

Abstract
Cytochrome P450 (CYP) 1B1 is implicated in vascular smooth muscle cell migration, proliferation, and hypertension. We assessed the contribution of CYP1B1 to angiotensin (Ang) II–induced abdominal aortic aneurysm (AAA). Male Apoe−/−/Cyp1b1+/+ and Apoe−/−/Cyp1b1−/− mice were infused with Ang II or its vehicle for 4 weeks; another group of Apoe−/−/Cyp1b1+/+ mice was coadministered the CYP1B1 inhibitor 2,3′,4,5′-tetramethoxystilbene (TMS) every third day for 4 weeks. On day 28 of Ang II infusion, AAAs were analyzed by ultrasound and ex vivo by Vernier calipers, mice were euthanized, and tissues were harvested. Ang II produced AAAs in Apoe−/−/Cyp1b1+/+ mice; mice treated with TMS or Apoe−/−/Cyp1b1−/− mice had reduced AAAs. Ang II enhanced infiltration of macrophages, T cells, and platelets and increased platelet-derived growth factor D, Pdgfrb, Itga2, and matrix metalloproteinases 2 and 9 expression in aortic lesions; these changes were inhibited in mice treated with TMS and in Apoe−/−/Cyp1b1−/− mice. Oxidative stress resulted in cyclooxygenase-2 expression in aortic lesions. These effects were minimized in Apoe−/−/Cyp1b1+/+ mice treated with TMS and in Apoe−/−/Cyp1b1−/− mice and by concurrent treatment with the superoxide scavenger 4-hydroxyl-2,2,6,6-tetramethylpiperidine-1-oxyl. CYP1B1 contributed to the development of Ang II–induced AAA and associated pathogenic events in mice, likely by enhancing oxidative stress and associated signaling events. Thus, CYP1B1 may serve as a target for therapeutic agents for AAA in males.
Aortic aneurysm is becoming a common malady worldwide.1 Aortic aneurysm manifests as either thoracic or abdominal aortic aneurysm (AAA).2 AAA has a high prevalence in aged men, whereas thoracic aortic aneurysm exhibits no sex propensity but has a greater prevalence in younger individuals.2 Irrespective of the type, it is well known that activation of the renin–angiotensin (Ang) system, with a consequent increase in the production of Ang II, promotes the development of aneurysms.1 It has been established that the expression of the Apoe gene is altered in patients with AAA,3 and that Apoe−/− mice receiving exogenous Ang II develop AAA, and are therefore used as a model for human AAA.4
AAA pathogenesis involves a series of events characterized by increased oxidative stress; inflammation caused by infiltration of immune cells resulting in production of cytokines/chemokines; apoptosis of smooth muscle cells; and activation of matrix metalloproteinase (MMP), resulting in degradation of elastic fibers and dilation of the aortic wall.5, 6 However, the underlying mechanism leading to this chain of events has not been established. Reactive oxygen species (ROS) produced by inflammatory cells and cells of the vasculature increase activity of one or more signaling molecules, including MMP, and cause lipid peroxidation and tissue damage.7, 8 ROSs are believed to be the major component of the mechanisms that contribute to AAA.7, 8 The metabolism of fatty acids by cyclooxygenase (COX), lipoxygenase, and cytochrome P450 (CYP) enzymes results in the generation of ROS,8, 9 and ROS generated via the activation of NADPH oxidase appears to play a major role in the development of AAA.10
Previously, we have shown that Ang II–induced vascular smooth muscle cell (VSMC) migration, proliferation and hypertrophy,11 and hypertension12 depend on CYP1B1,11, 12 a heme-thiolate monooxygenase that is expressed in extrahepatic tissues, including those in the cardiovascular system,13 and is involved in activating NADPH oxidase and generating ROS.11, 12, 13
These observations led us to the hypothesis that Ang II–induced AAA and associated pathogenesis are mediated by CYP1B1-generated ROS. To test this hypothesis, we examined the effect of 2,3′,4,5′-tetramethoxystilbene (TMS), a selective inhibitor of CYP1B1,14 and Cyp1b1 gene disruption on the development of Ang II–induced AAA and its pathogenesis in Apoe−/−/Cyp1b1+/+ male mice. Our study demonstrates for the first time that CYP1B1-generated oxidative stress and COX-2 expression most likely contribute to the inflammatory events characterized by copious infiltration of inflammatory cells and platelets. The resultant signaling events, via the expression of platelet-derived growth factor (PDGF) receptor β (Pdgfrb) and integrin α2 (Itga2) in the aorta, enhance MMP levels, resulting in the degradation of extracellular matrix (ECM) components and leading to Ang II–induced AAA.
Materials and Methods
Materials
Ang II was purchased from Bachem Americas, Inc. (Torrance, CA); dihydroethidium (DHE) was purchased from Molecular Probes (Thermo Fisher Scientific, Kalamazoo, MI); TMS was purchased from Cayman Chemical Company (Ann Arbor, MI); and dimethyl sulfoxide, Masson's trichrome for collagen staining, and the Elastic Staining Kit (HT25A) were purchased from Sigma-Aldrich (St. Louis, MO). A list of antibodies, their sources, and the concentrations used in this study are provided in Table 1. Appropriate isotype control antibodies from Abcam (Cambridge, UK) were also used for validating the specificity of the immunostaining, as follows: for α-smooth muscle actin, isotype control mouse IgG2a (ab18413); for PDGF-D, isotype control mouse IgG2b (ab170192); for F4/80, isotype control rat IgG2b (ab185797); for CD3, PDGF-A and -B, PDGFR-β, Itg-α2, COX-2, and MMP-2 and -9, isotype control rabbit IgG (ab27478); and for CD62p and PDGF-C, isotype control goat IgG (ab37373).
Table 1.
List of Antibodies for IHC and IF
| Antibody | Application | Primary antibody |
Blocking buffer | Secondary antibody |
|||
|---|---|---|---|---|---|---|---|
| Company & cat. # | Dilution | Host | Company & cat. # | Dilution | |||
| α-SMA | IHC | Sigma-Aldrich A2547 | 1:400 | Mouse | 5% BSA | Sigma-Aldrich A3682 | 1:150 |
| CD3 | IHC | Abcam (Cambridge, UK) ab5690 | 1:200 | Rabbit | 5% BSA | Abcam ab97051 | 1:200 |
| CD62p | IHC | Santa Cruz Biotechnology, Inc. (Dallas, TX) Sc6941 | 1:200 | Goat | 5% BSA | Abcam ab6741 | 1:200 |
| F4/80 | IHC | AbD Serotec (Raleigh, NC) MCA497R | 1:200 | Rat | Rabbit normal serum | AbD Serotec STAR72 | 1:200 |
| PDGFR-β | IHC | Sigma-Aldrich SAB4502146 | 1:100 | Rabbit | 5% BSA | Abcam ab97051 | 1:200 |
| Itg-α2 | IHC | Abcam ab133557 | 1:200 | Rabbit | 5% BSA | Abcam ab97051 | 1:200 |
| COX-2 | IHC | Abcam ab15191 | 1:200 | Rabbit | 5% BSA | Abcam ab97051 | 1:200 |
| PDGF-A | IHC | Santa Cruz Sc7958 | 1:200 | Rabbit | 5% BSA | Abcam ab97051 | 1:200 |
| PDGF-B | IHC | Santa Cruz Sc7878 | 1:200 | Rabbit | 5% BSA | Abcam ab97051 | 1:200 |
| PDGF-C | IHC | Santa Cruz Sc18228 | 1:200 | Goat | 5% BSA | Abcam ab6741 | 1:200 |
| PDGF-D | IHC | Santa Cruz Sc137030 | 1:200 | Mouse | 5% BSA | Sigma-Aldrich A3682 | 1:150 |
| MMP-2 | IF | Abcam ab37150 | 1:400 | Rabbit | 2% BSA/0.2% Tween 20 | Abcam ab150077 | 1:700 |
| MMP-9 | IF | Abcam ab38898 | 1:400 | Rabbit | 2% BSA/0.2% Tween 20 | Abcam ab150077 | 1:700 |
| CYP1B1 | WB | Santa Cruz Sc3882 | 1:500 | Rabbit | 5% Skim milk | GE Healthcare Bio-Sciences (Pittsburgh, PA) NA934V | 1:1000 |
| β-actin | WB | Santa Cruz Sc47778 | 1:1000 | Mouse | 5% Skim milk | GE Healthcare Bio-Sciences NA931V | 1:1000 |
BSA, bovine serum albumin; cat., catalog; COX, cyclooxygenase; CYP, cytochrome P450; IF, immunofluorescence; IHC, immunohistochemistry analysis; Itg-α2, integrin α2; MMP, matrix metalloproteinase; PDGFR-β, platelet-derived growth factor receptor β; SMA, smooth muscle actin; WB, Western blot.
Animals
Experiments were performed according to the protocols approved by The University of Tennessee Health Science Center (Memphis, TN) Institutional Animal Care and Use Committee, in accordance with the NIH Guide for the Care and Use of Laboratory Animals.15 Apoe−/−/Cyp1b1+/+ mice on a C57BL/6J background (purchased from The Jackson Laboratory, Bar Harbor, ME). Cyp1b1−/− mice (PMID 10051580) on a C57BL/6J background were backcrossed 10 generations to the C57BL/6J background, and brother–sister mice were mated to obtain homozygous Apoe+/+/Cyp1b1−/− mice on a C57BL/6J background. The genotype of these mice was confirmed in 4-week-old animals.
Apoe−/−/Cyp1b1−/− mice were generated in our laboratory at The University of Tennessee Health Science Center by breeding Apoe−/−/Cyp1b1+/+ mice on a C57BL/6J background with Apoe+/+/Cyp1b1−/− mice on a C57BL/6J background.16 Apoe−/−/Cyp1b1−/− mice and Apoe−/−/Cyp1b1+/+ mice generated from eight brother–sister matings were used in our experiments. All animals, including the Apoe−/−/Cyp1b1−/− mice, were normal, and we did not observe any differences in their phenotypes, including their movements, eyesight, or fertility. Animals were routinely genotyped using the following primers: ApoE forward-1, 5′-GCCTAGCCGAGGGAGAGCCG-3′; ApoE forward-2, 5′-TGTGACTTGGGAGCTCTGCAGC-3′; ApoE reverse, 5′-GCCGCCCCGACTGCATCT-3′, as described17; CYP1B1-3 forward, 5′-TTTGCCTGTCACCATTCCAC-3′; CYP1B1-3 reverse, 5′-ACGACTTGGGCTTAATGGTC-3′; NEO-1, 5′-TGAATGAACTGCAGGACGAG-3′; and NEO-2, 5′-CCACAGTCGATGAATCCAGA-3′, as described.18 Their genotypes were confirmed by PCR analysis. Male Apoe−/−/Cyp1b1+/+ and Apoe−/−/Cyp1b1−/− mice, approximately 16 weeks of age and weighing 28 to 32 g, were used in all experiments.
Ang II–Induced Aneurysm in Apoe−/−/Cyp1b1+/+ and Apoe−/−/Cyp1b1−/− Mice
Mice were anesthetized with 1.5% isoflurane, and Alzet miniosmotic pumps (model 2004; Durect Corporation, Cupertino, CA) were implanted s.c. to infuse 700 ng/kg per minute of Ang II or saline for 7 or 28 days. This dose of Ang II was based on those from pilot studies conducted in our laboratory to determine the dose of Ang II optimum for inducing AAA. This dose was chosen as it resulted in optimum lesion formation, coupled with a reduced prevalence of rupture (≤10%). A separate group of Apoe−/−/Cyp1b1+/+ mice receiving Ang II infusion were administered 300 μg/kg of selective inhibitor of CYP1B1, TMS or its vehicle (dimethyl sulfoxide), i.p. every third day for the duration of the experimental period. In another series of experiments, Apoe−/−/Cyp1b1+/+ mice infused with Ang II or its vehicle were administered 2 mmol/L 4-hydroxyl-2,2,6,6-tetramethylpiperidine-1-oxyl (tempol) (Sigma-Aldrich) in drinking water for 28 days. At the end of the experiment, mice were euthanized with a cocktail of 87 mg/kg of ketamine and 13 mg/kg of xylazine i.p., and their tissues were harvested.
Ultrasound Imaging of the Abdominal Aorta and ex Vivo Measurement of AAA with Vernier Calipers
Core body temperature was maintained at 37°C, and heart and respiration rates were monitored continuously with a pulse oximeter. Images of the aorta were taken using Vevo 2100 ultrasound equipment (VisualSonics Inc., Toronto, ON, Canada). Briefly, the mice were placed on a platform in the supine position. B-mode images were taken in short and parasternal long axis views and analyzed using Vevo 2100 software version 3.4 (VisualSonics Inc.). The mean of measurements from each animal was used for calculating the aortic diameter (in millimeters). Aortic measurements by ultrasound analysis to determine aneurysm lesion were further corroborated using ex vivo analysis by measurement of the diameter of the aorta with a Vernier caliper on sacrifice of the animal. A lesion was classified as AAA if the diameter of the aorta in a mouse from any treatment group (measured ex vivo using a Vernier caliper) was 50% greater than the mean aorta diameter of the animals from the vehicle control for the respective genotypes. Ultrasound analysis was performed on five animals per group. Ex vivo analysis was performed on all animals, namely, 20 per group.
IHC Analysis
Aortae, along with the perivascular tissue, were dissected and placed in optimal cutting temperature compound (Sakura Finetek USA Inc., Torrance, CA). Serial transverse cryosections (10 μm thick) were cut at −20°C with a Leica CM1850 cryostat (Leica Microsystems, Inc., Buffalo Grove, IL). Histopathologic and immunohistochemistry analyses of collagen, elastin, and α smooth muscle actin were performed to assess the integrity of extracellular matrix, as described.19 Tissue sections were also processed to determine the infiltration of CD3+ T cells and F4/80+ monocytes/macrophages, as described.19 Immunohistochemistry analysis was also performed to determine the infiltration of CD62p+ platelets, expression of PDGF-A to -D, PDGFR-β, Itg-α2, and COX-2 (Table 1).
Sections were fixed in formalin for 10 minutes. After three washes in phosphate-buffered saline (PBS), slides were incubated in 0.3% hydrogen peroxide for 10 minutes to block endogenous peroxidase activity. The sections were then rinsed once in PBS and blocked in 5% bovine serum albumin for 45 minutes. Primary antibodies were diluted in background-reducing diluent (catalog number S3022; Dako Cytomation, Carpintaria, CA), and sections were incubated with the respective primary antibodies overnight at 4°C in a humid chamber. Sections were then washed three times in PBS the following day and incubated with the secondary antibody for 1 hour, washed three times in PBS, and incubated in diaminobenzidine (catalog number D4293; Sigma-Aldrich), which was prepared according to the manufacturer's instructions, followed by washing with deionized distilled water. All sections were counterstained with hematoxylin (catalog number 245-651; Thermo Fisher Scientific), dehydrated, mounted, and viewed with an inverted system microscope (model BX41; Olympus America, Inc., Center Valley, PA) and imaged with a SPOT Insight 2 Mp Firewire digital camera (Diagnostic Instruments Inc., Sterling Heights, MI). Images were quantified for the intensity of the diaminobenzidine-stained area by drawing regions of interest to include the adventitial area to determine the number of positive pixels per area in two locations in each section (three sections per slide) of each slide representative of each animal in a treatment group (n = 5) by ImageJ software version 1.42 (NIH, Bethesda, MD). CD3+ T cells, F4/80+ macrophages, and CD62p+ platelets were counted manually from a total of six images in high-power fields (original magnification, ×40) for each replicate (three sections per slide; two images per section) from the different treatment groups of n = 5 each. Appropriate controls for the secondary antibody staining, diaminobenzidine staining, and isotype control antibody staining in Ang II–induced aortic sections from Apoe−/−/Cyp1b1+/+ mice were included in the experiments to validate the specificity of the immunostaining (Supplemental Figure S1).
Detection of Immunofluorescence by Confocal Microscopy
Slides with the aorta sections were air-dried and fixed in cold acetone for 10 minutes. Sections were rehydrated in 1X PBS for 15 minutes, followed by blocking (1X PBS/2% bovine serum albumin/0.1% Tween 20) for 30 minutes in a humid chamber. Slides were briefly washed using wash buffer (1X PBS/0.5% bovine serum albumin/0.1% Tween 20). The respective primary antibodies were diluted in background-reducing diluent (Dako Cytomation), applied to the sections, and allowed to incubate at room temperature for 1 hour, washed three times for 5 minutes each, and incubated with the diluted secondary antibody for 45 minutes at room temperature in a light-protected humid chamber. Further washing was performed in the dark, followed by incubation with the nuclear stain (DAPI) for 10 minutes, washed once more, and allowed to air dry, and then the coverslip was placed on sections using Vectashield HardSet Mounting Medium (catalog number H-1400; Vector Laboratories, Burlingame, CA). Images were taken using FV10-ASW viewer software version 4.1 (Olympus America Inc.), and fluorescence staining was quantified as region of interest pixel intensity in the adventitia (two images per section; three sections per slide) of the treatment groups (n = 5) relative to the control groups (n = 5) using ImageJ software version 1.42. Sections from Apoe−/−/Cyp1b1+/+ mice incubated with the appropriate rabbit IgG isotype control antibody and then labeled with the secondary antibody were included in the experiments to validate the specificity of the immunofluorescence staining.
Histologic Analysis of Vascular Remodeling
Histologic evaluation of aortae was performed by staining with hematoxylin and eosin (Sigma-Aldrich). Masson's trichrome staining (catalog number HT-15; Sigma-Aldrich) for collagen was performed on the aorta sections as per the manufacturer's instructions, with some modifications as previously described.19 Additionally, elastin staining for the aorta sections was performed by the modified Verhoeff–van Gieson staining method, according to the manufacturer's instructions. Briefly, slides with the aorta sections were air-dried and fixed in 10% formalin for 10 minutes, followed by rinsing using deionized distilled water. The slides were immersed in the working elastin solution for 10 minutes and then rinsed thoroughly using deionized distilled water. Slides were then differentiated in the working ferric chloride solution for 4.5 minutes, followed by rinsing in deionized distilled water to remove excess stain. The slides were dipped in 95% alcohol three times; rinsed with deionized distilled water; and then incubated in van Gieson's solution for 2 minutes, dehydrated, cleared, and coverslipped using Permount (CAS 68240-09-5; Thermo Fisher Scientific) before the images were obtained. Since no antibodies were used in histologic staining, the tissues from the controls and the treatment groups were compared to determine differences, which were then quantitated. Positive staining of collagen was quantified by the number of positive pixels per area from a total of two images in high-power field (×40) per section (three sections per slide) of each treatment group (n = 5) by using ImageJ software version 1.42. Quantification of elastin fiber breaks per high-power field (×40) per replicate (three sections per slide) of each treatment group of (n = 5) in control and treatment groups of mice (n = 5) was performed manually.20
Measurement of Oxidative Stress
ROS Production
To measure vascular ROS production, sections of aorta were exposed to DHE (catalog number D11347; Life Technologies, Carlsbad, NY), according to a previously published validated method.21 Briefly, aorta sections were incubated in PBS for 30 minutes at 37°C and then encircled with the hydrophobic pen. DHE (5 μmol/L) was topically applied, followed by incubation at 37°C in the dark for 30 minutes. Sections were rinsed in PBS, and oxidized DHE fluorescence was detected using a 585-nm filter with an inverted system microscope (model DP71; Olympus America Inc.). Images were obtained with an Olympus digital camera, and the fluorescence staining was quantified by a drawing of a region of interest to include adventitia and aorta wall. The region of interest pixel intensity (two images per section; three sections per slide) of the treatment groups (n = 5) relative to the control groups (n = 5) was determined using ImageJ software version 1.42.
Measurement of NADPH Oxidase Activity
NADPH oxidase activity was determined in cardiac homogenates by assessing lucigenin (N,N′-dimethyl-9,9′-biacridinium dinitrate) enhanced chemiluminescence, as described previously.22 Briefly, hearts were harvested and snap-frozen in liquid N2 and stored at −80°C until use. Tissue was ground to a fine powder in liquid N2, homogenized, and sonicated in lysis buffer containing protease inhibitors (20 mmol/L phosphate buffer, 1 mmol/L EGTA, 10 μg/mL of aprotinin, 0.5 μg/mL leupeptin, 0.7 μg/mL of pepstatin, 0.5 mmol/L phenylmethylsulfonyl fluoride, and 150 mmol/L sucrose). Centrifugation of the samples was performed at 3000 × g for 10 minutes at 4°C, and the protein contents of the supernatants were determined by the Bradford method. Equal amounts of protein were combined with a reaction mixture containing 5 μmol/L lucigenin (final concentration) and 100 μmol/L NADPH (final concentration) (catalog number N1630; Sigma-Aldrich), and luminescence was measured every minute for 10 minutes by using a luminometer (model TD-20/20; Turner BioSystems, Sunnyvale, CA). Lysis buffer was used as a blank and subtracted from each reading; activity was expressed as arbitrary units.
Measurement of CYP1B1 Activity
CYP1B1 activity was determined in cardiac homogenates using the P450-Glo assay kit (catalog number V8762; Promega Corporation, Madison, WI), as described previously.12 Briefly, tissue samples were homogenized (2 × 3 minutes) in ice-cold 100 mmol/L potassium phosphate buffer (pH 7.4) using the TissueLyser II (Qiagen, Valencia, CA). Centrifugation was performed at 10,000 × g for 20 minutes at 4°C. Protein contents in the samples were determined by the Bradford method, 500 μg of protein was added to a reaction mixture containing 20 μmol/L L-CEE substrate and 100 mmol/L potassium phosphate buffer (pH 7.4) and incubated at 37°C for 10 minutes, 100 μmol/L of NADPH (final concentration) was added, and the solution was further incubated at 37°C for 45 minutes. An equal volume of luciferin detection reagent was added to the samples, mixed for 10 seconds, and allowed to incubate at room temperature for 20 minutes. Luminescence was measured using a luminometer (model TD-20/20; Turner BioSystems). Potassium phosphate buffer was used as a blank and subtracted from each reading; CYP1B1 activity was expressed as relative luminescence units. For measuring CYP1B1 protein levels in the heart, Western blot analysis was performed by homogenizing the heart tissue in radioimmunoprecipitation assay lysis buffer. Protein content was determined by the Bradford method, and 50 μg of protein was loaded and resolved on 8% SDS-polyacrylamide gels, after which Western blot analysis was performed as described.7 Blots were probed with anti-CYP1B1 antibody (Table 1). Band intensity was measured with ImageJ software version 1.42 (NIH).
Measurement of Plasma 8-Isoprostane Levels
Total 8-isoprostane levels in the plasma were measured using the 8-Isoprostane Express EIA Assay Kit (catalog number 516351; Cayman Chemical), as per the manufacturer's instructions. Plasma samples were first purified using the 8-Isoprostane Purification Kit (catalog number 501110; Cayman Chemical) before the assay was performed. Data were expressed as the plasma levels of total 8-isoprostane, in picograms per milliliter.
Flow Cytometric Analysis of CD192 Expression
Blood was collected in lithium-heparin Microtainer tubes (Becton, Dickinson and Company, Franklin Lakes, NJ) by cardiac puncture before the mice were sacrificed. A 100-μL aliquot of whole blood was transferred to a 5-mL polystyrene tube (Becton, Dickinson and Company). The CD3-PE, CD11b-AF488 (BD Biosciences, San Jose, CA), and CD192-AF700 (R&D Systems, Minneapolis, MN) antibodies were diluted in fluorescence-activated cell-sorting buffer (PBS with 1% bovine serum albumin and 0.05% sodium azide) and stained for surface markers CD3, CD11b, and CD192 for 45 minutes, followed by a further 10-minute incubation after the addition of 1X RBC Lysis Buffer (eBioscience, San Diego, CA). Cells were washed twice using fluorescence-activated cell-sorting buffer by centrifugation at 300 × g for 10 minutes, and the supernatant was discarded; cells were resuspended in 300 μL of fluorescence-activated cell-sorting buffer, and data were collected on a BD LSR II flow cytometer (Becton, Dickinson, and Company). The levels of expression of CD192 on the CD3 and CD11b positively stained populations of cells (staining for the respective antibodies) were analyzed using FlowJo software version 10 (Tree Star, Ashland, OR). Data are presented as the total number of CD11b+ or CD3+ cells expressing CD192+. All data were normalized against the values obtained for the respective fluorescence (of all of the antibodies used in the cocktail), minus 1 controls (fluorescence of the particular antibody).
Real-Time PCR Analysis
Aortae were harvested on sacrifice of the animals and placed in RNAlater solution (Life Technologies), and total RNA was extracted with the TRIzol method.23 Two micrograms of purified RNA was reverse-transcribed using AffinityScript III qPCR cDNA Synthesis Kit (catalog number 600559; Agilent Technologies, Santa Clara, CA). Primers were designed using the Universal Probe Library Assay Design Center (Roche Life Science, Indianapolis, IN) (Table 2). mRNA expression levels of Pdgfd, Pdgfrb, Itga2, Mmp2, and Mmp9 were analyzed by real-time RT-PCR. Real-time quantitative PCR in 96-well plates with a LightCycler 480 System (F. Hoffmann-La Roche AG, Basel, Switzerland) using an LC480 or SYBR Green Master Mix and probe from Universal Probe Library (Roche Life Science), at a concentration of 10 μmol/L, a final reaction volume of 10 μL, and the following conditions: 95°C for 5 minutes for activation, 45 cycles at 95°C for 10 seconds, 60°C for 60 seconds, and 72°C for 10 seconds for amplification. The transcription level was normalized to ribosomal protein S19 gene. Cyp1b1 mRNA primers specific for Cyp1b1−/− mice were designed as described.24 All samples were analyzed in triplicate. The relative mRNA levels were normalized to the reference gene mRNA in the same sample of cDNA. The 2−ΔΔCt comparative quantitative method was used for calculating fold-changes in gene expression for the normalized data.25 The ΔΔCt values and error differences are provided along with the fold-change values to enable accurate interpretation of the data.
Table 2.
List of Primer Sequences
| Gene | Primer sequence |
|
|---|---|---|
| Left | Right | |
| Mmp2 | 5′-TAACCTGGATGCCGTCGT-3′ | 5′-TTCAGGTAATAAGCACCCTTGAA-3′ |
| Mmp9 | 5′-ATCCAGTATCTGTATGGTCGTG-3′ | 5′-GCTGTGGTTCAGTTGTGGTG-3′ |
| Pdgfd | 5′-TGTATCTGGACACCCCTCATT-3′ | 5′-GCCTGTCCAGGTCCACTTT-3′ |
| Pdgfrb | 5′-CACTTCCTCCACCATCTCCT-3′ | 5′-CTGCTTGCTGTGGCTCTTCT-3′ |
| Itga2 | 5′-ACTTCCGGCATACGAAAGAA-3′ | 5′-TCAGCCAGCAGGTGATGTTA-3′ |
| Cyp1b124 | 5′-ACATCCCCAAGAATACGGTC-3′ | 5′-TAGACAGTTCCTCACCGATG-3′ |
Statistical Analysis
For calculating the significance of the difference between the incidences of aneurysm formation (n = 20, ex vivo), the Fisher exact test was used. For all other studies, experimental data obtained for n = 5 were checked for assumptions of two-way analysis of variance using the Kolmogorov-Smirnov test, which is a stringent measure of determining normality and variance. When our data passed the test, we used two-way analysis of variance followed by the Bonferroni multiple-comparisons test to determine significant differences in the analysis of our data. Where our data did not satisfy the assumptions needed for two-way analysis of variance, we analyzed our data using the nonparametric Kruskal-Wallis test, followed by the Dunn multiple-comparisons test to determine significant differences. All statistical analyses were performed using Prism software version 5.0 (GraphPad Software, Inc., San Diego, CA). Results are expressed as means ± SEM. P < 0.05 was considered statistically significant. All comparisons were made between Apoe−/−/Cyp1b1+/+ mice treated with Ang II alone or Ang II and concurrent treatment with TMS, or Apoe−/−/Cyp1b1−/− mice treated with Ang II.
Results
Treatment with TMS or Cyp1b1 Gene Disruption Minimizes Ang II–Induced AAA in Apoe−/−/Cyp1b1+/+ Mice
Ang II infusion for 28 days in Apoe−/−/Cyp1b1+/+ mice increased aortic diameter, as determined from images obtained by ultrasound analysis conducted 1 day before sacrifice of the animals. Images and ex vivo measurements of the aortae were also obtained on the day after sacrifice of the mice. Ang II infusion increased aortic diameter and induced the formation of AAAs (70% prevalence) in Apoe−/−/Cyp1b1+/+ mice, with a rupture rate of 10% to 15%; both were minimized by concurrent treatment with TMS or by Cyp1b1 gene disruption (Figure 1 and Supplemental Figure S2).
Figure 1.

Angiotensin (Ang) II–induced formation of abdominal aortic aneurysm in Apoe−/−/Cyp1b1+/+ mice is alleviated by treatment with 2,3′,4,5′-tetramethoxystilbene [TMS; but not by its vehicle dimethyl sulfoxide (DMSO)] or by Cyp1b1 gene disruption. A: Representative ultrasound B-mode image of aorta in the short axis (blue dotted lines denote circumference of the aorta; lines indicate diameter), and respective aortae isolated with heart from euthanized mice after 28 days of Ang II infusion. B: Prevalences of aneurysms formed, as defined by a 50% increase in the aortic diameter in relation to the mean aortic diameter in the vehicle control group (Veh) for the respective genotype. Data are expressed as means. n = 20 (B). ∗∗P < 0.01, ∗∗∗P < 0.001 (Fisher exact test).
TMS Prevents Ang II–Induced Increase in Cardiac CYP1B1 Activity but Not Its Protein Levels or Aortic mRNA Expression
Ang II infusion for 28 days increased cardiac CYP1B1 activity in Apoe−/−/Cyp1b1+/+ mice (activity was measured in the heart because of the limited amount of aorta available for CYP1B1 assay). Treatment of these mice with TMS prevented Ang II–induced increase in CYP1B1 activity (Supplemental Figure S3A); however, CYP1B1 protein expression was not altered by treatment with TMS (Supplemental Figure S3B). Cyp1b1 mRNA expression measured in the aortae of Apoe−/−/Cyp1b1+/+ mice was not altered by Ang II and was absent in Apoe−/−/Cyp1b1−/− mice (Supplemental Figure S3C).
TMS or Cyp1b1 Gene Disruption Prevents Aortic Inflammatory Cell Infiltration in Ang II–Induced AAA
To determine the contribution of CYP1B1 to inflammatory cell infiltration associated with Ang II–induced AAA, we examined the localization of F4/80+ macrophages and CD3+ T lymphocytes. Since platelets also contribute to inflammation in AAA,26 we determined the localization of CD62p+-activated platelets in aortic sections from Apoe−/−/Cyp1b1+/+ mice treated with TMS and from Apoe−/−/Cyp1b1−/− mice. Ang II infusion for 28 days increased the accumulation of F4/80+ macrophages, CD3+ T cells, and CD62p+-activated platelets at the lesion site in the aortae of Apoe−/−/Cyp1b1+/+ mice but not in mice concurrently receiving TMS or in Apoe−/−/Cyp1b1−/− mice (Figure 2). Flow cytometry analysis of circulating CD11b+ monocytes and CD3+ T cells in blood revealed that Ang II–induced expression of the proinflammatory marker C-C chemokine receptor (CCR)-2 (CD192) in CD11b+ monocytes and CD3+ T cells in Apoe−/−/Cyp1b1+/+ mice but not in those concurrently treated with TMS or in Apoe−/−/Cyp1b1−/− mice (Supplemental Figures S4 and S5).
Figure 2.

2,3′,4,5′-Tetramethoxystilbene (TMS) or Cyp1b1 gene disruption reduces inflammatory cell infiltration and platelets in the aorta of Apoe−/−/Cyp1b1+/+ mice infused with angiotensin (Ang) II for 28 days. A–D: F4/80+ macrophages, CD3+ T cells, and CD62p+ platelets staining in the aorta (A). The respective quantified data is shown in B–D. Boxed areas in A are shown at higher magnification below. Arrows indicate F4/80+, CD3+, and CD62p+ cells. Data are expressed as means ± SEM. n = 5 per group. ∗∗P < 0.01, ∗∗∗P < 0.001 (two-way analysis of variance followed by the Bonferroni multiple-comparisons test). Scale bars: 50 μm (boxed areas); 200 μm (main images, all other groups); 400 μm (main image, Apoe−/−/Cyp1b1+/+ Ang II group). Original magnification: ×4 (main image, Apoe−/−/Cyp1b1+/+ Ang II group); ×10 (main images, all other groups); ×40 (boxed areas). Veh, vehicle.
TMS or Cyp1b1 Gene Disruption Prevents Aortic Protein Expression of PDGF-D and Pdgfrb and Itga2 mRNA Expression in Ang II–Induced AAA
PDGF is produced by various cell types, and its expression is altered in AAA.27, 28 It is also associated with enhanced leukocyte recruitment and MMP expression.29, 30, 31, 32 Therefore we assessed the role of CYP1B1 in the expression of different isoforms of PDGF. Ang II for 28 days selectively increased the expression of PDGF-D isoforms at the lesion site in the aortae of Apoe−/−/Cyp1b1+/+ mice, but not in mice receiving TMS or in Apoe−/−/Cyp1b1−/− mice (Figure 3, A and B). Moreover, a decreased mRNA expression of Pdgfd was observed in the aortae of Apoe−/−/Cyp1b1+/+ mice receiving Ang II infusion but not in Apoe−/−/Cyp1b1−/− mice (Figure 3C). PDGF-D is known to signal via Pdgfrb.30 Itga2 is a component of the Itg-α2 β1 subunit, a major collagen receptor found in VSMCs33 and in activated platelets.34 Hence, we determined the expression of Pdgfrb and Itga2 mRNA in aortae from Apoe−/−/Cyp1b1+/+ and Apoe−/−/Cyp1b1−/− mice receiving Ang II. We also assessed protein expression of PDGFR-β and Itg-α2 in the aorta tissue. Pdgfrb and Itga2 mRNA expression was down-regulated in the aortae of Apoe−/−/Cyp1b1−/− mice infused with Ang II (Figure 3, D and E). These findings were also reflected in the protein expression of these markers (Supplemental Figure S6).
Figure 3.

Expression of platelet-derived growth factor (PDGF)-D protein is inhibited in Apoe−/−/Cyp1b1+/+ mice treated with 2,3′,4,5′-tetramethoxystilbene (TMS) or by Cyp1b1 gene disruption after 28 days of angiotensin (Ang) II infusion, and expression of Pdgfd, Pdgfrb, and Itga2 in aortae of Apoe−/−/Cyp1b1+/+ mice and their down-regulation in aortae from Apoe−/−/Cyp1b1−/− mice infused with Ang II. A and B: PDGF-A to -D staining in the aorta (A), and the quantified data (B). Boxed areas in A are shown at higher magnification below. C–E: Fold-changes in Pdgfd, Pdgfrb, and Itga2 mRNA expression in aorta of Ang II–infused Apoe−/−/Cyp1b1+/+ and Apoe−/−/Cyp1b1−/− mice. mRNA expression was normalized against the reference S19 mRNA. Corresponding means ± SD ΔCT values for Apoe−/−/Cyp1b1+/+ Ang II versus Apoe−/−/Cyp1b1−/−, for reference: Pdgfd, 13 ± 0.7 versus 15.6 ± 0.4; Pdgfrb, 2.9 ± 0.05 versus 6.9 ± 0.03; and Itga2, 11.7 ± 0.1 versus 19.4 ± 0.2. Data are expressed as means ± SEM (B) or as means (C–E). n = 5 per group. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 (Kruskal-Wallis test followed by the Dunn multiple-comparisons test). Scale bars: 50 μm (boxed areas); 200 μm (main images, all other groups); 400 μm (main image, Apoe−/−/Cyp1b1+/+ Ang II group). Original magnification: ×4 (main image, Apoe−/−/Cyp1b1+/+ Ang II group); ×10 (main images, all other groups); ×40 (boxed areas). CT, cycle threshold value; Veh, vehicle.
Treatment with TMS or Cyp1b1 Gene Disruption Minimizes MMP Expression and ECM Degradation in Ang II–Induced AAA in Apoe−/−/Cyp1b1+/+ Mice
Aneurysm lesions are characterized by degradation of the ECM components by proteolytic enzymes like MMP-2 and -935; therefore, we assessed the contribution of CYP1B1 in the expression of MMP and ECM components (collagen, elastin, and α smooth muscle actin) in AAA due to Ang II infusion for 28 days. Increased fluorescence intensity of MMP-2 and -9 staining was observed in the aorta sections from Apoe−/−/Cyp1b1+/+ mice infused with Ang II but not in mice concurrently treated with TMS or in Apoe−/−/Cyp1b1−/− mice (Figure 4, A and B). Corresponding mRNA levels of these MMPs were also increased in the aortae of Apoe−/−/Cyp1b1+/+ mice but not in Apoe−/−/Cyp1b1−/− mice treated with Ang II (Supplemental Figure S7). Also, decreased staining of collagen, elastin, and α smooth muscle actin (indices of ECM integrity) was observed in aortic sections from Apoe−/−/Cyp1b1+/+ mice but not in mice receiving TMS or in Apoe−/−/Cyp1b1−/− mice infused with Ang II (Figure 4, C–F).
Figure 4.

2,3′,4,5′-Tetramethoxystilbene (TMS) treatment or Cyp1b1 gene disruption reduces matrix metalloproteinase (MMP) expression and extracellular matrix (ECM) degradation in the aorta of Apoe−/−/Cyp1b1+/+ mice infused with angiotensin (Ang) II for 28 days. A and B: MMP-2 and -9 staining in the aorta (A), and the quantified data (B). C and D: Staining for ECM components collagen, elastin, and actin (C), and the quantified data (D–F). Boxed areas in C are shown at higher magnification below. Arrows indicate areas of break in the elastin fiber. Data are expressed as means ± SEM. n = 5 per group. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 (two-way analysis of variance followed by the Bonferroni multiple-comparisons test). Scale bars: 50 μm (boxed areas in C); 200 μm (main images of all other groups in A and C); 400 μm (main image of Apoe−/−/Cyp1b1+/+ Ang II group in A and C). Original magnification: ×4 (main image of Apoe−/−/Cyp1b1+/+ Ang II group in A and C); ×10 (main images of all other groups in A and C); ×40 (boxed areas in C). H&E, hematoxylin and eosin; R.U., relative units; SMA, smooth muscle actin; Veh, vehicle.
TMS or Cyp1b1 Gene Disruption Minimizes Increases in Aortic Oxidative Stress and COX-2 Expression in Ang II–Induced AAA
Oxidative stress has been implicated in the murine model of AAA.35 The infusion of Ang II for 28 days increased aortic ROSs, as indicated by enhanced oxidized DHE fluorescence intensity in AAA (Figure 5, A and B), and cardiac NADPH oxidase activity and increased plasma levels of 8-isoprostane, another index of oxidative stress,36 in Apoe−/−/Cyp1b1+/+ mice, which was inhibited by treatment with TMS or by Cyp1b1 gene disruption (Figure 5, C and D).
Figure 5.

2,3′,4,5′-Tetramethoxystilbene (TMS) treatment or Cyp1b1 gene disruption reduces oxidative stress in the aorta of Apoe−/−/Cyp1b1+/+ mice infused with angiotensin (Ang) II for 28 days. A and B: Photomicrographs of reactive oxygen species (ROS) production determined by 2-hydroxyethidium staining (A) and quantified as fluorescence of 2-hydroxyethidium (OHE) (R.U.) (B). C–F: Nicotinamide adenine dinucleotide phosphate oxidase activity measured in heart homogenate (C); total 8-isoprostane level in the plasma (D); and cyclooxygenase (COX)-2 staining (E), quantified as intensity of diaminobenzidine (F). Data are expressed as means ± SEM. n = 5 per group. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 (two-way analysis of variance followed by the Bonferroni multiple-comparisons test). Scale bars: 50 μm (E, all groups); 200 μm (A, all other groups); 400 μm (A, Apoe−/−/Cyp1b1+/+ Ang II group). Original magnification: ×4 (A, Apoe−/−/Cyp1b1+/+ Ang II group); ×10 (A, all other groups); ×40 (E, all groups). A.U., arbitrary units; NADPH, nicotinamide adenine dinucleotide phosphate; R.U., relative units; Veh, vehicle.
Ang II increases COX-2 expression in VSMCs via p38 mitogen-activated protein kinase,37 and this pathway been implicated in the pathogenesis of AAA.38 Therefore, we determined the contribution of CYP1B1 to the expression of COX-2 in Ang II–induced AAA. Ang II infusion increased COX-2 expression in aortic sections of Apoe−/−/Cyp1b1+/+ mice, which was attenuated by TMS treatment, and in Apoe−/−/Cyp1b1−/− mice (Figure 5, E and F).
Tempol Prevents Ang II–Induced AAA, Oxidative Stress, and COX-2 Expression in Apoe−/−/Cyp1b1+/+ Mice
To further determine whether oxidative stress contributes to Ang II–induced CYP1B1-mediated AAA and associated pathogenic events, we examined the effect of the superoxide dismutase mimetic tempol.39 Administering tempol in the drinking water to Apoe−/−/Cyp1b1+/+ mice infused with Ang II for 28 days prevented AAAs, with a corresponding decrease in oxidative stress, as seen by oxidized DHE fluorescence and a reduction in 8-isoprostane plasma levels in these animals. Tempol also reduced the expression of COX-2 in the aortic sections of Apoe−/−/Cyp1b1+/+ mice infused with Ang II (Figure 6).
Figure 6.

Tempol inhibits angiotensin (Ang) II–induced abdominal aortic aneurysm, reactive oxygen species (ROS) production in the aorta, plasma levels of 8-isoprostane, and cyclooxygenase (COX)-2 expression in Apoe−/−/Cyp1b1+/+ mice. A: Representative photomicrographs of aorta of Apoe−/−/Cyp1b1+/+ mice infused with Ang II for 28 days, receiving drinking water alone or with tempol. B–F: Corresponding photomicrographs/quantified data for ROS production and COX-2 expression (B–E), and total 8-isoprostane levels in the plasma (F). Data are expressed as means ± SEM. n = 5 per group. ∗P < 0.05, ∗∗∗P < 0.001 (Kruskal-Wallis test followed by the Dunn multiple-comparisons test). Scale bars = 50 μm. Original magnification, ×40. OHE, 2-hydroxyethidium; R.U., relative units.
Cyp1b1 Gene Disruption Minimizes an Increase in Aortic Diameter, Inflammation, and Oxidative Stress in Aorta of Apoe−/−/Cyp1b1+/+ Mice after 7 Days of Ang II Infusion
To further validate that the proinflammatory changes and oxidative stress observed in AAA are mediated via CYP1B1 and are not merely the consequences of aortic lesions, we performed short-term infusion of Ang II for 7 days in Apoe−/−/Cyp1b1+/+ and Apoe−/−/Cyp1b−/− mice and then assessed the aortic tissue for the development of pathologic changes. Ang II infusion for 7 days resulted in a 50% prevalence of AAA formation (defined as a 50% increase in aortic diameter compared to the mean diameter in the vehicle control group for the respective genotype) only in Apoe−/−/Cyp1b1+/+ mice. No incident of rupture was observed; however, Ang II infusion for 7 days caused a significant increase in the aortic diameter only in Apoe−/−/Cyp1b1+/+ mice (Figure 7A and Supplemental Figure S8A). This increase was, however, less in comparison to the increase in aortic diameter observed in Apoe−/−/Cyp1b1+/+ mice after 28 days of Ang II infusion. Moreover, there was increased F4/80+ macrophage infiltration, enhanced degradation of elastin fibers, and increased oxidative stress and MMP-2 expression in the aortas of Ang II–infused Apoe−/−/Cyp1b1+/+ mice; these changes were not observed in the aortas from Ang II–infused Apoe−/−/Cyp1b1−/− mice (Figure 7B and Supplemental Figure S8, B–D). No significant differences in the expression of collagen, CD3+, and CD62p+ cell infiltration and MMP-9 expression in the aorta were observed after Ang II infusion for 7 days in Apoe−/−/Cyp1b1+/+ and Apoe−/−/Cyp1b1−/− mice (Supplemental Figure S9).
Figure 7.

Cyp1b1 gene disruption minimizes an increase in aortic diameter, inflammation, and oxidative stress in aorta of Apoe−/−/Cyp1b1+/+ mice after 7 days of angiotensin (Ang) II infusion. Apoe−/−/Cyp1b1+/+ and Apoe−/−/Cyp1b1−/− mice were infused with Ang II for 7 days. A: Photomicrographs of ex vivo intact and hematoxylin and eosin–stained aorta. B: Representative photomicrographs of F4/80+, elastin fiber staining, 2-hydroxyethidium (OHE), and matrix metalloproteinase (MMP)-2 immunofluorescence staining. Arrows indicate F4/80+ cell staining. n = 4 per group. Scale bars: 200 μm (A); 50 μm (B). Original magnification, ×10 (A); ×40 (B).
Discussion
This study demonstrates for the first time that CYP1B1-dependent Ang II–induced AAA and associated pathogenic events, including infiltration of inflammatory cells, activated platelets, expression of COX-2 and PDGF-D and its associated signaling molecules Pdgfrb and Itga2, and increased MMP-2 and -9 expression are associated with the degradation of matrix components of vessel wall in male Apoe−/−/Cyp1b1+/+ mice These events are most likely mediated via CYP1B1-dependent oxidative stress.
The infusion of Ang II for 28 days in Apoe−/−/Cyp1b1+/+ mice produced predominantly AAAs at a prevalence of approximately 70%, similar to observations made by other investigators.40 Thoracic aortic aneurysms were observed in only approximately 20% of animals. The administration of the selective CYP1B1 inhibitor TMS14 or Cyp1b1 gene disruption (Apoe−/−/Cyp1b1−/−) minimized Ang II–induced aortic aneurysms in these mice, suggesting that CYP1B1 is crucial for the development of AAA. Ang II did not alter Cyp1b1 mRNA expression in the aortae of Apoe−/−/CYP1b1+/+ mice. Ang II increased cardiac CYP1B1 activity that was inhibited by TMS, without alteration in CYP1B1 protein expression in Apoe−/−/Cyp1b1+/+ mice. This increase in cardiac CYP1B1 activity in Ang II–infused Apoe−/−/Cyp1b1+/+ mice could have been due to biochemical modification of this enzyme, but this hypothesis remains to be tested. Although we have previously shown that CYP1B1 mediates Ang II–induced hypertension in male mice,12 it has already been established that in Apoe−/− male mice the development of Ang II–induced aneurysm is independent of increased blood pressure.40 Therefore, we believe that protection against Ang II–induced AAA mediated by CYP1B1 is independent of hypertension.
Ang II–induced AAA is associated with transmural migration of macrophages and T cells.5 In the present study, infiltration of CD3+ T cells and F4/80+ macrophages in lesion sites was reduced by TMS treatment or by CYP1B1 gene disruption, suggesting that CYP1B1 contributes to these pathogenic events in AAA. CYP1B1 is expressed in nonhepatic tissues, including cardiovascular tissues and blood cells.41, 42 We observed that circulating monocytes (CD11b+) and T cells (CD3+) from Ang II–infused Apoe−/−Cyp1b1+/+ mice had enhanced expression of CCR2 (CD192+), which was diminished in mice treated with TMS or in Apoe−/−/Cyp1b1−/− mice. CCR2 expression is associated with recruitment of monocytes and T cells to the site of inflammation.43 Although CCR2 expression in the monocytes is an indicator of a proinflammatory M1 phenotype, in T cells CCR2 expression is associated with a stable effector memory T-cell subtype44 and, in combination with CCR5, has an increased propensity to produce MMP-9.45 This enhanced expression of CCR2 in the circulating monocytes and T cells suggests a proinflammatory/memory phenotype, and CYP1B1 in these cells could contribute to the expression of CCR2.
An important finding in our study was that increase in platelet infiltration at the site of the lesion in Ang II–induced AAA in Apoe−/−Cyp1b1+/+ mice was also decreased by TMS or by Cyp1b1 gene disruption. The accelerated accumulation of platelets at the lesion site provides hemostatic protection initially, but, as the lesion progresses, platelets via production of proinflammatory mediators like PDGF could contribute to aneurysm formation.26, 27 Although platelets are the main source of PDGF, other cell types, including VSMCs, endothelial cells, and inflammatory cells, also produce this agent.28, 32, 46 Therefore, the contribution of CYP1B1 to the expression of the four different isoforms of PDGF was assessed at the site of the lesion in Ang II–induced AAA. We observed increased expression of only PDGF-D in the aorta of Ang II-infused Apoe−/−Cyp1b1+/+ mice, especially in the area of platelet infiltration, namely the thrombus, that was prevented by treatment with TMS or by Cyp1b1 gene disruption. The assessment of PDGF expression at earlier time points after Ang II administration before a fully developed thrombus is formed could allow for the delineation of the kinetics of platelet infiltration and associated signaling events. PDGF-D is a relatively novel member of PDGFs, which signal via Pdgfrb.29, 30 It stimulates VSMC migration and proliferation, monocytes/macrophage recruitment, and increased secretion of MMPs in various cell types.46, 47 Although we did not assess the involvement of VSMCs specifically in this study, we did observe enhanced macrophage and T-cell infiltration and MMP expression at the site of aortic lesion. PDGF also activates Itga2,48 which is crucial for platelet aggregation/adhesion.49 In the present study, PDGF-D expression was associated with increased Pdgfrb and Itga2 mRNA expression in aorta of Apoe−/−Cyp1b1+/+ mice, which was markedly reduced in Apoe−/−/Cyp1b1−/− mice infused with Ang II. Protein expression of PDGFR-β and Itg-α2 was also decreased in Ang II–infused Apoe−/−/Cyp1b1+/+ mice treated with TMS. Therefore, PDGF-D, via its receptors Pdgfrb and Itga2, most likely by up-regulating MMPs, contributes to the CYP1B1-mediated pathogenesis of Ang II–induced AAA. Supporting this view was our demonstration that Ang II–induced AAA in Apoe−/−/Cyp1b1+/+ mice was associated with increased expression of MMP-2 and -9 in aorta and was attenuated by TMS treatment or by Cyp1b1 gene disruption. Alterations in the microstructural organization of a healthy elastic arterial wall, resulting in degradation of collagen, elastin, and actin, cause pathologic dilation of the vessel.5 We observed enhanced degradation of the ECM components, which may have been facilitated by the enhanced MMP expression observed at the lesion site. However, further studies are required to delineate the relative contribution of CYP1B1-dependent PDGF-D, Pdgfrb, and Itga2, as well as MMP-2 and -9 expression in different cell types.
The activation of MMPs is associated with increased oxidative stress.18, 38 We have previously reported that Ang II generates ROSs from arachidonic acid via CYP1B1.11 CYP1B1 can metabolize various substrates, including steroids, retinoic acid, and fatty acids.48, 50 In the present study, Ang II–induced AAA in Apoe−/−/Cyp1b1+/+ mice was associated with oxidative stress, as indicated by increased cardiac NADPH oxidase, production of ROSs in the lesion site in aortae, and increased plasma levels of 8-isoprostane, all of which were minimized by TMS or in Apoe−/−/Cyp1b1−/− mice. Collectively, these observations suggest that the generation of oxidative stress and lipid peroxides by CYP1B1 contributes to the development of Ang II–induced AAA. However, to provide direct evidence to implicate ROSs in CYP1B1-mediated AAA development, it would be necessary to determine whether increasing ROSs in Apoe−/−/Cyp1b1−/− mice increases Ang II–induced AAA. We plan to address this in our future studies, using agents that directly increase production of ROSs with and without tempol in Apoe−/−/Cyp1b1−/− mice. Moreover, recently it has been reported that isoketals, which are formed via the isoprostane pathway of lipid peroxidation, activate T cells by modifying dendritic cell proteins.51 Therefore, it is possible that CYP1B1 might also participate in the generation of isoketals, but this hypothesis needs to be determined.
Ang II–induced oxidative stress increases COX-2 expression in various cell types in the cardiovascular system52, 53 and plays a role in Ang II–induced AAA.37, 54 Our results demonstrating that an increase in COX-2 expression in Ang II–induced AAA in Apoe−/−/Cyp1b1+/+ mice was prevented by TMS or Cyp1b1 gene disruption suggests that CYP1B1 acts upstream of COX-2 in minimizing AAA, which could be due to ROSs generated by CYP1B1. Supporting this view was our finding that the administration of tempol attenuated the expression of COX-2, which could occur via p38 MAPK, as reported in previous studies.38, 55 Whether oxidative stress generated by Ang II through CYP1B1 increases COX-2 expression in a parallel manner or sequential pathway remains to be determined.
Finally, although we observed an increase in aortic diameter in Apoe−/−/Cyp1b1+/+ mice after 7 days of Ang II infusion, this increase was less compared to that observed at the 28-day time point. Also, the enhanced dilation of aorta was characterized with proinflammatory changes and enhanced oxidative stress only in Apoe−/−/Cyp1b1+/+ but not in Apoe−/−/Cyp1b1−/− mice infused with Ang II for a short period of 7 days. This finding validates our hypothesis that the pathophysiologic changes in AAA are dependent on CYP1B1 and are not the consequence of AAA lesion.
In conclusion, this study provides the first evidence that CYP1B1 contributes to Ang II–induced AAA and associated pathogenesis, including infiltration of proinflammatory cells and platelets to the site of aortic lesions, in male Apoe−/−/Cyp1b1+/+ mice via generation of ROSs. CYP1B1-generated oxidative stress also appears to regulate the expression of COX-2, which has been implicated in Ang II–induced AAA. The likely mechanism by which oxidative stress generated via CYP1B1 regulates the activity of these signaling molecules in male mice is depicted in Figure 8. Since testosterone has been implicated in Ang II–induced AAA,56 and because CYP1B1-generated testosterone metabolite 6β-hydroxytestosterone mediates Ang II–induced oxidative stress in male mice,57, 58 further studies are being conducted to determine whether this metabolite of testosterone contributes to Ang II–induced AAA. In contrast, in female mice, the CYP1B1-generated estradiol metabolite 2-methoxyestradiol is protective against Ang II hypertension, and estradiol ameliorates Ang II–induced AAA.59, 60 Our preliminary results indicate that CYP1B1 and 2-methoxyestradiol are protective against Ang II–induced AAA in female Apoe−/− mice (Thirunavukkarasu and Malik, unpublished work). Therefore, our study has important translational implications and suggests that the development of selective inhibitors of CYP1B1, such as TMS, could be useful in treating AAA associated with increased activity of the renin–Ang system in males, but could be detrimental in females.
Figure 8.

Schematic diagram illustrating the possible mechanism of cytochrome P450 (CYP) 1B1–dependent angiotensin (Ang) II–induced abdominal aortic aneurysm formation. Ang II via CYP1B1-mediated reactive oxygen species (ROS) generation and cyclooxygenase (COX)-2 expression, and inflammatory changes, characterized by enhanced infiltration of CD192+ monocytes and T cells, are accompanied by increased platelet infiltration and activation. These events, combined with the actions of Ang II to induce vascular smooth muscle cell proliferation and migration, result in elevated platelet-derived growth factor (PDGF)-D expression by various cell types, and enhanced signaling via Itga2 and Pdgfrb, resulting in enhanced matrix metalloproteinase (MMP) expression, which by acting on the medial layer causes degradation of extracellular matrix (ECM) components, leading to generation of aneurysm.
Acknowledgment
We thank Dr. David Armbruster for editorial assistance.
Footnotes
Supported by NIH National Heart, Lung, and Blood Institute grant R01HL079109-09 (K.U.M.) and Department of Veterans Affairs grant BX001193-02 (D.D.B.).
Disclosures: None declared.
The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the National Heart, Lung and Blood Institute. This work was earlier submitted and chosen for a travel award and oral presentation at the American Society for Investigative Pathology Mini Symposium sessions (Vascular Biology and Molecular Mechanisms of Diseases) on Experimental Biology, March 28 to April 1, 2015, Boston, Massachusetts.
Supplemental material for this article can be found at http://dx.doi.org/10.1016/j.ajpath.2016.04.005.
Supplemental Data
Supplemental Figure S1.

Controls for immunohistochemistry (IHC) analysis and immunofluorescence staining. A and B: Representative photomicrographs of secondary antibody only and diaminobenzidine (DAB) controls (A) and respective isotype controls (B) for F4/80, CD3, CD62p, platelet-derived growth factor (PDGF)-D, PDGF receptor β (PDGFR-β), integrin (Itg)-α2, cyclooxygenase (COX)-2, and α smooth muscle actin (SMA) IHC staining in aorta from angiotensin (Ang) II–induced Apoe−/−/Cyp1b1+/+ mice. C: Immunofluorescence staining in aorta from Ang II–induced Apoe−/−/Cyp1b1+/+ mice for rabbit polyclonal IgG isotype control antibody for matrix metalloproteinase (MMP)-2 and -9 antibodies. Scale bars = 50 μm. Original magnification, ×40.
Supplemental Figure S2.

Formation of abdominal aortic aneurysm in Apoe−/−/Cyp1b1+/+ mice infused with angiotensin (Ang) II for 28 days is alleviated by treatment with 2,3′,4,5′-tetramethoxystilbene (TMS) but not by its vehicle (Veh) dimethyl sulfoxide or by Cyp1b1 gene disruption. A and B: Quantitated data for the ultrasound B-mode images in short axis (A, Figure 1A), and corresponding quantified data (B) for ex vivo measurements of aortic diameter by Vernier caliper. C: Representative ultrasound B-mode image of aorta in the long axis. n = 5 for each group. ∗P < 0.05, ∗∗∗P < 0.001 (two-way analysis of variance followed by the Bonferroni multiple-comparisons test).
Supplemental Figure S3.
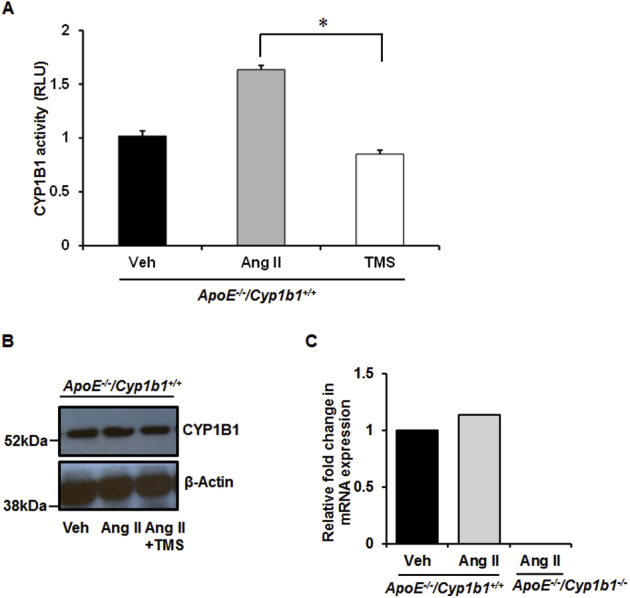
Angiotensin (Ang) II infusion increases cardiac cytochrome P450 (CYP) 1B1 activity in Apoe−/−/Cyp1b1+/+ mice without altering Cyp1b1 gene expression in aorta. Apoe−/−/Cyp1b1+/+ and Apoe−/−/Cyp1b1−/− mice were infused with Ang II for 28 days. A and B: Cardiac tissue was collected to measure CYP1B1 activity using the P450-Glo assay kit (catalog number V8762; Promega Corporation, Madison, WI) and expressed as relative luminescence units (RLU) (A) and CYP1B1 protein expression by Western blot analysis (B). C:Cyp1b1 mRNA expression in aorta of Apoe−/−/Cyp1b1+/+ and Apoe−/−/Cyp1b1−/− mice was determined by real-rime quantitative PCR. Cyp1b1 mRNA expression was normalized against the reference gene S19. Corresponding means ± SD ΔCT values for Apoe−/−/Cyp1b1+/+ vehicle (Veh) versus Apoe−/−/Cyp1b1+/+Ang II: 6.3 ± 0.94 versus 6.47 ± 1.1. Data are expressed as means ± SEM fold-changes in gene expression for the normalized data. n = 5 per group of experiments. ∗P < 0.05 (two-way analysis of variance followed by the Bonferroni multiple-comparisons test). TMS, 2,3′,4,5′-tetramethoxystilbene.
Supplemental Figure S4.

2,3′,4,5′-Tetramethoxystilbene (TMS) treatment or Cyp1b1 gene disruption reduces angiotensin (Ang) II–induced increase in CD192 expression [C-C chemokine receptor 2 (CCR2)] in CD11b+ monocytes in Apoe−/−/Cyp1b1+/+ mice. Apoe−/−/Cyp1b1+/+ and Apoe−/−/Cyp1b1−/− mice were infused with vehicle (Veh), or Ang II, or Ang II along with TMS injections for 28 days. Blood was collected, peripheral blood mononuclear cells were stained with fluorescence-tagged antibodies to the cell-surface markers, and data were collected by flow cytometry. A: CD192 (CCR2) expression in CD11b+ monocytes is presented as a dot plot with CD11b intensity on the x axis and CD192 intensity on the y axis. Boxed areas indicate CD11b+CD192+ cells. B: The quantitated data for the CD11b+CD192+ cells are presented as a bar graph. Data are expressed as means ± SEM. n = 5 per experimental group. ∗P < 0.05 (Kruskal-Wallis test followed by the Dunn multiple-comparisons test).
Supplemental Figure S5.

2,3′,4,5′-Tetramethoxystilbene (TMS) or Cyp1b1 gene disruption reduces angiotensin (Ang) II–induced increase in CD192 expression [chemokine receptor 2 (CCR2)] in CD3+ T lymphocytes in Apoe−/−/Cyp1b1+/+ mice. Apoe−/−/Cyp1b1+/+ and Apoe−/−/Cyp1b1−/− mice were infused with vehicle (Veh), or Ang II, or Ang II along with TMS injections for 28 days. Blood was collected and peripheral blood mononuclear cells were stained with fluorescence-tagged antibodies to the cell-surface markers; data were collected by flow cytometry. A: CD192 (CCR2) expression in CD3+ T lymphocytes is presented as a contour plot (threshold set at 2%) with CD3 intensity on the x axis and CD192 intensity on the y axis. B: These data are plotted in histogram form. Data are expressed as means ± SEM. n = 5 per experimental group. ∗P < 0.05 (Kruskal-Wallis test followed by the Dunn multiple-comparisons test).
Supplemental Figure S6.

2,3′,4,5′-Tetramethoxystilbene (TMS) treatment or Cyp1b1 gene disruption inhibits expression of platelet-derived growth factor receptor β (PDGFR-β) and integrin (Itg)-α2 in aorta of Apoe−/−/Cyp1b1+/+ mice infused with angiotensin (Ang) II. Apoe−/−/Cyp1b1+/+ and Apoe−/−/Cyp1b1−/− mice were infused with vehicle (Veh), or Ang II, or II along with TMS injection for 28 days. A–C: IHC images of PDGFR-β- and Itg-α2–stained sections of aorta (A), and respective quantified data (B and C). Data are expressed as means ± SEM. n = 5 per experimental group. ∗∗P < 0.01 (two-way analysis of variance followed by the Bonferroni multiple-comparisons test). Scale bars = 50 μm. Original magnification, ×40.
Supplemental Figure S7.
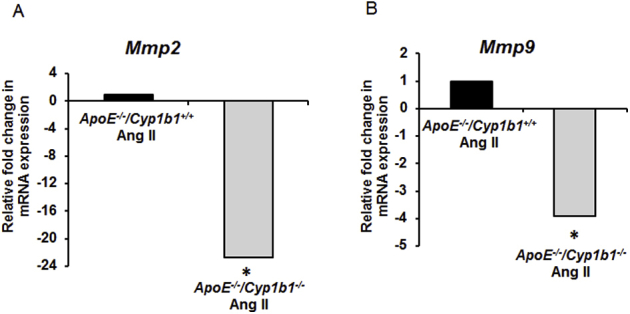
Cyp1b1 gene disruption down-regulates Mmp2 and -9 gene expression in the aorta of Apoe−/−/Cyp1b1+/+ mice infused with angiotensin (Ang) II. Apoe−/−/Cyp1b1+/+ and Apoe−/−/Cypb11−/− mice were infused with Ang II for 28 days. A and B:Mmp2 (A) and Mmp9 (B) mRNA expression in aorta of Ang II–infused Apoe−/−/Cyp1b1+/+ and Apoe−/−/Cyp1b1−/− mice was determined by real-time quantitative PCR. Mmp2 and -9 mRNA expression values were normalized against the reference gene S19. Corresponding means ± SD ΔCT values for Apoe−/−/Cyp1b1+/+ Ang II versus Apoe−/−/Cyp1b1−/−Ang II are provided for reference: Mmp2, 3.8 ± 0.01 versus 8.3 ± 1.7; and Mmp9, 6.5 ± 1.3 versus 8.4 ± 1.1. Data are expressed as fold-changes in gene expression for the normalized data. n = 5 per experimental group. ∗P < 0.05 (Kruskal-Wallis test followed by the Dunn multiple-comparisons test).
Supplemental Figure S8.

Cyp1b1 gene disruption minimizes an increase in aortic diameter and inflammatory and oxidative stress in aorta of Apoe−/−/Cyp1b1+/+ mice after 7 days of angiotensin (Ang) II infusion. Apoe−/−/Cyp1b1+/+ and Apoe−/−/Cypb11−/− mice were infused with Ang II for 7 days. A–E: Quantitated data for aortic diameter (A), F4/80+ cellular infiltration (B), elastin fiber break (C), fluorescence of matrix metalloproteinase (MMP)-2 expression (R.U.) (D), and fluorescence of 2-hydroxyethidium (OHE) expression (R.U.) (E). Data are expressed as means ± SEM. n = 4 per group. ∗P < 0.05, ∗∗P < 0.01 (Kruskal-Wallis test followed by the Dunn multiple-comparison test).
Supplemental Figure S9.
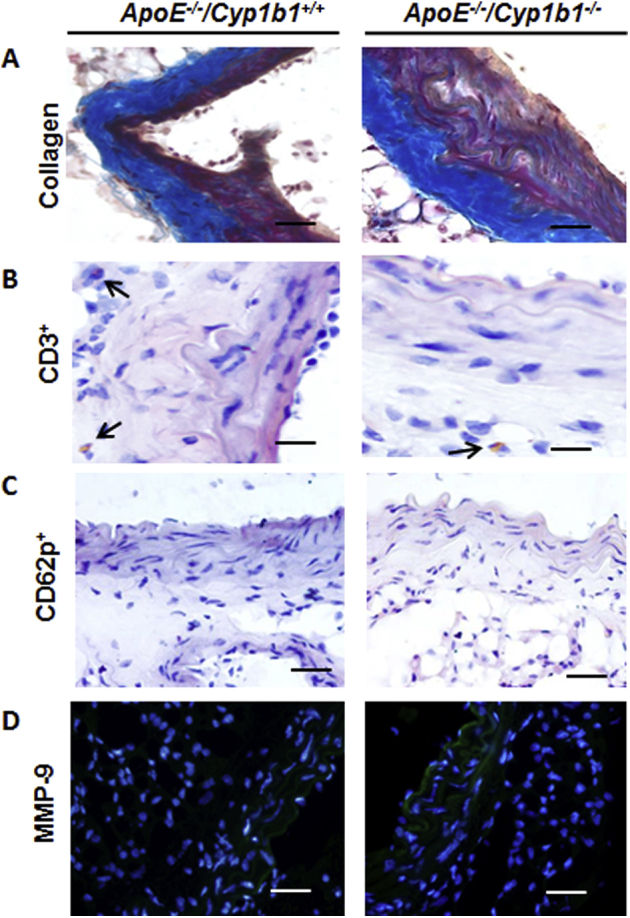
Cyp1b1 gene disruption does not alter aortic wall collagen expression, CD3+ T-cell and CD62p+ platelet infiltration, and matrix metalloproteinase (MMP)-9 expression in aorta of Apoe−/−/Cyp1b1+/+ and Apoe−/−/Cyp1b1+/+ mice after 7 days of angiotensin (Ang) II infusion. Apoe−/−/Cyp1b1+/+ and Apoe−/−/Cypb11−/− mice were infused with Ang II for 7 days. Photomicrographs of Masson's trichrome staining for collagen (A), CD3+ T cells (B), CD62p+ platelets (C), and MMP-9 (D). Arrows point to CD3+ T cells. n = 4. Scale bars = 50 μm. Original magnification, ×40.
References
- 1.Lu H., Rateri D.L., Bruemmer D., Cassis L.A., Daugherty A. Involvement of the renin–angiotensin system in abdominal and thoracic aortic aneurysms. Clin Sci. 2012;123:531–543. doi: 10.1042/CS20120097. [DOI] [PubMed] [Google Scholar]
- 2.Guo D.C., Papke C.L., He R., Milewicz D.M. Pathogenesis of thoracic and abdominal aortic aneurysms. Ann N Y Acad Sci. 2006;1085:339–352. doi: 10.1196/annals.1383.013. [DOI] [PubMed] [Google Scholar]
- 3.Tung W.S., Lee J.K., Thompson R.W. Simultaneous analysis of 1176 gene products in normal human aorta and abdominal aortic aneurysms using a membrane-based complementary DNA expression array. J Vasc Surg. 2001;34:143–150. doi: 10.1067/mva.2001.113310. [DOI] [PubMed] [Google Scholar]
- 4.Daugherty A., Manning M.W., Cassis L.A. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest. 2000;105:1605–1612. doi: 10.1172/JCI7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ailawadi G., Eliason J.L., Upchurch G.R., Jr. Current concepts in the pathogenesis of abdominal aortic aneurysm. J Vasc Surg. 2003;38:584–588. doi: 10.1016/s0741-5214(03)00324-0. [DOI] [PubMed] [Google Scholar]
- 6.Golledge J., Tsao P.S., Dalman R.L., Norman P.E. Circulating markers of abdominal aortic aneurysm presence and progression. Circulation. 2008;118:2382–2392. doi: 10.1161/CIRCULATIONAHA.108.802074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rajagopalan S., Meng X.P., Ramasamy S., Harrison D.G., Galis Z.S. Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. Implications for atherosclerotic plaque stability. J Clin Invest. 1996;98:2572–2579. doi: 10.1172/JCI119076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Touyz R.M. Reactive oxygen species and angiotensin II signaling in vascular cells: implications in cardiovascular disease. Braz J Med Biol Res. 2004;37:1263–1273. doi: 10.1590/s0100-879x2004000800018. [DOI] [PubMed] [Google Scholar]
- 9.Paravicini T.M., Touyz R.M. NADPH oxidases, reactive oxygen species, and hypertension: clinical implications and therapeutic possibilities. Diabetes Care. 2008;31:S170–S180. doi: 10.2337/dc08-s247. [DOI] [PubMed] [Google Scholar]
- 10.Griendling K.K., Sorescu D., Ushio-Fukai M. NAD(P)H Oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 11.Yaghini F.A., Song C.Y., Lavrentyev E.N., Ghafoor H.U.B., Fang X.R., Estes A.M., Campbell W.B., Malik K.U. Angiotensin II-induced vascular smooth muscle cell migration and growth are mediated by cytochrome p450 1B1-dependent superoxide generation. Hypertension. 2010;55:1461–1467. doi: 10.1161/HYPERTENSIONAHA.110.150029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jennings B.L., Sahan-Firat S., Estes A.M., Das K., Farjana N., Fang X.R., Gonzalez F.J., Malik K.U. Cytochrome P450 1b1 contributes to angiotensin II-induced hypertension and associated pathophysiology. Hypertension. 2010;56:667–674. doi: 10.1161/HYPERTENSIONAHA.110.154518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elbekai R.H., El-Kadi A.O. Cytochrome P450 enzymes: central players in cardiovascular health and disease. Pharmacol Ther. 2006;112:564–587. doi: 10.1016/j.pharmthera.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 14.Chun Y.J., Kim S., Kim D., Lee S.K., Guengerich F.P. A new selective and potent inhibitor of human cytochrome P450 1B1 and its application to antimutagenesis. Cancer Res. 2001;61:8164–8170. [PubMed] [Google Scholar]
- 15.Committee for the Update of the Guide for the Care and Use of Laboratory Animals. National Research Council . Eighth Edition. National Academies Press; Washington, DC: 2011. Guide for the Care and Use of Laboratory Animals. [Google Scholar]
- 16.Song C.Y., Ghafoor K., Ghafoor H.U., Khan N.S., Thirunavukkarasu S., Jennings B.L., Estes A.M., Zaidi S., Bridges D., Tso P., Gonzalez F.J., Malik K.U. Cytochrome P450 1B1 contributes to the development of atherosclerosis and hypertension in apolipoprotein E–deficient mice. Hypertension. 2016;67:206–213. doi: 10.1161/HYPERTENSIONAHA.115.06427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palinski W., Ord V.A., Plump A.S., Breslow J.L., Steinberg D., Witztum J.L. ApoE-deficient mice are a model of lipoprotein oxidation in atherogenesis. Demonstration of oxidation-specific epitopes in lesions and high titers of autoantibodies to malondialdehyde-lysine in serum. Arterioscler Thromb. 1994;14:605–616. doi: 10.1161/01.atv.14.4.605. [DOI] [PubMed] [Google Scholar]
- 18.Castro D.J., Baird W.M., Pereira C.B., Giovanini J., Lohr C.V., Fischer K.A., Yu Z., Gonzalez F.J., Krueger S.K., Williams D.E. Fetal mouse Cyp1b1 and transplacental carcinogenesis from maternal exposure to dibenzo(a,l) pyrene. Cancer Prev Res (Phila) 2008;1:128–134. doi: 10.1158/1940-6207.CAPR-07-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khan N.S., Song C.Y., Jennings B.L., Estes A.M., Fang X.R., Bonventre J.V., Malik K.U. Cytosolic phospholipase A2[alpha] is critical for angiotensin II–induced hypertension and associated cardiovascular pathophysiology. Hypertension. 2015;65:784–792. doi: 10.1161/HYPERTENSIONAHA.114.04803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gallo E.M., Loch D.C., Habashi J.P., Calderon J.F., Chen Y., Bedja D., van Erp C., Gerber E.E., Parker S.J., Sauls K., Judge D.P., Cooke S.K., Lindsay M.E., Rouf R., Myers L., ap Rhys C.M., Kent K.C., Norris R.A., Huso D.L., Dietz H.C. Angiotensin II-dependent TGF-beta signaling contributes to Loeys-Dietz syndrome vascular pathogenesis. J Clin Invest. 2014;124:448–460. doi: 10.1172/JCI69666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller F.J., Jr., Gutterman D.D., Rios C.D., Heistad D.D., Davidson B.L. Superoxide production in vascular smooth muscle contributes to oxidative stress and impaired relaxation in atherosclerosis. Circ Res. 1998;82:1298–1305. doi: 10.1161/01.res.82.12.1298. [DOI] [PubMed] [Google Scholar]
- 22.Yoshida K., Kobayashi N., Ohno T., Fukushima H., Matsuoka H. Cardioprotective effect of angiotensin II type 1 receptor antagonist associated with bradykinin-endothelial nitric oxide synthase and oxidative stress in Dahl salt-sensitive hypertensive rats. J Hypertens. 2007;25:1633–1642. doi: 10.1097/HJH.0b013e32814db89f. [DOI] [PubMed] [Google Scholar]
- 23.Chomczynski P. A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. Biotechniques. 1993;15:532–534. 6-7. [PubMed] [Google Scholar]
- 24.Uno S., Dalton T.P., Dragin N., Curran C.P., Derkenne S., Miller M.L., Shertzer H.G., Gonzalez F.J., Nebert D.W. Oral benzo[a]pyrene in Cyp1 knockout mouse lines: CYP1A1 important in detoxication, CYP1B1 metabolism required for immune damage independent of total-body burden and clearance rate. Mol Pharmacol. 2006;69:1103–1114. doi: 10.1124/mol.105.021501. [DOI] [PubMed] [Google Scholar]
- 25.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 26.Owens A.P., Edwards T.L., Antoniak S., Geddings J.E., Jahangir E., Wei W.-Q., Denny J.C., Boulaftali Y., Bergmeier W., Daugherty A., Sampson U.K.A., Mackman N. Platelet inhibitors reduce rupture in a mouse model of established abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 2015;35:2033–2041. doi: 10.1161/ATVBAHA.115.305537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanazawa S., Miyake T., Kakinuma T., Tanemoto K., Tsunoda T., Kikuchi K. The expression of platelet-derived growth factor and connective tissue growth factor in different types of abdominal aortic aneurysms. J Cardiovasc Surg (Torino) 2005;46:271–278. [PubMed] [Google Scholar]
- 28.Pontén A., Bergsten Folestad E., Pietras K., Eriksson U. Platelet-derived growth factor D induces cardiac fibrosis and proliferation of vascular smooth muscle cells in heart-specific transgenic mice. Circ Res. 2005;97:1036–1045. doi: 10.1161/01.RES.0000190590.31545.d4. [DOI] [PubMed] [Google Scholar]
- 29.He C., Medley S.C., Hu T., Hinsdale M.E., Lupu F., Virmani R., Olson L.E. PDGFR[beta] signalling regulates local inflammation and synergizes with hypercholesterolaemia to promote atherosclerosis. Nat Commun. 2015;6:7770. doi: 10.1038/ncomms8770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karvinen H., Rutanen J., Leppänen O., Lach R., Levonen A.L., Eriksson U., Ylä-Herttuala S. PDGF-C and -D and their receptors PDGFR-[alpha] and PDGFR-[beta] in atherosclerotic human arteries. Eur J Clin Invest. 2009;39:320–327. doi: 10.1111/j.1365-2362.2009.02095.x. [DOI] [PubMed] [Google Scholar]
- 31.Sano H., Sudo T., Yokode M., Murayama T., Kataoka H., Takakura N., Nishikawa S., Nishikawa S.-I., Kita T. Functional blockade of platelet-derived growth factor receptor-[beta] but not of receptor-[alpha] prevents vascular smooth muscle cell accumulation in fibrous cap lesions in apolipoprotein E–deficient mice. Circulation. 2001;103:2955–2960. doi: 10.1161/01.cir.103.24.2955. [DOI] [PubMed] [Google Scholar]
- 32.Raines E.W. PDGF and cardiovascular disease. Cytokine Growth Factor Rev. 2004;15:237–254. doi: 10.1016/j.cytogfr.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 33.Lee R.T., Berditchevski F., Cheng G.C., Hemler M.E. Integrin-mediated collagen matrix reorganization by cultured human vascular smooth muscle cells. Circ Res. 1995;76:209–214. doi: 10.1161/01.res.76.2.209. [DOI] [PubMed] [Google Scholar]
- 34.Kunicki T.J. The influence of platelet collagen receptor polymorphisms in hemostasis and thrombotic disease. Arterioscler Thromb Vasc Biol. 2002;22:14–20. doi: 10.1161/hq0102.100458. [DOI] [PubMed] [Google Scholar]
- 35.Xiong W., Mactaggart J., Knispel R., Worth J., Zhu Z., Li Y., Sun Y., Baxter B.T., Johanning J. Inhibition of reactive oxygen species attenuates aneurysm formation in a murine model. Atherosclerosis. 2009;202:128–134. doi: 10.1016/j.atherosclerosis.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cracowski J.L., Durand T., Bessard G. Isoprostanes as a biomarker of lipid peroxidation in humans: physiology, pharmacology and clinical implications. Trends Pharmacol Sci. 2002;23:360–366. doi: 10.1016/s0165-6147(02)02053-9. [DOI] [PubMed] [Google Scholar]
- 37.Ohnaka K., Numaguchi K., Yamakawa T., Inagami T. Induction of cyclooxygenase-2 by angiotensin II in cultured rat vascular smooth muscle cells. Hypertension. 2000;35:68–75. doi: 10.1161/01.hyp.35.1.68. [DOI] [PubMed] [Google Scholar]
- 38.McCormick M.L., Gavrila D., Weintraub N.L. Role of oxidative stress in the pathogenesis of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2007;27:461–469. doi: 10.1161/01.ATV.0000257552.94483.14. [DOI] [PubMed] [Google Scholar]
- 39.Wilcox C.S. Effects of tempol and redox-cycling nitroxides in models of oxidative stress. Pharmacol Ther. 2010;126:119–145. doi: 10.1016/j.pharmthera.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cassis L.A., Gupte M., Thayer S., Zhang X., Charnigo R., Howatt D.A., Rateri D.L., Daugherty A. ANG II infusion promotes abdominal aortic aneurysms independent of increased blood pressure in hypercholesterolemic mice. Am J Physiol Heart Circ Physiol. 2009;296:H1660–H1665. doi: 10.1152/ajpheart.00028.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Finnström N., Thörn M., Lööf L., Rane A. Independent patterns of cytochrome P 450 gene expression in liver and blood in patients with suspected liver disease. Eur J Clin Pharmacol. 2001;57:403–409. doi: 10.1007/s002280100318. [DOI] [PubMed] [Google Scholar]
- 42.Korashy H.M., El-Kadi A.O. The role of aryl hydrocarbon receptor in the pathogenesis of cardiovascular diseases. Drug Metab Rev. 2006;38:411–450. doi: 10.1080/03602530600632063. [DOI] [PubMed] [Google Scholar]
- 43.Mack M., Cihak J., Simonis C., Luckow B., Proudfoot A.E.I., Plachý Jí, Brühl H., Frink M., Anders H.-J., Vielhauer V., Pfirstinger J., Stangassinger M., Schlöndorff D. Expression and characterization of the chemokine receptors CCR2 and CCR5 in mice. J Immunol. 2001;166:4697–4704. doi: 10.4049/jimmunol.166.7.4697. [DOI] [PubMed] [Google Scholar]
- 44.Zhang H.H., Song K., Rabin R.L., Hill B.J., Perfetto S.P., Roederer M., Douek D.C., Siegel R.M., Farber J.M. CCR2 identifies a stable population of human effector memory CD4+ T cells equipped for rapid recall response. J Immunol. 2010;185:6646–6663. doi: 10.4049/jimmunol.0904156. [DOI] [PubMed] [Google Scholar]
- 45.Sato W., Tomita A., Ichikawa D., Lin Y., Kishida H., Miyake S., Ogawa M., Okamoto T., Murata M., Kuroiwa Y., Aranami T., Yamamura T. CCR2(+)CCR5(+) T cells produce matrix metalloproteinase-9 and osteopontin in the pathogenesis of multiple sclerosis. J Immunol. 2012;189:5057–5065. doi: 10.4049/jimmunol.1202026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu Q., Hou X., Xia J., Qian X., Miele L., Sarkar F.H., Wang Z. Emerging roles of PDGF-D in EMT progression during tumorigenesis. Cancer Treat Rev. 2012;39:640–646. doi: 10.1016/j.ctrv.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen J., Han Y., Lin C., Zhen Y., Song X., Teng S., Chen C., Chen Y., Zhang Y., Hui R. PDGF-D contributes to neointimal hyperplasia in rat model of vessel injury. Biochem Biophys Res Commun. 2005;329:976–983. doi: 10.1016/j.bbrc.2005.02.062. [DOI] [PubMed] [Google Scholar]
- 48.Choudhary D., Jansson I., Stoilov I., Sarfarazi M., Schenkman J.B. Metabolism of retinoids and arachidonic acid by human and mouse cytochrome P450 1b1. Drug Metab Dispos. 2004;32:840–847. doi: 10.1124/dmd.32.8.840. [DOI] [PubMed] [Google Scholar]
- 49.Choi W.S., Jeon O.H., Kim H.H., Kim D.S. MMP-2 regulates human platelet activation by interacting with integrin alphaIIbbeta3. J Thromb Haemost. 2008;6:517–523. doi: 10.1111/j.1538-7836.2007.02871.x. [DOI] [PubMed] [Google Scholar]
- 50.Hayes C.L., Spink D.C., Spink B.C., Cao J.Q., Walker N.J., Sutter T.R. 17 beta-estradiol hydroxylation catalyzed by human cytochrome P450 1B1. Proc Natl Acad Sci U S A. 1996;93:9776–9781. doi: 10.1073/pnas.93.18.9776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kirabo A., Fontana V., de Faria A.P., Loperena R., Galindo C.L., Wu J., Bikineyeva A.T., Dikalov S., Xiao L., Chen W., Saleh M.A., Trott D.W., Itani H.A., Vinh A., Amarnath V., Amarnath K., Guzik T.J., Bernstein K.E., Shen X.Z., Shyr Y., Chen S.C., Mernaugh R.L., Laffer C.L., Elijovich F., Davies S.S., Moreno H., Madhur M.S., Roberts J., 2nd, Harrison D.G. DC isoketal-modified proteins activate T cells and promote hypertension. J Clin Invest. 2014;124:4642–4656. doi: 10.1172/JCI74084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rocca B., Secchiero P., Ciabattoni G., Ranelletti F.O., Catani L., Guidotti L., Melloni E., Maggiano N., Zauli G., Patrono C. Cyclooxygenase-2 expression is induced during human megakaryopoiesis and characterizes newly formed platelets. Proc Natl Acad Sci U S A. 2002;99:7634–7639. doi: 10.1073/pnas.112202999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martinez-Gonzalez J., Badimon L. Mechanisms underlying the cardiovascular effects of COX-inhibition: benefits and risks. Curr Pharm Des. 2007;13:2215–2227. doi: 10.2174/138161207781368774. [DOI] [PubMed] [Google Scholar]
- 54.Gitlin J.M., Trivedi D.B., Langenbach R., Loftin C.D. Genetic deficiency of cyclooxygenase-2 attenuates abdominal aortic aneurysm formation in mice. Cardiovasc Res. 2007;73:227–236. doi: 10.1016/j.cardiores.2006.10.015. [DOI] [PubMed] [Google Scholar]
- 55.Ushio-Fukai M., Alexander R.W., Akers M., Griendling K.K. p38 mitogen-activated protein kinase is a critical component of the redox-sensitive signaling pathways activated by angiotensin II: role in vascular smooth muscle cell hypertrophy. J Biol Chem. 1998;273:15022–15029. doi: 10.1074/jbc.273.24.15022. [DOI] [PubMed] [Google Scholar]
- 56.Zhang X., Thatcher S., Wu C., Daugherty A., Cassis L.A. Castration of male mice prevents the progression of established angiotensin II-induced abdominal aortic aneurysms. J Vasc Surg. 2015;61:767–776. doi: 10.1016/j.jvs.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pingili A.K., Kara M., Khan N.S., Estes A.M., Lin Z., Li W., Gonzalez F.J., Malik K.U. 6beta-hydroxytestosterone, a cytochrome P450 1B1 metabolite of testosterone, contributes to angiotensin II-induced hypertension and its pathogenesis in male mice. Hypertension. 2015;65:1279–1287. doi: 10.1161/HYPERTENSIONAHA.115.05396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pingili A.K., Thirunavukkarasu S., Kara M., Brand D.D., Katsurada A., Majid D.S., Navar L.G., Gonzalez F.J., Malik K.U. 6[beta]-hydroxytestosterone, a cytochrome P450 1B1-testosterone–metabolite, mediates angiotensin II–induced renal dysfunction in male mice. Hypertension. 2016;67:916–926. doi: 10.1161/HYPERTENSIONAHA.115.06936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jennings B.L., George L.W., Pingili A.K., Khan N.S., Estes A.M., Fang X.R., Gonzalez F.J., Malik K.U. Estrogen metabolism by cytochrome P450 1B1 modulates the hypertensive effect of angiotensin II in female mice. Hypertension. 2014;64:134–140. doi: 10.1161/HYPERTENSIONAHA.114.03275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thatcher S., Zhang X., Woody S., Wang Y., Alsiraj Y., Charnigo R., Daugherty A., Cassis L. Exogenous 17-[beta] estradiol administration blunts progression of established angiotensin II-induced abdominal aortic aneurysms in female ovariectomized mice. Biol Sex Differ. 2015;6:1–11. doi: 10.1186/s13293-015-0030-1. [DOI] [PMC free article] [PubMed] [Google Scholar]