

Abstract
Although the range of biocatalysts available for the synthesis of enantiomerically pure chiral amines continues to expand, few existing methods provide access to secondary amines. To address this shortcoming, we have over‐expressed the gene for an (R)‐imine reductase [(R)‐IRED] from Streptomyces sp. GF3587 in Escherichia coli to create a recombinant whole‐cell biocatalyst for the asymmetric reduction of prochiral imines. The (R)‐IRED was screened against a panel of cyclic imines and two iminium ions and was shown to possess high catalytic activity and enantioselectivity. Preparative‐scale synthesis of the alkaloid (R)‐coniine (90 % yield; 99 % ee) from the imine precursor was performed on a gram‐scale. A homology model of the enzyme active site, based on the structure of a closely related (R)‐IRED from Streptomyces kanamyceticus, was constructed and used to identify potential amino acids as targets for mutagenesis.
Keywords: amines, asymmetric catalysis, biocatalysis, nitrogen heterocycles, reduction
Chiral amines are key structural motifs widely found in many natural products as well as pharmaceuticals, agrochemicals, and fine chemicals.1 Methods for their preparation in optically pure form include organo‐ and transition‐metal‐catalyzed asymmetric synthesis,2 although approaches based on classical resolution through crystallization or kinetic resolution3 often remain the method of choice in industry despite the maximum theoretical yield capped at 50 %.4 An alternative and attractive strategy for chiral amine synthesis is to employ enzymes, in particular biocatalysts the properties of which have been tailored and enhanced by protein engineering and directed evolution.5 Notable developments in this area include the use of monoamine oxidases (MAO‐N),6 phenylalanine ammonia lyases,7 and aminotransferases (transaminases, Scheme 1).8
Scheme 1.
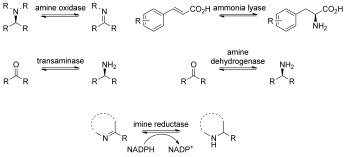
Reactions of enzymes employed in the synthesis of chiral amines, with imine reductase (bottom) representing a recent addition to available biocatalysts.
Additionally, an l‐amino‐acid dehydrogenase has recently been subjected to protein engineering and shown to catalyze the asymmetric reductive amination of ketones (amine dehydrogenases, Scheme 1).9 In a number of cases, these biocatalysts have been implemented in large‐scale manufacturing processes for the production of active pharmaceutical ingredients as well as their key intermediates.10
With the exception of MAO‐N, which can be used for the chemoenzymatic deracemization of amines with broad structural features, the majority of these biocatalysts generate only primary amines, with no direct access to secondary or tertiary amines. In contrast, the asymmetric reduction of imines provides an alternative method for generating these motifs. Such an approach has been explored by the development of chemocatalytic processes11 and also the creation of artificial metalloenzymes,12 which have been successfully used in tandem with biocatalysts in cascade reactions.13 However, the enzymatic equivalent of this process remains largely unexplored.14
Recently, an imine reductase [(S)‐IRED] from Streptomyces sp. GF3546 expressed in Escherichia coli 15 was shown to catalyze the enantioselective reduction of a range of different imines, including 2‐substituted cyclic imines, dihydroisoquinolines, β‐carbolines, and also iminium ions.16 This (S)‐IRED was found to be exclusively dependent on the cofactor NADPH as a source of hydride. The identification of enantiocomplementary enzymes that provide access to both enantiomers of target products remains a key challenge in biocatalysis, which has been addressed recently by the development of (R)‐selective transaminases17 and an (R)‐selective amine oxidase.18 Herein we report the heterologous expression and characterization of an enantiocomplementary (R)‐IRED from Streptomyces sp. GF3587, which has been previously described and found to act on 2‐methyl‐1‐pyrroline 1 a, giving some indication of substrate preference.19 In this communication, the (R)‐IRED is shown to possess a wide substrate scope and has been applied to the synthesis of the alkaloid coniine 4 i on a gram scale. Additionally, the imine reductase displayed significantly higher catalytic activity than previously reported IREDs, with a preference for reducing 6‐membered ring piperideine substrates, which may provide insight into its natural substrate.
To explore the substrate scope of the (R)‐IRED, a panel of cyclic imines was synthesized and screened for activity. With the exception of 1 a, all 2‐substituted imines were prepared by a previously reported method20 involving the addition of an organometallic Grignard reagent to an N‐Boc‐protected lactam (Boc=tert‐butoxycarbonyl), followed by deprotection of the amine, which undergoes spontaneous cyclization to give the free‐base cyclic imine. This flexible approach allowed access to cyclic imines of varying ring sizes with a range of substituents. The imines were isolated as their hydrochloride salts to enhance their stability as well as water solubility.
Since the only previously reported substrate for the (R)‐IRED was 1 a, which gave excellent conversion (>98 %) and ee (>98 %), our initial focus was directed towards screening a range of 2‐substituted pyrrolines. Biotransformations using resting E. coli cells expressing the (R)‐IRED were conducted at substrate concentrations of 5 mm with added glucose to aid cofactor recycling (see Supporting Information for the conditions used for cultivation of the recombinant strain and expression of the (R)‐IRED). Negative controls using cells harboring the empty plasmid vector were run in parallel to ensure that any observed activity occurred as a result of the IRED biocatalysts.
The (R)‐IRED was shown to catalyze the reduction of pyrrolines containing both aliphatic and aromatic substituents at C‐2 (Table 1). In addition to the reduction of 1 a to (R)‐2 a [from which the designation as (R)‐IRED was derived] the phenyl‐ and cyclohexyl‐substituted pyrrolines 1 b and 1 e, respectively, were fully converted after 24 h; however, a significant reduction in enantioselectivity was observed with these bulkier substituents. When a para‐methoxy substituent was introduced on the benzene ring, conversion fell to 20 % but the reaction proceeded with excellent selectivity to give (S)‐2 c in >98 % ee. Under the same conditions, the (R)‐IRED gave significantly higher conversions than the previously reported (S)‐IRED for pyrroline substrates 1 a–d.
Table 1.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
To further investigate the substrate scope of the (R)‐IRED, whole‐cell biotransformations were performed with larger 6‐ and 7‐membered‐ring cyclic imine scaffolds. Tetrahydroazepines 3 a and 3 c were both converted with excellent selectivity. The (R)‐IRED was found to display particularly high activity towards 6‐membered rings, yielding 2‐substituted piperidines, which are important scaffolds in many biologically significant compounds.21 The high activity towards this class of compound is apparent if comparing the conversions of paramethoxyphenyl‐substituted homologues 1 c, 3 c, and 5 c (conversions=20, 50, and ≥98 %, respectively). The biocatalytic reduction of a large panel of piperideines 5 a–m was performed to fully gauge the effects of varying substituents (Table 2).
Table 2.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Excellent conversion was observed after 24 h with piperideines 5 a–l. Reduced conversion of 54 % was observed with benzyl‐substituted derivative 5 m and detectable conversion was also observed with the bulky 1‐naphthyl‐substituted 5 n, suggestive of an enzyme possessing a large active site. For substrates bearing phenyl derivatives, the introduction of a substituent on the ring led to an increase in enantioselectivity. Both electron‐donating and electron‐withdrawing groups were readily accepted, with high conversion and ee observed. Interestingly, a moderate decrease in selectivity was observed if moving the position of the methoxy‐substituent from para to meta and ortho, signaling an increasing contribution of a competing binding mode in which the opposite face of the imine is exposed for reduction by the NADPH. The (R)‐IRED readily accepted other heterocyclic motifs including a thiophene ring (5 l).
With simple alkyl and alkenyl substituents, the (R)‐IRED showed excellent enantioselectivity. In addition, reduction of α,β‐unsaturated imine 5 j proceeded exclusively at the carbon–nitrogen double bond with no reduction of the alkene observed. Reduction of n‐propyl‐substituted piperideine 5 i resulted in the formation of the more active (R)‐enantiomer of the natural product coniine in >98 % ee. To demonstrate the application of the (R)‐IRED biocatalyst for preparative‐scale synthesis of chiral amines, the reduction of 5 j was performed on a 1.0 g (25 mm) scale, yielding (R)‐coniine 6 i (90 % yield, >98 % ee), which was isolated as its hydrochloride salt. In addition to simple 2‐substituted piperideines, 3,4‐dihydroisoquinolines 7 and 7 a were also confirmed to be substrates for the enzyme. Significantly, the corresponding N‐methyl iminium derivatives 9 and 9 a were also reduced, with chiral amine 10 a produced with comparable selectivity (74 % ee), albeit with reduced conversion, demonstrating the potential application of this biocatalyst for the synthesis of tertiary chiral amines.
Biotransformations of several substrates conducted with the isolated enzyme, coupled with an NADPH cofactor recycling system using glucose dehydrogenase 2 from Bacillus megaterium,22 showed no significant change in enantioselectivity if compared to their whole‐cell equivalents, with the exception of dihydroisoquinoline 7 a for which unusually the selectivity appeared to diminish to give (R)‐8 a in 47 % ee (see Supporting Information). As the whole‐cell biocatalyst requires only the addition of glucose for cofactor recycling and negates the necessity of using large amounts of NADPH or an additional cofactor recycling system, this approach was used for any preparative biotransformations.
Kinetic parameters were determined for representative substrates (Table 3). Kinetic constants with the previously reported (S)‐IRED from Streptomyces sp. GF3546 have been described with substrates 1 a, 3 a, 5 a, 7, 7 a, and 9 (i.e., k cat=0.024 s−1, 0.039 s−1, 0.137 s−1, 0.445 s−1, 0.040 s−1, and 0.483 s−1, respectively).16 Excluding 7, significantly higher k cat values and lower Michaelis constants (K m) were observed for the (R)‐IRED described herein (Table 3). The previously reported substrate 2‐methyl‐1‐pyrroline 1 a had a notably lower k cat/K m value (catalytic efficiency) than several piperideine substrates tested. In particular, we observed a greater than sixty‐fold increase in k cat/Km for 2‐methyl‐1‐piperideine 5 a over pyrroline 1 a. This preference for 6‐ and 7‐membered ring systems is shared with the (S)‐IRED and may allude to the natural function of this class of enzyme. The (R)‐IRED also displayed greater activity towards simple alkyl‐ and alkenyl‐substituted imines compared with those bearing aromatic moieties.
Table 3.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
A selection of ketones and oximes were screened for activity with the (R)‐IRED, with no conversion detected (see Supporting Information). The absence of activity towards ketones suggests future potential applications of this enzyme for intermolecular reductive aminations, which remains a significant challenge in organic synthesis.
We have recently determined the structure of oxidoreductase Q1EQE0 from Streptomyces kanamyceticus,22 which shares 50 % sequence identity with the (R)‐IRED, and which also catalyzes the (R)‐selective reduction of imine substrates such as 2‐methyl‐1‐pyrroline 1 a. The structure of Q1EQE0 revealed a dimeric association in which two monomers associate very closely through domain swapping. The active site, containing NADPH, is a channel that traverses the width of the dimer. The active site also revealed residues which may be responsible for catalysis, including Asp187, which was proposed to be the catalytic residue for protonation of the imine in IRED‐mediated catalysis.22 The similarity between the two sequences gave us confidence to build a model of the (R)‐IRED (Figure 1 a) using the Phyre2 server.23 In the model, Asp187 in Q1EQE0 is conserved in the (R)‐IRED as Asp172 and many of the other residues within the region of the active site also appear to be conserved between the two enzymes (Figure 1 b).
Figure 1.
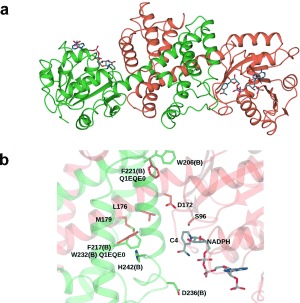
a) Homology model of the (R)‐IRED, shown in ribbon format, based on the known structure of Q1EQE0,22 with which it shares 50 % sequence identity and 70 % similarity. Subunit A is shown in green; subunit B in coral. NADPH is shown bound at the dimer interface in cylinder format, with carbon atoms in grey. b) Detail of active site, illustrating conservation of active residues between Q1EQE0 and (R)‐IRED using (R)‐IRED numbering. All labeled residues are conserved, except Trp206 (replaced by Phe in Q1EQE0) and Phe217 (Trp232), both shown in red, and Met122 (Leu137) and Ala245 (Thr258), not shown, in the active‐site region close to the NADPH nicotinamide ring. Asp172, which, as Asp187 in Q1EQE0 was suggested to be a catalytic residue,22 was selected for mutational studies.
The role of Asp172 in the catalytic mechanism was further probed by generation of the Asp172Ala and Asp172Lys point mutants by site‐directed mutagenesis. Interestingly, both mutants retained catalytic activity although conversions were generally lower, for example, for 5 c the conversion was reduced from 92 % for the wild‐type isolated protein to 40 % and 72 % for the Asp172Ala and Asp172Lys mutants, respectively. Remarkably, reduction of 5 a by the mutants showed no change in enantioselectivity whereas for 7 a the ee value was increased to 81 % for the Asp172Ala variant (Table 4). These initial studies highlight the importance of this residue as a hotspot for mutagenesis to improve conversions and/or enantioselectivity.
Table 4.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
In summary, the gene for Streptomyces sp. GF3587 (R)‐IRED was overexpressed in E. coli to produce a recombinant whole‐cell biocatalyst possessing broad substrate scope, suitable for the preparation of chiral cyclic secondary amines with broad structural features and with two examples of tertiary amine synthesis also demonstrated. The activity of the (R)‐IRED was shown to be orders of magnitude higher than previously reported imine reductases, which is significant for potential industrial applications.14 Although wild‐type IREDs appear to possess relatively broad substrate specificity, further engineering through directed evolution and rational design, using the protein model presented herein, will undoubtedly lead to more active biocatalysts.25 Moreover, the opportunity to combine IREDs with existing biocatalysts in cascade reactions26 should result in greater application of these enzymes in asymmetric synthesis.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
Acknowledgements
The research leading to these results received support from the EPSRC (to S.H. and S.F.), AstraZeneca (to S.H.), Pfizer (to S.P.F.), the industrial affiliates of the Centre of Excellence for Biocatalysis, Biotransformations and Biomanufacture (CoEBio3, to H.M. and E.W.), the European Union’s Seventh Framework Programme FP7/2007‐2013 under grant agreement no. 266025 (BIONEXGEN, to F. L.) and EFPIA companies’ in kind contribution for the Innovative Medicine Initiative under Grant Agreement No. 115360 (Chemical manufacturing methods for the 21st century pharmaceuticals industries, CHEM21, to F.L.).
References
- 1. Welsch M. E., Snyder S. A., Stockwell B. R., Curr. Opin. Chem. Biol. 2010, 14, 347–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Silverio D. L., Torker S., Pilyugina T., Vieira E. M., Snapper M. L., Haeffner F., Hoveyda A. H., Nature 2013, 494, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Binanzer M., Hsieh S. Y., Bode J. W., J. Am. Chem. Soc. 2011, 133, 19698–19701. [DOI] [PubMed] [Google Scholar]
- 4. Breuer M., Ditrich K., Habicher T., Hauer B., Kesseler M., Sturmer R., Zelinski T., Angew. Chem. Int. Ed. 2004, 43, 788–824; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 806–843. [Google Scholar]
- 5. Bornscheuer U. T., Huisman G. W., Kazlauskas R. J., Lutz S., Moore J. C., Robins K., Nature 2012, 485, 185–194. [DOI] [PubMed] [Google Scholar]
- 6. Ghislieri D., Houghton D., Green A. P., Willies S. C., Turner N. J., Acs Catal. 2013, 3, 2869–2872; [Google Scholar]; Ghislieri D., Green A. P., Pontini M., Willies S. C., Rowles I., Frank A., Grogan G., Turner N. J., J. Am. Chem. Soc. 2013, 135, 10863–10869. [DOI] [PubMed] [Google Scholar]
- 7. Turner N. J., Curr. Opin. Chem. Biol. 2011, 15, 234–240. [DOI] [PubMed] [Google Scholar]
- 8. Koszelewski D., Lavandera I., Clay D., Guebitz G. M., Rozzell D., Kroutil W., Angew. Chem. Int. Ed. 2008, 47, 9337–9340; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 9477–9480; [Google Scholar]; Koszelewski D., Lavandera I., Clay D., Rozzell D., Kroutil W., Adv. Synth. Catal. 2008, 350, 2761–2766. [Google Scholar]
- 9. Abrahamson M. J., Vazquez‐Figueroa E., Woodall N. B., Moore J. C., Bommarius A. S., Angew. Chem. Int. Ed. 2012, 51, 3969–3972; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 4036–4040. [Google Scholar]
- 10. Li T., Liang J., Ambrogelly A., Brennan T., Gloor G., Huisman G., Lalonde J., Lekhal A., Mijts B., Muley S., Newman L., Tobin M., Wong G., Zaks A., Zhang X., J. Am. Chem. Soc. 2012, 134, 6467–6472; [DOI] [PubMed] [Google Scholar]; Savile C. K., Janey J. M., Mundorff E. C., Moore J. C., Tam S., Jarvis W. R., Colbeck J. C., Krebber A., Fleitz F. J., Brands J., Devine P. N., Huisman G. W., Hughes G. J., Science 2010, 329, 305–309; [DOI] [PubMed] [Google Scholar]; de Lange B., Hyett D. J., Maas P. J. D., Mink D., van Assema F. B. J., Sereinig N., de Vries A. H. M., de Vries J. G., ChemCatChem 2011, 3, 289–292. [Google Scholar]
- 11. Chang M. X., Li W., Hou G. H., Zhang X. M., Adv. Synth. Catal. 2010, 352, 3121–3125; [Google Scholar]; Chang M. X., Li W., Zhang X. M., Angew. Chem. Int. Ed. 2011, 50, 10679–10681; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 10867–10869; [Google Scholar]; Uematsu N., Fujii A., Hashiguchi S., Ikariya T., Noyori R., J. Am. Chem. Soc. 1996, 118, 4916–4917. [Google Scholar]
- 12. Dürrenberger M., Heinisch T., Wilson Y. M., Rossel T., Nogueira E., Knörr L., Mutschler A., Kersten K., Zimbron M. J., Pierron J., Schirmer T., Ward T. R., Angew. Chem. Int. Ed. 2011, 50, 3026–3029; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 3082–3085. [Google Scholar]
- 13. Köhler V., Wilson Y. M., Durrenberger M., Ghislieri D., Churakova E., Quinto T., Knorr L., Häussinger D., Hollmann F., Turner N. J., Ward T. R., Nat. Chem. 2013, 5, 93–99. [DOI] [PubMed] [Google Scholar]
- 14. Vaijayanthi T., Chadha A., Tetrahedron: Asymmetry 2008, 19, 93–96; [Google Scholar]; Muramatsu H., Mihara H., Kakutani R., Yasuda M., Ueda M., Kurihara T., Esaki N., J. Biol. Chem. 2005, 280, 5329–5335; [DOI] [PubMed] [Google Scholar]; Espinoza‐Moraga M., Petta T., Vasquez‐Vasquez M., Laurie V. F., Moraes L. A. B., Santos L. S., Tetrahedron: Asymmetry 2010, 21, 1988–1992; [Google Scholar]; Mirabal‐Gallardo Y., Soriano M. D. C., Santos L. S., Tetrahedron: Asymmetry 2013, 24, 440–443. [Google Scholar]
- 15. Mitsukura K., Kuramoto T., Yoshida T., Kimoto N., Yamamoto H., Nagasawa T., Appl. Microbiol. Biotechnol. 2013, 97, 8079–8086. [DOI] [PubMed] [Google Scholar]
- 16. Leipold F., Hussain S., Ghislieri D., Turner N. J., ChemCatChem 2013, 5, 3505–3508. [Google Scholar]
- 17. Höhne M., Schätzle S., Jochens H., Robins K., Bornscheuer U. T., Nat. Chem. Biol. 2010, 6, 807–813. [DOI] [PubMed] [Google Scholar]
- 18. Heath R. S., Pontini M., Bechi B., Turner N. J., ChemCatChem 2014, 6, 996–1002. [Google Scholar]
- 19. Mitsukura K., Suzuki M., Shinoda S., Kuramoto T., Yoshida T., Nagasawa T., Biosci. Biotechnol. Biochem. 2011, 75, 1778–1782. [DOI] [PubMed] [Google Scholar]
- 20. Williams G. D., Pike R. A., Wade C. E., Wills M., Org. Lett. 2003, 5, 4227–4230. [DOI] [PubMed] [Google Scholar]
- 21. Ren H., Wulff W. D., Org. Lett. 2013, 15, 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nagao T., Mitamura T., Wang X. H., Negoro S., Yomo T., Urabe I., Okada H., J. Bacteriol. 1992, 174, 5013–5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rodríguez‐Mata M., Frank A., Wells E., Leipold F., Turner N. J., Hart S., Turkenburg J. P., Grogan G., ChemBioChem 2013, 14, 1372–1379. [DOI] [PubMed] [Google Scholar]
- 24. Kelley L. A., Sternberg M. J. E., Nat. Protoc. 2009, 4, 363–371. [DOI] [PubMed] [Google Scholar]
- 25. Turner N. J., Nat. Chem. Biol. 2009, 5, 567–573. [DOI] [PubMed] [Google Scholar]
- 26. Schrittwieser J. H., Groenendaal B., Resch V., Ghislieri D., Wallner S., Fischereder E.‐M., Fuchs E., Grischek B., Sattler J. H., Macheroux P., Turner N. J., Kroutil W., Angew. Chem. Int. Ed. 2014, 53, 3731–3734; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3805–3809; [Google Scholar]; O’Reilly E., Iglesias C., Ghislieri D., Hopwood J., Galman J. L., Lloyd R. C., Turner N. J., Angew. Chem. Int. Ed. 2014, 53, 2447–2450; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 2479–2482. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information