

Abstract
The transcription factor CREB (cAMP-response element binding protein) regulates differentiation, migration, survival and activity-dependent gene expression in the developing and mature nervous system. However, its specific role in the proliferation of embryonic neural progenitors is still not completely understood. Here we investigated how CREB regulates proliferation of mouse embryonic neural progenitors by a conditional mutant lacking Creb gene in neural progenitors. In parallel, we explored possible compensatory effects by the genetic ablation of another member of the same gene family, the cAMP-responsive element modulator (Crem). We show that CREB loss differentially impaired the proliferation, clonogenic potential and self-renewal of precursors derived from the ganglionic eminence (GE), in comparison to those derived from the cortex. This phenotype was associated with a specific reduction of histone acetylation in the GE of CREB mutant mice, and this reduction was rescued in vivo by inhibition of histone deacetylation. These observations indicate that the impaired proliferation could be caused by a reduced acetyltransferase activity in Creb conditional knock-out mice. These findings support a crucial role of CREB in controlling embryonic neurogenesis and propose a novel mechanism by which CREB regulates embryonic neural development.
Keywords: CREB, CBP, Ganglionic eminence, proliferation, histone acetylation
Abbreviations
- BIO
6-bromoindirubin-30-oxime
- BrdU
bromodeoxyuridine
- CBP
CREB-binding protein
- CREB
cAMP-response element binding protein
- CREM
cAMP-responsive element modulator
- d.p.c
days post coitum
- Ctr
control
- DM
double mutant
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- FGF-2
fibroblast growth factor-2
- FACS
fluorescence activated cell sorting
- GE
ganglionic eminence
- HD
Huntingtons disease
- HDAC
histone deacetylases
- IHC
immunohistochemistry
- IZ
intermediate zone
- PCNA
Proliferating Cell Nuclear Antigen
- PH3
phosphohistone H3
- PI
propidium iodide
- PP
pre-plate
- Shh
Sonic hedgehog
- SVZ
subventricular zone
- TSA
trichostatin A
- VZ
ventricular zone
Introduction
Tight regulation of proliferation and differentiation is essential for brain development.1-3 Timing of cell fate specification and differentiation is especially achieved by the combination and integration of multiple transcriptional programs,4 although epigenetic mechanisms are also emerging.2,5 Severe deficits in neurogenesis during embryonic development are exhibited by multiple neurodevelopmental disorders including Down syndrome, lissencephaly, microcephaly, autism, and epilepsy6-8 prompting to a better understanding of the complex mechanisms regulating embryonic neurogenesis.
The cAMP-response element binding protein (CREB) belongs to a family of transcription factors implicated in the regulation of several cellular processes in the developing brain.9 CREB affects neurogenesis at different levels for example, by regulation of migration, differentiation, maintenance and survival.9-12 The genetic ablation of CREB in specific cellular contexts has highlighted its cell-autonomous and cell-specific functions.12,13 So far the role of CREB in survival and differentiation of neural progenitors has been extensively investigated in the adult brain.14-16 During embryonic development the specific loss of CREB in neural progenitors achieved by the Cre/LoxP system results in increased apoptosis but only when also the cAMP-responsive element modulator (CREM) was lost, due to its compensatory effects.12 Although in the initial analysis of these models no effects on proliferation were reported, recent evidence have shown that CREB is involved in the regulation of neural progenitor cell proliferation in culture in a global Creb knockout mice.17–19 However, it is poorly understood how the loss of CREB reduces progenitor proliferation and whether CREB plays a similar role in vivo.
Here we investigated the cell autonomous role of CREB signaling in mouse embryonic neural progenitors. For this analysis we took advantage of a mouse model based on the conditional ablation of Creb under the control of the NestinCre transgene in combination with Crem global knock-out to exclude possible compensatory roles due to CREM.12 Using this experimental system we show that at embryonic day (E) 13.5 whereas in the cortex the number of neural progenitors is unaltered, in the ganglionic eminence (GE) CREB ablation leads to its rapid decrease. A similar effect was observed in neurosphere cultures. This analysis revealed that CREB loss impaired the proliferation, clonogenic potential and self-renewal of precursors derived from the GE but not from the cortical counterpart. This phenotype was associated with a reduction of histone acetylation in the GE but not in the cortical anlage. Moreover, inhibition of histone deacetylation in vivo rescued proliferation in vitro. These observations indicate that the context-specific impairment of proliferation could be caused by a reduction of histone acetyltransferase activity in Creb conditional knockout mice.
Results
Lack of CREB impairs the proliferation of GE but not of cortical precursors
The function of CREB on the regulation of neural precursor activity has mainly been focused on neuronal maturation and differentiation.10-14 Recent evidence has shown that CREB also regulates proliferation of neural precursors derived from the embryonic GE.17 However, it is unknown whether this role depends on a cell autonomous or non cell-autonomous mechanism and whether CREM, the transcription factor usually compensating CREB loss in the brain, has a similar role.12
To investigate the cell autonomous function of CREB signaling in the early embryonic neurogenesis we have taken advantage of a conditional approach by which the Creb gene is specifically ablated in neuronal and glial progenitors after expression of the NestinCre transgene.12 In particular, we analyzed proliferation and neural stem cell activity in cultures isolated from the E13.5 GE of Crem+/−; Crebfl/fl (control), Crem−/−; Crebfl/fl (Crem−/−), CrebNestinCre; Crem+/+ (Creb−/−) and CrebNestinCre; Crem−/− double mutant (DM) mouse embryos. Consistent with previous observations,17 we found that 7 day old neurospheres obtained from E13.5 Creb−/− and DM GE contained fewer cells than the counterpart established from control and Crem−/− embryos (Fig. 1A), suggesting that the mutation affected proliferation. Indeed, clonal analysis revealed a decrease both in the number (Fig. 1B) and in the size of the clones generated from the dissociated GE of Creb−/− and DM embryos (Fig. 1C), although the single mutation of Creb resulted in a less penetrant phenotype with more variation among individual mutant cultures than in the DM counterparts. Moreover, besides the change in size, clones originating from Creb mutant precursors did not display spheric morphology (Fig. 1C), suggesting that loss of CREB also affects cell-cell adhesion, as previously proposed.11
Figure 1.
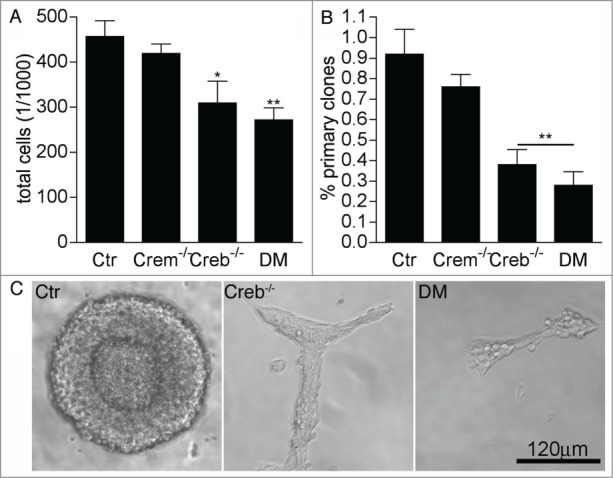
Loss of CREB and CREM impairs the proliferation of striatal precursors. (A–B) Quantitative analysis of total cells in high (A) and clonal (B) density neurosphere cultures established from dissociated GE of E13.5 embryos with the indicated genotypes. Data represent means ± SEM in at least 3 independent experiments. Asterisks indicate significant differences versus control (Ctr; ANOVA) * P <0 .05; ** P<0 .01. (C) Representative images illustrating clone size and morphology.
Next, to investigate the mechanisms of impaired neural stem cell proliferation in Creb mutants, we purified the clone-forming cells. To this end we used flow cytometry to isolate cells expressing high levels of EGFR (EGFRhigh cells) from cultures of the E13.5 GE grown in the presence of exogenous FGF2 for 24 hours. We have previously shown that at least one in 5 EGFRhigh cells displays stem cell characteristics i.e. long-term self-renewal and multipotentiality.20–22 Independent of the genotype, after 24 hours after exposure to exogenous FGF2, cultures derived from the dissociated GE contained similar numbers of EGFRhigh cells (Fig. 2A and B) showing that the defect in proliferation is not due to impaired EGFR expression. In contrast, Creb−/− and DM cultures exposed to FGF2 for 48 hours displayed a significantly higher number of EGFRhigh cells than the control or Crem−/− counterparts (Fig. 2C). This effect did not reflect a change in cell viability as shown by the number of cells incorporating propidium iodide (PI) (% PI positive cells: control: 5.62 ± 1.08; Crem−/−: 6.05 ± 1.03; Creb−/−: 6.06 ± 0.71; DM: 5.08 ± 1.00). Taken together these data indicate that the viability and the number of clone-forming neural precursors isolated from the GE of Creb−/− and DM E13.5 embryos is not compromised. Furthermore, the increase in EGFR expression was transient, as it was not observed if the culture medium was replaced with fresh one 24 hours after dissection (Fig. 2D), or after 5 d in the continuous presence of FGF2 (Fig. 2E). Together, these data indicate that factors present in the culture medium other than FGF2 promote the upregulation of EGFR expression in CREB- deficient precursors. Immunofluorescence revealed a decrease in the expression of Ki67 and bromodeoxyuridine (BrdU) incorporation, suggesting a defect in proliferation of EGFRhigh cells isolated from Creb−/− and DM mice. This defect was reflected by a smaller fold increase in cell number after the first passage (Fig. 2G). However, the percentage decrease in immunopositive cells for either marker was similar, suggesting that CREB ablation affects the number of proliferating precursors rather than cell cycle kinetics. In addition, although the number of cells expressing the early neuronal marker Tuj1 was increased, the expression of Nestin in EGFRhigh cells isolated from DM and control embryos was not significantly changed (Fig. 2F). However in mutant cultures the total number of EGFRhigh cells is transiently increased and this excess of neuroblasts could be a consequence of lingering high levels of EGFR expression during lineage progression. Together these data indicate that, compared to the control counterpart, EGFRhigh cells proliferate less and display characteristics of more differentiated neuronal progenitors.
Figure 2.

Effect of CREB and CREM on number and cycling rate of EGFRhigh precursors. (A) Representative FACS plots illustrating EGFRhigh cells in DIV1 control (Ctr) and DM cultures, pre-treated with EGF before staining with EGF-Alexa488, as negative control, or left untreated as indicated. (B–E) Quantitative analysis of EGFRhigh cells in the cultures established from the GE of E13.5 embryos with the indicated genotype measured after the indicated day in vitro (DIV). In (D) the culture medium was replaced 24 hours after plating. (F) Quantitative analysis of BrdU incorporation and expression of the given markers in EGFRhigh cells isolated from DIV2 cultures of Ctr and DM GE. Data represent means ± SEM in at least 3 independent experiments. Asterisks indicate significant differences vs. Ctr (Student's t-test) *P <0 .05; **P <0 .01. (G) Fold increase in cell number from DIV0 to DIV5 neurosphere cultures established from dissociated E13.5 GE cells of embryos with the indicated genotypes.
Both EGFRhigh and EGFRlow cells sorted from 2 day-old Creb−/− and DM cultures displayed a defect in primary clone formation (Fig. 3A and B), proliferation potential (Fig. 3C) and self-renewal (Fig. 3D). Consistent with our previous analyses, independent of EGFR expression CREM ablation did not affect the number of clone forming precursors and clone size (Fig. 3A-C). However, primary clones obtained from Crem−/− precursors gave rise to fewer secondary clones (Fig. 3D), indicating a defect in self-renewal. Thus, ablation of CREB impacts the proliferation rate and self-renewal of neural stem cells in the GE, whereas deletion of CREM alone impacts only their ability to self-renew. A defect in primary clone formation (Fig. 3E and F) and differentiation (Fig. S1) was also observed if cells were maintained for longer time (3-5 days) in culture before FACS analysis and isolation. However, at these later time-points proliferation potential and self-renewal were unaffected (Fig. 3G and H). Taken together, these results indicate that lack of CREB impairs the ability of highly neurogenic and self-renewing precursors to undergo clone formation. The proliferation of less neurogenic and self-renewing secondary progenitors is instead less affected, especially in high-density cultures.
Figure 3.
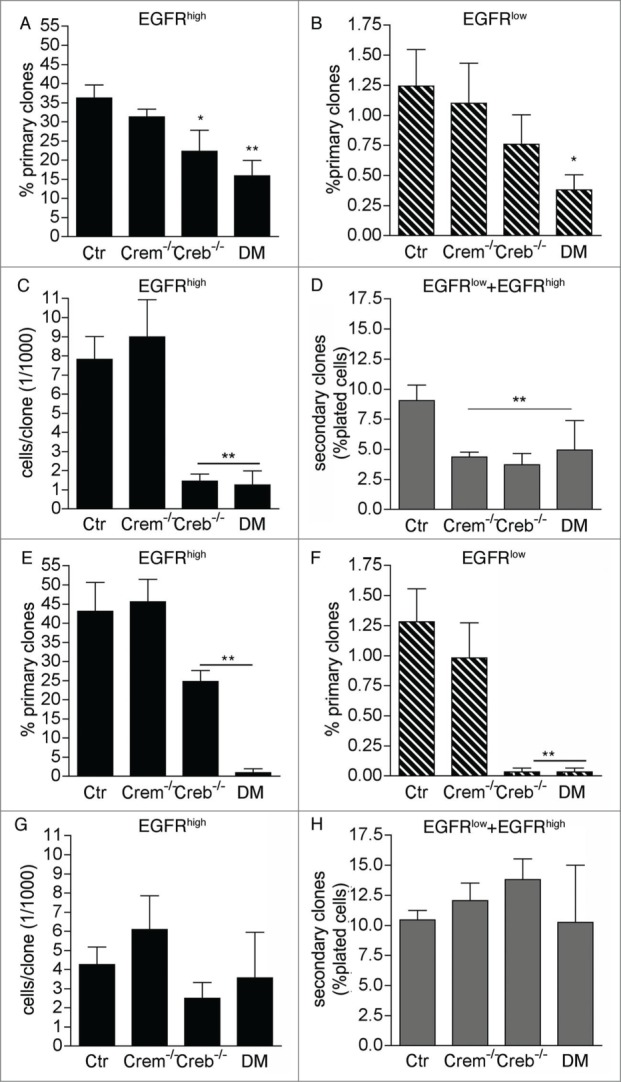
Effect of CREB and CREM on the ability of precursors isolated from the E13.5 GE to form primary and secondary clones. Analysis of clones obtained from EGFRhigh and/or EGFRlow cells sorted at DIV1 and DIV2 (A and D) and DIV 3-5 (E and H) from cultures of the GE from E13.5 embryos with the given genotypes. Graphs illustrate quantitative analyses of primary (A, B, E, F) and secondary (D and H) clones as well as clone size (C and G). Data represent means ± SEM in at least 3 independent experiments. Asterisks indicate significant differences versus Ctr (ANOVA) *P <0 .05; **P <0 .01.
The effect of CREM and CREB on proliferation is modulated by regional cues
To establish whether CREB regulates neural precursor proliferation in a region-specific manner, we next investigated the clonogenic activity of cells isolated from the E13.5 cortical anlage by a similar FACS-based procedure. Cortical EGFRhigh cells from single mutant Creb−/− and Crem−/− formed clones at a rate comparable to controls (Fig. S2A). Although DM EGFRhigh precursors (Fig. S2A), Creb−/− and DM EGFRlow cells displayed a decreased ability of forming clones, this effect was not significant (Fig. S2B). Moreover, in either group of precursors, the size and the ability of undergoing secondary clone formation was not affected (data not shown). In contrast to the GE, 48 hours after dissection control, Crem−/−, Creb−/− and DM cultures contained similar numbers of EGFRhigh cells (Fig. S2C). Together these data show that absence of CREB per se is not a prerequisite for clone formation in neural precursors, and the inability to proliferate in vitro shown by high self-renewing and neurogenic mutant precursors isolated from the GE rather reflect a specific change in this cell population.
The functional relevance of CREB–dependent gene expression in neural precursors is supported by CREB being phosphorylated on Serine-133 in the germinal layer,17 a modification crucial for stimulus-dependent CREB activity.23 Our analysis of phosphorylated CREB protein at E13.5 in the germinal zones of the GE and cortex confirmed these previous observations showing phospho-CREB immunoreactivity also in these regions characterized by high density of cells actively cycling and dividing, as indicated by Ki67 (Fig. S3A-C) and BrdU immunoreactivity (Fig. S3D-G). Moreover, double immunofluorescence of control E13.5 telencephalic sections with antibodies specific to a third proliferative marker, i.e., the proliferating cell nuclear antigen (PCNA), and phospho-CREB showed that phospho-CREB is also expressed in precursors undergoing proliferation in the germinal zone of the cortex and especially of the GE. Notably, at E13.5 phospho-CREB immunoreactivity was stronger and more widespread in the GE than in the cortical counterpart (Fig. 4) suggesting that CREB activation is more tightly associated to the proliferation of neural precursors in the GE than in the cortex. A similar analysis of DM littermate embryo showed that by E13.5 phospho-CREB immunoreactivity was strongly decreased in both regions with only a few immunopositive cells observed mostly at the apical side of the germinal areas (Fig. 4).
Figure 4.
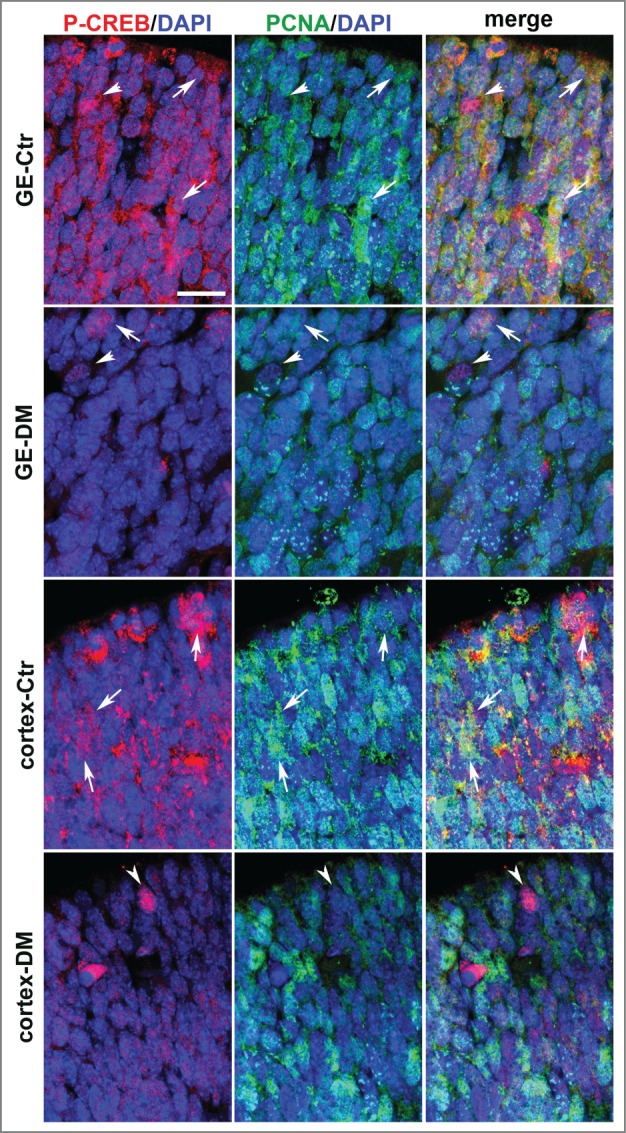
Analysis of phospho-CREB and PCNA expression in the coronal sections of the E13.5 telencephalon. Representative confocal micrographs illustrating the germinal regions of the GE and cortex from control (Ctr) and DM embryos upon double immunostaining with phospho-CREB (P-CREB, red), PCNA (green) and DAPI (blue) counterstaining of the nuclei. The apical side is shown on the upper side. Arrows and arrowheads indicate double and single immunopositive cells, respectively. Scale bar is 30 μM.
To further investigate the specific role of CREB/CREM loss in the proliferation of neural progenitors in the embryonic brain, we quantified the number of cells in S-phase by BrdU incorporation in control and DM E13.5 embryos (Fig. 5). Pregnant females were given a single intraperitoneal injection of BrdU and embryos at E13.5 were collected and analyzed after 2 hours. This analysis revealed a small but significant decrease in the number of BrdU+ cells within the apical VZ of the GE in DM embryos but not within the cortical anlage supporting that the number of cells in the S-phase is reduced in DM mice (Fig. 5 Ea, Ec). The VZ of the embryonic GE includes radial glia precursors that undergo mitosis at the apical border, and a newly identified subset of precursors undergoing sub-apical mitosis.24 We therefore, analyzed phosphohistone H3 (PH3) positive mitotic cells in the GE of control and DM embryos. Consistent with our previous observations, loss of CREB and CREM did not affect the number of cortical mitotic cells, whereas, compared to control, the number of PH3+ cells within the germinal region of DM embryos was reduced. Moreover, the genotype specifically affected precursors undergoing sub-apical division in the VZ.
Figure 5.
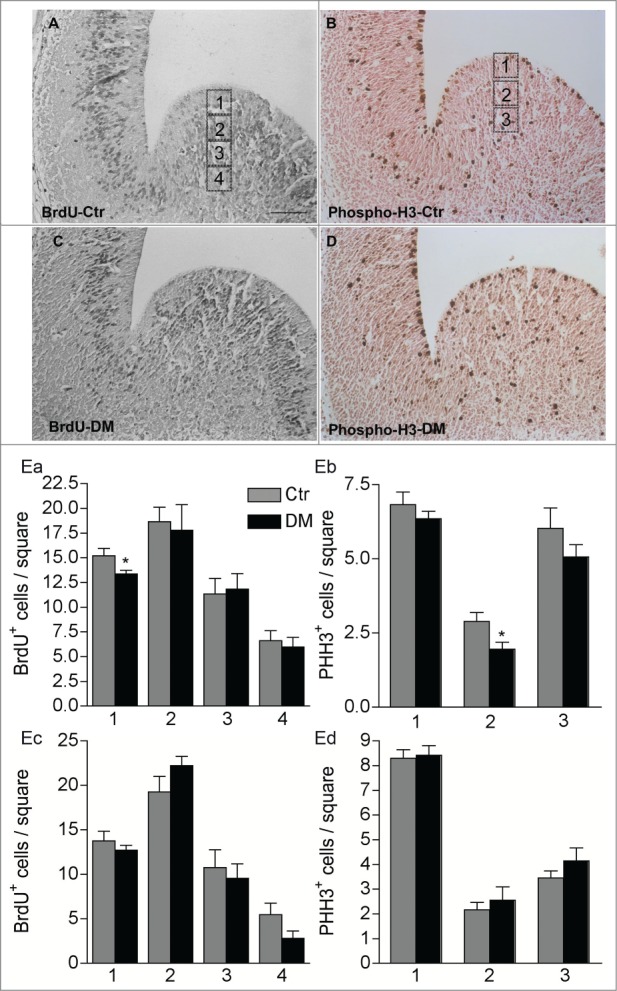
Ablation of Creb and Crem leads to reduced proliferation in the GE but not in the cortex of E13.5 embryos. (A–D) Representative micrographs of coronal telencephalic sections obtained from control (Ctr; A and B) and DM (C and D) embryos upon immunostaining with BrdU (A and C) and phospho-H3 (B and D) antibodies. (E) Quantitative analyses of the immunostaining with BrdU (Ea and Ec) and PH3 (Eb and Ec) antibodies in the GE (Ea and Eb) and in the cortex (Ec and Ed). Scale bar is 60 μm; N = 3–4. Asterisks indicate significant differences vs. control (Ctr) counterpart (Student's t-test) *P < 0 .05.
Taken together these data indicate that, in vitro as in vivo, CREB regulates neural progenitors proliferation in a region-specific manner decreasing the proliferation of dividing precursors in the GE but not in the cortex.
Lack of CREB and CREM differentially affects the ability of precursors isolated from the GE to respond to environmental signals
The expression of EGFR and clone formation in neural precursors can be independently affected by a plethora of different signals, among others Wnt and Sonic hedgehog (Shh),25 prominent in the developing dorsal and ventral telencephalon, respectively. We had found that GE primary precursors were more sensitive to CREB ablation than cortical precursors, indicating that the effect of CREB signaling interacts with ventral-dorsal cues. To investigate whether control and mutant precursors similarly respond to environmental cues we next determined the effect of Wnt and Shh signaling on EGFR expression and clone in the various genetic landscapes. To this end, we cultured dissociated GE precursors for 2 d with either 6-bromoindirubin-30-oxime (BIO), a GSK inhibitor that functions as an agonist of canonical Wnt signaling, or with Shh before sorting the cells based on EGFR expression and investigating the effect of the treatments on the number of EGFRhigh cells (Fig. S4A), primary (Fig. S4B) and secondary (Fig. S4C) clone formation. This analysis revealed that whereas exposure to Shh did not significantly affect EGFR expression, treatment with BIO increased the number of EGFRhigh cells in control and Crem−/− cultures (Fig. S4A). This treatment also effectively increased the ability of DM precursors to generate primary (Fig. S4B) and secondary clones (Fig. S4C). Both BIO and Shh treatments rescued self-renewal in Creb−/− but not in Crem−/− single mutant precursors (Fig. S4C), whereas neither treatment affected primary clone formation in either genotypes (Fig. S4B).
Thus, the ability of neural precursors to upregulate EGFR expression and undergo clone formation in response to Wnt and Shh signaling is dependent on the genotype.
Inhibition of histone acetylation rescues the defective clone formation of GE precursors lacking CREB
Our data indicate that CREB deficiency could differentially modulate proliferation of neural precursors by context-specific genetic programs and environmental cues. Notably CREB can modulate histone acetylation and expression of its target genes by binding the transcriptional coactivator and acetyl transferase CREB-Binding Protein (CBP).9 Hence, to test whether CREB loss results in decreased histone acetylation, we analyzed the acetylation of histone H3 and H4 in cell lysates of the GE and cortical anlage dissected from control, Creb−/− and DM E13.5 embryos. Interestingly, the acetylation levels of these histones decreased in both regions (Fig. 6). However, the decrease in acetylation was more prominent in GE than in cortical extracts. In addition, acetylated H3 in the GE was more affected than H4.
Figure 6.
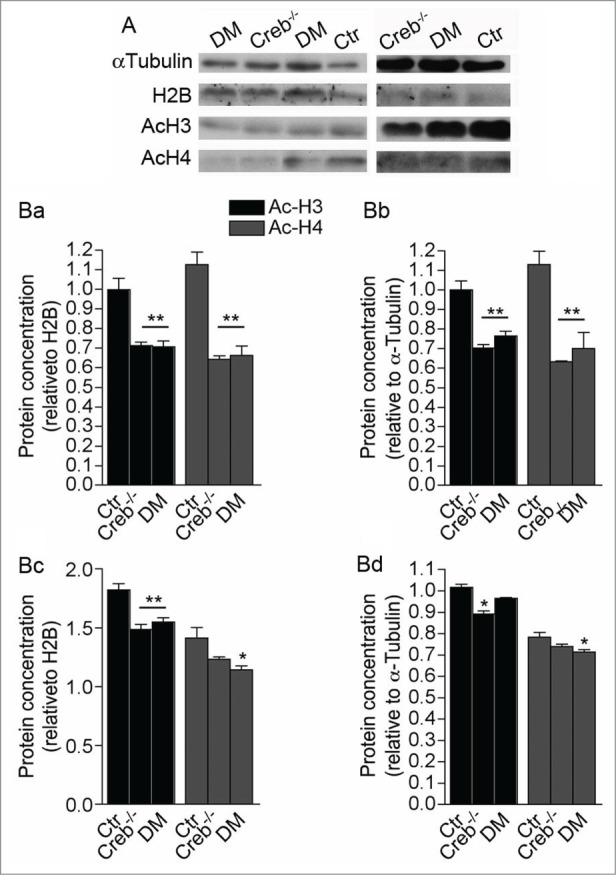
Ablation of CREB and CREM results in decreased histone acetylation in the E13.5 telencephalon. (A) Western blot analysis of GE (left column panels) and cortical protein extracts (right column panels) showing analysis of αlpha-tubulin, total histone H2B, acetylated histones H3 and H4. (B) Quantification of western blot analysis of the GE (Ba and Bb) and cortex (Bc and Bd). Bands were normalized against H2B (Ba and Bc) and α Tubulin (Bb and Bd). Note that ablation of CREB leads to a stronger alteration of histone acetylation in the GE (Ba and Bb) than in the cortex (Bc and Bd). Asterisks indicate significantly different from the respective control (Ctr) (ANOVA) * P < 0.05; **P < 0.01.
To explore the functional relevance of these histone modifications, we next investigated how the inhibitor of class I and II histone deacetylases (HDACs) trichostatin A (TSA), affects neural precursor proliferation. Time-mated pregnant females were injected every 12 hours with 12.5 μg TSA at 11.5 d.p.c. and embryos were sacrificed at E13.5. In comparison with controls (Fig. 7A) acetylated H4 and acetylated H3 displayed a more intense immunostaining especially at the contour of the nuclei upon TSA treatment. The treatment had a similar effect on the distribution but not on the intensity (Fig. 7A) of H3 immunoreactivity. These observations are in agreement with previous studies showing that TSA results in redistribution of acetylated H3 and H4.26,27 Thereafter, dissociated GE cells were plated in FGF-2 medium and sorted by FACS at DIV2 (Fig. 7B). Clonal analysis revealed that independent of EGFR expression, the treatment completely rescued the impairment of Creb and/or Crem mutant precursors to form primary clone in terms of number (Fig. 7B) and clone size (not shown). Importantly, the treatment did not significantly affect clone number in control or Crem mutant progenitors (see for comparison Figures 3A, B and 7A, B). Taken together, these data indicate that the impaired clone formation in GE precursors can be ascribed to altered acetylation patterns in these cells and can be rescued by preventing histone deacetylation.
Figure 7.

Blocking histone deacetylation rescues the ability of neural precursors to form clones. (A) Representative micrographs of coronal telencephalic sections obtained from control E13.5 embryos showing the effect of TSA on the levels and distribution of acetylated histone 3 (AcH3), acetylated histone 4 (AcH4) and histone 3 (H3) immunoreactivity in the GE and in the cortex. Pregnant females were injected with TSA at 11 d.p.c. and every 12h for 48h before analysis. V = Ventricle. Scale bar is 30 mM. (B) Quantitative analyses of the number of primary clones obtained from EGFRhigh and EGFRlow cells isolated at DIV 1 cultures established from E13.5 embryos with the indicated genotypes after TSA treatment.
Discussion
Our study based on conditional genetic mutants showing no overt defects in brain development dissects the context-specific role of CREB in neural progenitors proliferation. The data presented here support the following conclusions. First, loss of CREB and CREM impairs neural precursor proliferation and self-renewal. Some of these deficits are also evident in Crem single mutant. Second, CREB plays a region-specific role in vivo, as proliferation and histone acetylation of GE precursors are more impaired than cortical precursors at this initial stage. Third, Wnt and Shh signaling stimulation, can both ameliorate the clonogenic deficits of Creb mutant neural precursors, indicating that multiple subtle changes rather than the lack of individual molecular components contribute to this phenotype. However, the extent of the improvement elicited by each signal varies and the understanding of the mechanisms underlying the altered response to Wnt and Shh will require the identification of downstream targets. Finally, we show that histone acetylation is reduced in Creb mutants and its restoration rescues the clone formation deficit, suggesting that CREB is required for CBP histone acetyl transferase activity. Differently from previous approaches our study is based on mutant mice in which CREB is specifically ablated in neural progenitors cells, allowing us to dissect the cell-autonomous effects of CREB in vivo. Whereas the ablation of CREB dramatically affected the ability on GE precursors to proliferate in vitro, especially in clonal cultures, the mutation in vivo specifically affected progenitors undergoing sub-apical division in the GE. Indeed, our analysis of phospho-CREB and PCNA expression in the germinal areas of the cortex and GE highlighted a differential pattern of CREB activation in proliferating cells between the two regions that could account for the differential sensitivity of GE and cortex to Creb conditional ablation. Progenitors undergoing sub-apical division in the GE include basal radial glia progenitors, which in the murine telencephalon are abundant in the GE but not in the cortical anlage. This population has been associated with rapid proliferation and expansion of the progenitors, which is necessary to increase neuronal output. Indeed, basal radial glia progenitors are abundant in the cortex of gyrencephalic species.24 Consistent with the hypothesis that the proliferation of these progenitors is affected in Creb mutant mice, we found a decrease in the number of highly proliferating and neurogenic clonogenic precursors in mutant embryos. Despite the loss of CREB impaired the proliferation of these precursors also in vivo, previous analysis of these mutants have essentially highlighted a massive deficit in neuronal survival rather than in the telencephalic neurogenesis,12 suggesting that environmental factors may compensate the loss of CREB.
Besides the consequence of CREB ablation on neural precursors, we could investigate Crem single mutants and show a novel function of CREM in self-renewal. Nevertheless CREM does not compensate loss of CREB in neural precursor proliferation, as it does in neuronal survival and stimulus-dependent gene expression in other contexts.12,28 This finding is in line with the similar phenotypes of Creb mutant mice in catecholaminergic neurons either lacking only CREB or both CREB and CREM13,29 and highlights context-specific compensatory mechanisms triggered in absence of CREB.
Previous studies have established a role for histone acetylation in the regulation of neural progenitor differentiation in the postnatal hippocampus and in neural precursor cultures.30-32 Our results point to a specific role of CREB-dependent gene expression in cortical and GE precursors and suggest a model in which CREB controls the number of highly proliferative and neurogenic precursors in the developing GE by chromatin acetylation status (Fig. 8). Interestingly, differential biological effects in the cortex and GE upon manipulation of histone acetylation levels have been previously reported in developing mouse brain.33,34 HDAC inhibition by TSA in utero resulted in increased proliferation of precursors in the VZ and SVZ of the GE, a decrease in neurogenesis, and a corresponding reduction in GE- derived GABA-positive cells undergoing tangential migration in the cortex.33 In contrast, in utero HDAC inhibition resulted in increases in neural precursors and neurogenesis in the cortex, despite the fact that acetylation levels of histone H3 increased in both GE and cortex. Although we did not detect a change in the cortex of Creb and DM mutants, the results seen in the GE are consistent with previous data.33 Increased levels of histone acetylation, either through CREB-mediated recruitment of CBP or the inhibition of HDACs with TSA, increased the ability of GE neural precursors to undergo in vitro proliferation and clone formation. Several transcriptional regulators recruit histone acetylation modifying enzymes to gene promoters, e.g. the transcriptional repressors REST and TLX are capable of recruiting HDACs by indirect and direct binding, respectively.35-37 While REST regulates the expression of genes associated with neuronal differentiation,38 TLX controls the proliferation of neural precursors in both the embryonic and postnatal brain39,40 and it promotes cell cycle entry of neural stem cells.41 Besides HDAC recruitment, transcription factors can also recruit histone acetyl transferases. In particular, serine 133-phosphorylated CREB binds CBP, which displays lysine acetyltransferase activity.42 In mature neurons impaired CBP activity has been mainly associated to transcriptional repression and neuronal death. Notably the polyglutamine expansion in the mutant protein huntingtin, accounting for Huntington´s disease (HD), results in the interaction with the acetyltransferase domain of CBP and inhibition of its activity that leads to neurodegeneration.43 Moreover HDAC inhibitors arrest neurodegeneration in cellular and animal models of HD.44
Figure 8.
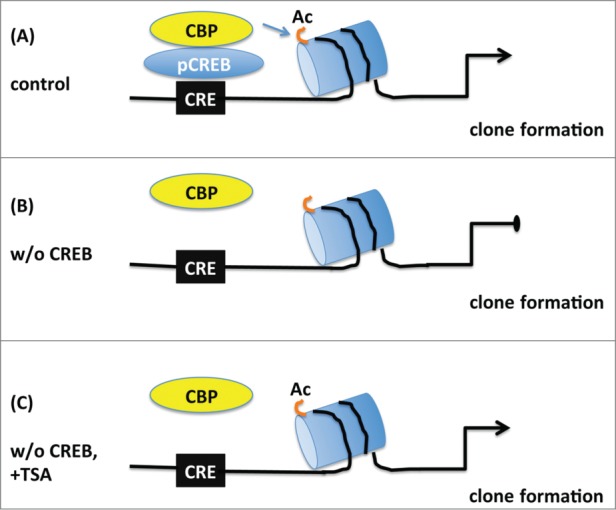
Schematic model depicting the proposed role of CREB in the proliferation of embryonic neural progenitors. (A) Under normal conditions phosphorylated CREB (pCREB) binds to cAMP-responsive element (CRE) and recruits CBP promoting histone acetylation (Ac) of targets genes which in turn lead to clone formation. (B) Upon CREB ablation, CBP is not capable of maintaining histone acetylation and clonogenic activity of neural progenitors is impaired. (C) Treatment with the histone deacetylase inhibitor TSA prevents the loss of histone acetylation and rescues the deficit in clone formation observed in the absence of CREB suggesting that CREB regulates proliferation essentially by modification of chromatin structure.
Here we show that HDAC inhibition rescues the deficits of neural proliferation caused by CREB loss, prompting to further studies addressing whether impaired neuronal survival can be rescued by HDAC inhibitors in neuron-specific conditional Creb mutant mice. Notably, TSA rescues memory deficits and transcription of selective CREB/CBP target genes in models of HD.45 Conditional loss of CBP attenuates the neurogenic response to an enriched environment in the adult dentate gyrus, indicating that CBP is crucial for the transmission of environmental cues known to control neurogenesis.46 The analysis of CBP heterozygous mice shows decreased CBP histone acetylation at promoters of neuronal and glial genes, but TSA treatment rescued these deficits.8
In summary, we provide the first evidence that CREB regulates proliferation of GE neural precursors by promoting chromatin activation. These results show novel implications of impaired CREB-dependent programs during neurogenesis in vivo and represent an important advance for in the understanding of neurogenesis and neurodevelopmental disorders.
Materials and Methods
Ethical statement
All experimental procedures involving animals were carried out in strict accordance with the guidelines provided by the Regierungspräsidium in Karlsruhe and the local authorities at the University of Heidelberg. All experimental protocols were approved by the “Regierungspräsidium” in Karlsruhe. Every effort was made to minimize the number of animals used and their suffering.
Mice
Experimental mice were obtained by crossing the following either Creb+/fl; Crem+/−; NestinCre+/Tg or Crebfl/fl; Crem+/−; NestinCre+/+ males with were crossed with Crebfl/f; Crem−/−; NestinCre+/+ or Creb+/fl; Crem−/−; NestinCre+/Tg females. Crebfl/fl; Crem−/−; NestinCre+/−, abbreviated as CrebNesCre; Crem−/− are indicated in the text as DM. The analysis of the genotype was performed as previously described.12
Culture of primary embryonic neural precursors
Neurosphere cultures: Following mechanical dissociation, cells were plated at a density of 105 cells/ml in 24 well plates in culture medium consisting of ice cold Euromed-N basal serum free culture medium, Penicillin-Streptomycin (100 U/ml), glutamine (2 mM) and 2% B27 (Gibco). Human recombinant EGF and FGF-2 (Peprotech) were added to the culture medium at a concentration of 20 ng/ml and 10 ng/ml, respectively with or without GDF15 (10 ng/ml, R&D). Cells were fed with 1/2 the volume of fresh culture medium every 4 d precursors were allowed to proliferate in suspension for 7 days, before mechanical dissociation and counting.
Clonal cultures: Using FACS automated cell deposition one EGFRhigh and 10 EGFRlow cells were plated at a density of one cell per well in 96 well/plates in culture medium supplemented with EGF and FGF-2. Clones were scored after 7 d.
Secondary clone formation: Single spheres from clonal cultures were mechanically dissociated, and replated in 96 well plates in EGF and FGF-2 supplemented culture medium at a density of 103 cells per well. Secondary neurospheres were scored after 7 d.
Fluorescence activated cell sorting (FACS)
Sorting was performed FACSAria sorter and a FACSVantage (Becton Dickinson) as previously described.20 Briefly, after mechanical dissociation cells were suspended in ice-cold sorting medium: Euromed-N basal medium/Leibovitz medium (Gibco) (1:1) containing 100 U/ml Penicillin/Streptomycin, 2 mM L-glutamine, 2% B27, 1% FCS, 0.6% Glucose, 1 mg/ml Propidium Iodide, 10 ng/ml FGF-2 and 20 U/ml DNase. To detect EGFR, cells were stained with 20 ng/ml EGF-Alexa 488 (Molecular Probes). Cells were labeled after being plated at a density of 105 cells/ml in FGF-2 culture medium, for 24 or 48 hours. FACS gates were set using cells stained with propidium iodide (PI) only and cells that had been incubated with 20 ng/ml EGF (Sigma) for 15–30 minutes at 37°C prior to staining with EGF-Alexa. EGFRlow and EGFRhigh populations were gated on the basis of a 20-fold decrease or increase in fluorescence levels with respect to the more fluorescent control cells.
Antibodies
The following antibodies were used at the indicated dilution: mouse monoclonal α-Bromodeoxyuridin (BrdU) 1:10 (Roche, USA); α-BrdU dilution 1:100 (DAKO, M0744); rabbit polyclonal α-phosphohistone H3 (PH3) 1:500 (Upstate, 06–570), α-PH3 1:30 (Santa Cruz), α-Ki67 1:200 (Abcam, ab15580), PCNA 1:500 (DAKO, M0879), α-phosphoCREB 1:1000 or 1:100 (Upstate, 06–519), α-H2B 1:200 (Santa Cruz), α-Ac.H3 H3K9/18ac 1:1000 (Upstate, 07–593), α-Ac.H3 H3K27ac 1:500 (Abcam, ab4729), α-Ac.H4K5/8/12/16 1:1000 or 1:500 (Upstate, 06–866), mouse monoclonal: α−β-tubulin type III (Tuj1, 1:400, Sigma); α-Tubulin 1:50000 (Sigma); rat monoclonal α-Nestin 1:500 (PharMingen).
Histological analysis
For immunohistochemistry (IHC), embryos were fixed in 4% paraformaldehyde, pH 7.2 overnight, processed for paraffin sections and sectioned at 7 μm. The sections were incubated in citrate buffer, pH 6.0, and boiled in a microwave oven. The primary antibodies were incubated overnight at 4°C. Biotin-conjugated secondary antibody was diluted 1:400 in PBS and detection was performed using the avidin-biotin system (Vector Laboratories, PK-6100) with the VECTOR peroxidase kit. The staining was developed with diaminobenzidine (DAB) and H2O2 (Sigma, D4293). For immunofluorescence, Alexa 488 anti-rabbit antibody (Invitrogen, A-21206), anti-mouse (A-11029) and Alexa Fluor® 594 anti-rabbit (A-21203) were used (1:100).
Cell counts
Left and right hemispheres of 3 to 5 nonadjacent, 7-μm-thick sections per brain region (GE and cortex) were photographed. Sections at comparable rostrocaudal levels were selected for each genotype before performing the immunostaining. Sections were visualized on a Nikon (Melville, NY) microscope and photographed at 40x magnification with a high-resolution digital camera (Zeiss AxioCam) and by Leica-SP8 confocal microscope. Photographs were imported into Photoshop 5.0 (Adobe Systems, San Jose, CA), and composites of each section were aligned. For BrdU immunostaining different subregions of the cerebral wall were counted. These subregions were defined by the distance between the ventricle and the apical border of the abventricular band formed by BrdU labeled cells in S phase. This distance was used to define the side of the squared area used to count immunopositive cells. Two adjacent regions in line with the orientation of the ventricular zone (VZ) included the entire VZ (subregion 1 and subregion 2). The same area was used to count cells in the subventricular zone (SVZ) (subregion 3). In the cortex subregion 3 included also the pre-plate (PP) and the intermediate zone (IZ). The subregion 4 comprised the CP in the cortex and the striatum in the GE. PH3 labeling was used to distinguish cells proliferating in the VZ and SVZ. PH3 immunostaining was performed on adjacent sections, immunopositive cells were counted using the same criteria as for BrdU. However, dividing apically PH3 immunostained cells were counted separately from remaining mitotic cells in subregion 1.
TSA injections
Time-mated pregnant females were injected intraperitoneally every 12 hours at 11.5 d post coitum (d.p.c.) with either vehicle (8% ethanol in 16 PBS) or TSA (Sigma, Calbiochem, Schwalbach, Germany; 0.5 mg/kg body weight) dissolved in vehicle (100 mg/ml). Females were sacrificed at 13.5 d.p.c. after a total of 5 injections.
Statistical analysis
The means and standard errors of at least 3 independent experiments were calculated and statistical significance tests (t-test or one-way ANOVA, followed by a Bonferroni posthoc test) were performed using Prism 4.03 (GraphPad Inc., USA).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Prof. Günther Schütz and Prof. Joachim Kirsch for support and infrastructure and Katharina Sowodniok for technical assistance. We also thank Dr. Haikun Liu and Dr. Holger Bierhoff for providing useful antibodies.
Funding
FC and GH-W were supported by the program Adult Stammzellen I by the Landesstiftung Baden Württemberg. RP and BL were supported by the program for medical genome research with financial support from the German Federal Ministry for Education and Research (BMBF) under the support code 01GS08141 and the Alfried Krupp Prize. KLT was supported by the Deutsche Forschungsgemeinschaft (SFB488, Teilprojekt B9).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Gotz M, Huttner WB. The cell biology of neurogenesis. Nat Rev Mol Cell Biol 2005; 6:777-88; PMID:16314867; http://dx.doi.org/ 10.1038/nrm1739 [DOI] [PubMed] [Google Scholar]
- 2. Martynoga B, Drechsel D, Guillemot F. Molecular control of neurogenesis: a view from the mammalian cerebral cortex. Cold Spring Harb Perspect Biol 2012; 4:a008359 PMID:23028117; http://dx.doi.org/ 10.1101/cshperspect.a008359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Miller FD, Gauthier AS. Timing is everything: making neurons versus glia in the developing cortex. Neuron 2007; 54:357-69; PMID:17481390; http://dx.doi.org/ 10.1016/j.neuron.2007.04.019 [DOI] [PubMed] [Google Scholar]
- 4. Ernsberger U. Regulation of gene expression during early neuronal differentiation: evidence for patterns conserved across neuron populations and vertebrate classes. Cell Tissue Res 2012; 348:1-27; PMID:22437873; http://dx.doi.org/ 10.1007/s00441-012-1367-y [DOI] [PubMed] [Google Scholar]
- 5. Mohamed Ariff I, Mitra A, Basu A. Epigenetic regulation of self-renewal and fate determination in neural stem cells. J Neurosci Res 2012; 90:529-39; PMID:22183977; http://dx.doi.org/ 10.1002/jnr.22804 [DOI] [PubMed] [Google Scholar]
- 6. MacDonald JL, Roskams AJ. Epigenetic regulation of nervous system development by DNA methylation and histone deacetylation. Prog Neurobiol 2009; 88:170-83; PMID:19554713 [DOI] [PubMed] [Google Scholar]
- 7. Ronan JL, Wu W, Crabtree GR. From neural development to cognition: unexpected roles for chromatin. Nat Rev Genet 2013; 14:347-59; PMID:23568486; http://dx.doi.org/ 10.1038/nrg3413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang J, Weaver IC, Gauthier-Fisher A, Wang H, He L, Yeomans J, Wondisford F, Kaplan DR, Miller FD. CBP histone acetyltransferase activity regulates embryonic neural differentiation in the normal and Rubinstein-Taybi syndrome brain. Dev Cell 2010; 18:114-25; PMID:20152182; http://dx.doi.org/ 10.1016/j.devcel.2009.10.023 [DOI] [PubMed] [Google Scholar]
- 9. Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron 2002; 35:605-23; PMID:12194863; http://dx.doi.org/ 10.1016/S0896-6273(02)00828-0 [DOI] [PubMed] [Google Scholar]
- 10. Aguado F, Diaz-Ruiz C, Parlato R, Martinez A, Carmona MA, Bleckmann S, Ureña JM, Burgaya F, del Río JA, Schütz G, et al. The CREB/CREM transcription factors negatively regulate early synaptogenesis and spontaneous network activity. J Neurosci 2009; 29:328-33; PMID:19144833; http://dx.doi.org/ 10.1523/JNEUROSCI.5252-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Diaz-Ruiz C, Parlato R, Aguado F, Urena JM, Burgaya F, Martinez A, Carmona MA, Kreiner G, Bleckmann S, Del Río JA, et al. Regulation of neural migration by the CREB/CREM transcription factors and altered Dab1 levels in CREB/CREM mutants. Mol Cell Neurosci 2008; 39:519-28; PMID:18786638; http://dx.doi.org/ 10.1016/j.mcn.2008.07.019 [DOI] [PubMed] [Google Scholar]
- 12. Mantamadiotis T, Lemberger T, Bleckmann SC, Kern H, Kretz O, Martin Villalba A, Tronche F, Kellendonk C, Gau D, Kapfhammer J, et al. Disruption of CREB function in brain leads to neurodegeneration. Nat Genet 2002; 31:47-54; PMID:11967539; http://dx.doi.org/ 10.1038/ng882 [DOI] [PubMed] [Google Scholar]
- 13. Parlato R, Otto C, Begus Y, Stotz S, Schutz G. Specific ablation of the transcription factor CREB in sympathetic neurons surprisingly protects against developmentally regulated apoptosis. Development 2007; 134:1663-70; PMID:17376811; http://dx.doi.org/ 10.1242/dev.02838 [DOI] [PubMed] [Google Scholar]
- 14. Giachino C, De Marchis S, Giampietro C, Parlato R, Perroteau I, Schutz G, Fasolo A, Peretto P. cAMP response element-binding protein regulates differentiation and survival of newborn neurons in the olfactory bulb. J Neurosci 2005; 25:10105-18; PMID:16267218; http://dx.doi.org/ 10.1523/JNEUROSCI.3512-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Merz K, Herold S, Lie DC. CREB in adult neurogenesis—master and partner in the development of adult-born neurons? Eur J Neurosci 2011; 33:1078-86; PMID:21395851; http://dx.doi.org/ 10.1111/j.1460-9568.2011.07606.x [DOI] [PubMed] [Google Scholar]
- 16. Schwarz TJ, Ebert B, Lie DC. Stem cell maintenance in the adult mammalian hippocampus: a matter of signal integration? Dev Neurobiol 2012; 72:1006-15; PMID:22488809; http://dx.doi.org/ 10.1002/dneu.22026 [DOI] [PubMed] [Google Scholar]
- 17. Dworkin S, Malaterre J, Hollande F, Darcy PK, Ramsay RG, Mantamadiotis T. cAMP response element binding protein is required for mouse neural progenitor cell survival and expansion. Stem Cells 2009; 27:1347-57; PMID:19489105; http://dx.doi.org/ 10.1002/stem.56 [DOI] [PubMed] [Google Scholar]
- 18. Dworkin S, Mantamadiotis T. Targeting CREB signalling in neurogenesis. Expert Opin Ther Targets 2010; 14:869-79; PMID:20569094; http://dx.doi.org/ 10.1517/14728222.2010.501332 [DOI] [PubMed] [Google Scholar]
- 19. Iguchi H, Mitsui T, Ishida M, Kanba S, Arita J. cAMP response element-binding protein (CREB) is required for epidermal growth factor (EGF)-induced cell proliferation and serum response element activation in neural stem cells isolated from the forebrain subventricular zone of adult mice. Endocr J 2011; 58:747-59; PMID:21701076; http://dx.doi.org/ 10.1507/endocrj.K11E-104 [DOI] [PubMed] [Google Scholar]
- 20. Ciccolini F. Identification of two distinct types of multipotent neural precursors that appear sequentially during CNS development. Mol Cell Neurosci 2001; 17:895-907; PMID:11358486 [DOI] [PubMed] [Google Scholar]
- 21. Ciccolini F, Mandl C, Holzl-Wenig G, Kehlenbach A, Hellwig A. Prospective isolation of late development multipotent precursors whose migration is promoted by EGFR. Dev Biol 2005; 284:112-25; PMID:15950215; http://dx.doi.org/ 10.1016/j.ydbio.2005.05.007 [DOI] [PubMed] [Google Scholar]
- 22. Ciccolini F, Svendsen CN. Fibroblast growth factor 2 (FGF-2) promotes acquisition of epidermal growth factor (EGF) responsiveness in mouse striatal precursor cells: identification of neural precursors responding to both EGF and FGF-2. J Neurosci 1998; 18:7869-80; PMID:9742155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ginty DD, Kornhauser JM, Thompson MA, Bading H, Mayo KE, Takahashi JS, Greenberg ME. Regulation of CREB phosphorylation in the suprachiasmatic nucleus by light and a circadian clock. Science 1993; 260:238-41; PMID:8097062; http://dx.doi.org/ 10.1126/science.8097062 [DOI] [PubMed] [Google Scholar]
- 24. Pilz GA, Shitamukai A, Reillo I, Pacary E, Schwausch J, Stahl R, Ninkovic J, Snippert HJ, Clevers H, Godinho L, et al. Amplification of progenitors in the mammalian telencephalon includes a new radial glial cell type. Nat Commun 2013; 4:2125; PMID:23839311; http://dx.doi.org/ 10.1038/ncomms3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Viti J, Gulacsi A, Lillien L. Wnt regulation of progenitor maturation in the cortex depends on Shh or fibroblast growth factor 2. J Neurosci 2003; 23:5919-27; PMID:12843296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rada-Iglesias A, Enroth S, Ameur A, Koch CM, Clelland GK, Respuela-Alonso P, Wilcox S, Dovey OM, Ellis PD, Langford CF, et al. Butyrate mediates decrease of histone acetylation centered on transcription start sites and down-regulation of associated genes. Gen Res 2007; 17:708-19; PMID:17567991; http://dx.doi.org/ 10.1101/gr.5540007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Peixoto L, Abel T. The role of histone acetylation in memory formation and cognitive impairments. Neuropsychopharmacology 2013; 38:62-76; PMID:22669172; http://dx.doi.org/ 10.1038/npp.2012.86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lemberger T, Parkitna JR, Chai M, Schutz G, Engblom D. CREB has a context-dependent role in activity-regulated transcription and maintains neuronal cholesterol homeostasis. FASEB J 2008; 22:2872-9; PMID:18424767; http://dx.doi.org/ 10.1096/fj.08-107888 [DOI] [PubMed] [Google Scholar]
- 29. Parlato R, Rieker C, Turiault M, Tronche F, Schutz G. Survival of DA neurons is independent of CREM upregulation in absence of CREB. Genesis 2006; 44:454-64; PMID:16981198; http://dx.doi.org/ 10.1002/dvg.20236 [DOI] [PubMed] [Google Scholar]
- 30. Balasubramaniyan V, Boddeke E, Bakels R, Kust B, Kooistra S, Veneman A, Copray S. Effects of histone deacetylation inhibition on neuronal differentiation of embryonic mouse neural stem cells. Neuroscience 2006; 143:939-51; PMID:17084985; http://dx.doi.org/ 10.1016/j.neuroscience.2006.08.082 [DOI] [PubMed] [Google Scholar]
- 31. Hsieh J, Nakashima K, Kuwabara T, Mejia E, Gage FH. Histone deacetylase inhibition-mediated neuronal differentiation of multipotent adult neural progenitor cells. Proc Natl Acad Sci U S A 2004; 101:16659-64; PMID:15537713; http://dx.doi.org/ 10.1073/pnas.0407643101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Humphrey GW, Wang YH, Hirai T, Padmanabhan R, Panchision DM, Newell LF, McKay RD, Howard BH. Complementary roles for histone deacetylases 1, 2, and 3 in differentiation of pluripotent stem cells. Differentiation 2008; 76:348-56; PMID:18021260; http://dx.doi.org/ 10.1111/j.1432-0436.2007.00232.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shaked M, Weissmuller K, Svoboda H, Hortschansky P, Nishino N, Wolfl S, Tucker KL. Histone deacetylases control neurogenesis in embryonic brain by inhibition of BMP2/4 signaling. PloS one 2008; 3:e2668; PMID:18628975; http://dx.doi.org/ 10.1371/journal.pone.0002668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Scholl C, Weibetamuller K, Holenya P, Shaked-Rabi M, Tucker KL, Wolfl S. Distinct and overlapping gene regulatory networks in BMP- and HDAC-controlled cell fate determination in the embryonic forebrain. BMC genomics 2012; 13:298; PMID:22748179; http://dx.doi.org/ 10.1186/1471-2164-13-298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Humphrey GW, Wang Y, Russanova VR, Hirai T, Qin J, Nakatani Y, Howard BH. Stable histone deacetylase complexes distinguished by the presence of SANT domain proteins CoREST/kiaa0071 and Mta-L1. J Biol Chem 2001; 276:6817-24; PMID:11102443; http://dx.doi.org/ 10.1074/jbc.M007372200 [DOI] [PubMed] [Google Scholar]
- 36. Sun G, Yu RT, Evans RM, Shi Y. Orphan nuclear receptor TLX recruits histone deacetylases to repress transcription and regulate neural stem cell proliferation. Proc Natl Acad Sci U S A 2007; 104:15282-7; PMID:17873065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. You A, Tong JK, Grozinger CM, Schreiber SL. CoREST is an integral component of the CoREST- human histone deacetylase complex. Proc Natl Acad Sci U S A 2001; 98:1454-8; PMID:11171972; http://dx.doi.org/ 10.1073/pnas.98.4.1454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen ZF, Paquette AJ, Anderson DJ. NRSF/REST is required in vivo for repression of multiple neuronal target genes during embryogenesis. Nat Genet 1998; 20:136-42; PMID:9771705; http://dx.doi.org/ 10.1038/2431 [DOI] [PubMed] [Google Scholar]
- 39. Roy K, Kuznicki K, Wu Q, Sun Z, Bock D, Schutz G, Vranich N, Monaghan AP. The Tlx gene regulates the timing of neurogenesis in the cortex. J Neurosci 2004; 24:8333-45; PMID:15385616; http://dx.doi.org/ 10.1523/JNEUROSCI.1148-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shi Y, Chichung Lie D, Taupin P, Nakashima K, Ray J, Yu RT, Gage FH, Evans RM. Expression and function of orphan nuclear receptor TLX in adult neural stem cells. Nature 2004; 427:78-83; PMID:14702088; http://dx.doi.org/ 10.1038/nature02211 [DOI] [PubMed] [Google Scholar]
- 41. Obernier K, Simeonova I, Fila T, Mandl C, Holzl-Wenig G, Monaghan-Nichols P, Ciccolini F. Expression of Tlx in both stem cells and transit amplifying progenitors regulates stem cell activation and differentiation in the neonatal lateral subependymal zone. Stem Cells 2011; 29:1415-26; PMID:21714038; http://dx.doi.org/ 10.1002/stem.682 [DOI] [PubMed] [Google Scholar]
- 42. Chan HM, La Thangue NB. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J Cell Sci 2001; 114:2363-73; PMID:11559745 [DOI] [PubMed] [Google Scholar]
- 43. Nucifora FC, Jr., Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, et al. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science 2001; 291:2423-8; PMID:11264541; http://dx.doi.org/ 10.1126/science.1056784 [DOI] [PubMed] [Google Scholar]
- 44. Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in drosophila. Nature 2001; 413:739-43; PMID:11607033; http://dx.doi.org/ 10.1038/35099568 [DOI] [PubMed] [Google Scholar]
- 45. Giralt A, Puigdellivol M, Carreton O, Paoletti P, Valero J, Parra-Damas A, Saura CA, Alberch J, Ginés S. Long-term memory deficits in Huntington's disease are associated with reduced CBP histone acetylase activity. Hum Mol Genet 2012; 21:1203-16; PMID:22116937; http://dx.doi.org/ 10.1093/hmg/ddr552 [DOI] [PubMed] [Google Scholar]
- 46. Lopez-Atalaya JP, Ciccarelli A, Viosca J, Valor LM, Jimenez-Minchan M, Canals S, Giustetto M, Barco A. CBP is required for environmental enrichment-induced neurogenesis and cognitive enhancement. The EMBO J 2011; 30:4287-98; PMID:21847097; http://dx.doi.org/ 10.1038/emboj.2011.299 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.