently and selectively (4a-4l). Similar results were obtained with heterocyclic moieties, including those containing an O- (4m-n), S- (4o-p) or N-substituent (4q). Additions to α,β-unsaturated aldimines needed more forcing conditions (cf. 4r). Transformations with alkyl-substituted imines were equally efficient and considerably γ-selective but less diastereo- and enantioselective. This may be attributed to the diminished rates of addition to the more electron-rich aliphatic substrates, allowing for alternative (uncatalyzed) pathways with different kinetic attributes (i.e., different turnover limiting step) to become competitive; related arguments may apply to the reaction leading to p-dimethylaminoaryl-substituted 4i. Other than the processes with commercially available crotyl–B(pin) reagent Z-3, synthesis of 9 and especially that of chloro-substituted 10[14] underscore the method's reach and versatility.
Reactions are scalable (cf. 4k, Scheme 8), and the ligands and organoboron reagents are easy to access and manipulate. In most cases, the phosphinoyl group renders the substrates and products crystalline and isolable without chromatography and is removable under mild conditions at low cost.[2a] These characteristics compare favorably to the alternative strategies.[4] Homoallylic amines are precursors to an array of important enantiomerically enriched N-containing molecules, including β-amino acids.[15,16]
Scheme 8.

Products obtained from γ-, diastereo- and enantioselective additions of different organoboron reagents (>98% E for phenyl-substituted allylboronate and >98% Z for Cl-substituted reagent) to various aldimines. [a] 5.0 mol % 1b was used. [b] Reaction time, 24 h. [c] Reaction time, 5 h. [d] Reaction time, 4 h. [e] With 10 mol % 1b and Zn(OMe)2 after 8 h. [f] With 12.5 mol % 1b and 7.5 mol % Zn(OMe)2 was used. [g] At 50 °C, 6 h. [h] 10 mol % 1b and Zn(OMe)2, 24 h. See the Supporting Information for details.
The concepts elucidated here should find applications in the design of other efficient catalytic and stereoselective processes.
Supplementary Material
Acknowledgments
Financial support was provided by the NIH (GM-57212 and in part GM-59426). We are grateful to Daiichi Sankyo Co. for supporting H. M. as a Visiting Fellow, to E. M. Vieira, S. Torker and C. Qin for helpful discussions and to R. Morrison for experimental assistance.
References & Footnotes
- 1.For recent reviews on enantioselective synthesis through additions of allyl groups to ketones and imines, see: Yus M, González-Gómez JC, Foubelo F. Chem Rev. 2011;111:7774–7854. doi: 10.1021/cr1004474.
- 2.a) Silverio DL, Torker S, Pilyugina T, Vieira EM, Snapper ML, Haeffner F, Hoveyda AH. Nature. 2013;494:216–221. doi: 10.1038/nature11844. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wu H, Haeffner F, Hoveyda AH. J Am Chem Soc. 2014;136:3780–3783. doi: 10.1021/ja500374p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.For another example of α-selective catalytic enantioselective allyl addition to imines, see: Chakrabarti A, Konishi H, Yamaguchi M, Schneider U, Kobayashi S. Angew Chem Int Ed. 2010;49:1838–1841. doi: 10.1002/anie.200906308.
- 4.For catalytic diastereo- and enantioselective addition of allyl units to different types of aldimines, see: Fang X, Johannsen M, Yao S, Gathergood N, Hazell RG, Jørgensen KA. J Org Chem. 1999;64:4844–4849. doi: 10.1021/jo990238+.Gastner T, Ishitani H, Akiyama R, Kobayashi S. Angew Chem Int Ed. 2001;40:1896–1898.Ferraris D, Young B, Cox C, Dudding T, Drury WJ, III, Ryzhkov L, Taggi AE, Lectka T. J Am Chem Soc. 2002;124:67–77. doi: 10.1021/ja016838j.Tan KL, Jacobsen EN. Angew Chem Int Ed. 2007;46:1315–1317. doi: 10.1002/anie.200603354.Lou S, Moquist PN, Schaus SE. J Am Chem Soc. 2007;129:15398–15404. doi: 10.1021/ja075204v.Fujita M, Nagano T, Schneider U, Hamada T, Ogawa C, Kobayashi S. J Am Chem Soc. 2008;130:2914–2915. doi: 10.1021/ja710627x.Naodovic M, Wadamoto M, Yamamoto H. Eur J Org Chem. 2009:5129–5131. doi: 10.1002/ejoc.200900669. h) Ref. [3]; Momiyama N, Nashimoto H, Terada M. Org Lett. 2011;13:2126–2129. doi: 10.1021/ol200595b.Luo Y, Hepburn HB, Chotsaeng N, Lam HW. Angew Chem Int Ed. 2012;51:8309–8313. doi: 10.1002/anie.201204004.Chen TY, Tsutsumi R, Montgomery TP, Volchkov I, Krische MJ. J Am Chem Soc. 2015;137:1798–1801. doi: 10.1021/ja5130258.
- 5.For representative diastereoselective additions of allyl fragments with enantiomerically pure reagents and/or aldimines, respectively, see: Hanessian S, Yang RY. Tetrahedron Lett. 1996;37:5273–5276.Gao Y, Sato F. J Org Chem. 1995;60:8136–8137.Berger R, Rabbat PMA, Leighton JL. J Am Chem Soc. 2003;125:9596–9597. doi: 10.1021/ja035001g.Kobayashi S, Ogawa C, Konishi H, Sugiura M. J Am Chem Soc. 2003;125:6610–6611. doi: 10.1021/ja035061m.Reddy LR, Hu B, Prashad M, Prasad K. Org Lett. 2008;10:3109–3112. doi: 10.1021/ol800998u.Takahashi M, McLaughlin M, Micalizio GC. Angew Chem Int Ed. 2009;48:3648–3652. doi: 10.1002/anie.200900236.Feske MI, Santanilla AB, Leighton JL. Org Lett. 2010;12:688–691. doi: 10.1021/ol9026864.Liu M, Shen A, Sun XW, Deng F, Xu MH, Lin GQ. Chem Commun. 2010;46:8460–8462. doi: 10.1039/c0cc03230a.Arena G, Zill N, Salvadori J, Girard N, Mann A, Taddei M. Org Lett. 2011;13:2294–2297. doi: 10.1021/ol200557r.Guo T, Song R, Yuan BH, Chen XY, Sun XW, Lin GQ. Chem Commun. 2013;49:5402–5404. doi: 10.1039/c3cc42481b.Soares de Rogo Barros O, Sirvent JA, Foubelo F, Yus M. Chem Commun. 2014;50:6898–6901. doi: 10.1039/c4cc02317j.
- 6.a) Hancock KG, Kramer JD. J Am Chem Soc. 1973;95:6463–6465. [Google Scholar]; b) Hancock KG, Kramer JD. J Organomet Chem. 1974;64:C29–C31. [Google Scholar]; c) Henriksen U, Snyder JP, Halgren TA. J Org Chem. 1981;46:3767–3768. [Google Scholar]; d) Bühl M, von Ragué Schleyer P, Ibrahim MA, Clark T. J Am Chem Soc. 1991;113:2466–2471. [Google Scholar]; e) Bubnov YN, Gurskii ME, Gridnev ID, Ignatenko AV, Ustynyuk YA, Mstislavsky VI. J Organomet Chem. 1992;424:127–132. [Google Scholar]; f) Gridnev ID, Gursky ME, Bubnov YN. Organometallics. 1996;15:3696–3702. [Google Scholar]; g) Bubnov YN. Pure Appl Chem. 1987;59:895–906. [Google Scholar]; h) Choi JY, Kim CK, Kim CK, Lee I. J Phys Chem A. 2002;106:5709–5715. [Google Scholar]; i) Ess DH, Kister J, Chen M, Roush WR. Org Lett. 2009;11:5538–5541. doi: 10.1021/ol902364d. [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Gurskii ME, Belyakov PA, Lyssenko KA, Semenova AL, Bubnov YN. Russian Chem Bull Int Ed. 2014;63:480–486. [Google Scholar]
- 7.a) Zweifel G, Horng A. Synthesis. 1973:672–674. [Google Scholar]; b) Kramer GW, Brown HC. J Organomet Chem. 1977;132:9–27. [Google Scholar]; c) Zweifel G, Backlund SJ, Leung T. J Am Chem Soc. 1978;100:5561–5562. [Google Scholar]; d) Brown HC, Liotta R, Kramer GW. J Am Chem Soc. 1979;101:2966–2970. [Google Scholar]; e) Hoffmann RW, Zeiβ HJ. J Org Chem. 1981;46:1309–1314. [Google Scholar]; f) Wang KK, Nikam SS, Ho CD. J Org Chem. 1983;48:5376–5377. [Google Scholar]; g) Brown HC, Jadhav PK, Bhat KS. J Am Chem Soc. 1985;107:2564–2565. [Google Scholar]; h) Brown HC, Rangaishenvi MV, Jayaraman S. Organometallics. 1992;11:1948–1954. [Google Scholar]; i) Lombardo M, Morganti S, Tozzi M, Trombini C. Eur J Org Chem. 2002:2823–2830. [Google Scholar]; j) Burgos CH, Canales E, Matos K, Soderquist JA. J Am Chem Soc. 2005;127:8044–8049. doi: 10.1021/ja043612i. [DOI] [PubMed] [Google Scholar]; k) Gonzalez AZ, Soderquist JA. Org Lett. 2007;9:1081–1084. doi: 10.1021/ol070074g. [DOI] [PubMed] [Google Scholar]; l) Canales E, Gonzalez AZ, Soderquist JA. Angew Chem Int Ed. 2007;46:397–399. doi: 10.1002/anie.200603467. [DOI] [PubMed] [Google Scholar]; m) Fang GY, Aggarwal VK. Angew Chem Int Ed. 2007;46:359–362. doi: 10.1002/anie.200603659. [DOI] [PubMed] [Google Scholar]; n) Gonzalez AZ, Román JG, Alicea E, Canales E, Soderquist JA. J Am Chem Soc. 2009;131:1269–1273. doi: 10.1021/ja808360z. [DOI] [PubMed] [Google Scholar]; o) Chen JLY, Scott HK, Hesse MJ, Willis CL, Aggarwal VK. J Am Chem Soc. 2013;135:5316–5319. doi: 10.1021/ja401564z. [DOI] [PubMed] [Google Scholar]; p) Chen M, Roush WR. J Am Chem Soc. 2013;135:9512–9517. doi: 10.1021/ja4033633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.See the Supporting Information for details.
- 9.For representative reports where Lewis acid association with a B(pin) moiety has been proposed, see: Yamamoto H, Futatsugi K. Angew Chem Int Ed. 2005;44:1924–1942. doi: 10.1002/anie.200460394.Rauniyar V, Zhai H, Hall DG. J Am Chem Soc. 2008;130:8481–8490. doi: 10.1021/ja8016076.Barnett DS, Moquist PN, Schaus SE. Angew Chem Int Ed. 2009;48:8679–8682. doi: 10.1002/anie.200904715.Wang H, Jain P, Antilla JC, Houk KN. J Org Chem. 2013;78:1208–1215. doi: 10.1021/jo302787m.
- 10.Allylzinc reagents are likely not involved because such highly nucleophilic entities would not survive the reaction conditions, demonstrated previously to be acidic (see Ref. [2a]).
- 11.Catalyst screening indicated that aminophenol 1b generally delivers somewhat higher enantioselectivity compared to 1a.
- 12.Initial isomerization of rac-8 to E- and Z-3 (prior to the first γ-addition to generate the chiral allylboron iii) is unlikely, as in that case anti-4a would be formed with high diastereoselectivity. The stronger propensity of an aminophenol-based allylboron to undergo a 1,3-borotropic shift is almost certainly due to its more Lewis acidic boron atom.
- 13.Attempts to observe Z-i through NMR spectroscopy were unsuccessful, as only a minute fraction of the aminophenol present is converted to the active catalyst.
- 14.The chloro-substituted allylboron reagent was prepared by catalytic Z-selective cross-metathesis; see: Koh MJ, Nguyen TT, Zhang H, Schrock RR, Hoveyda AH. doi: 10.1038/nature17396.
- 15.Ramachandran PV, Burghardt TE. Chem Eur J. 2005;11:4387–4395. doi: 10.1002/chem.200401295. [DOI] [PubMed] [Google Scholar]
- 16.a) Liu M, Sibi MP. Tetrahedron. 2002;58:7991–8035. [Google Scholar]; b) Fuller AA, Chen B, Minter AR, Mapp AK. J Am Chem Soc. 2005;127:5376–5383. doi: 10.1021/ja0431713. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.