

Abstract
The Bcl2-associated anthanogene (BAG) 3 protein is a member of the BAG family of cochaperones, which supports multiple critical cellular processes, including critical structural roles supporting desmin and interactions with heat shock proteins and ubiquitin ligases intimately involved in protein quality control. The missense mutation P209L in exon 3 results in a primarily cardiac phenotype leading to skeletal muscle and cardiac complications. At least 10 other Bag3 mutations have been reported, nine resulting in a dilated cardiomyopathy for which no specific therapy is available. We generated αMHC-human Bag3 P209L transgenic mice and characterized the progressive cardiac phenotype in vivo to investigate its utility in modeling human disease, understand the underlying molecular mechanisms, and identify potential therapeutic targets. We identified a progressive heart failure by echocardiography and Doppler analysis and the presence of pre-amyloid oligomers at 1 year. Paralleling the pathogenesis of neurodegenerative diseases (eg, Parkinson disease), pre-amyloid oligomers–associated alterations in cardiac mitochondrial dynamics, haploinsufficiency of wild-type BAG3, and activation of p38 signaling were identified. Unexpectedly, increased numbers of activated cardiac fibroblasts were identified in Bag3 P209L Tg+ hearts without increased fibrosis. Together, these findings point to a previously undescribed therapeutic target that may have application to mutation-induced myofibrillar myopathies as well as other common causes of heart failure that commonly harbor misfolded proteins.
The Bcl2-associated athanogene (BAG) 3 protein is a member of the BAG family of cochaperones found primarily in skeletal and cardiac muscle. Within muscle cells, BAG3 colocalizes with desmin and α-actinin at the sarcomere Z-disk, a primary node where mechanical stimulation is translated to signal transduction pathways allowing the cell to respond to stress. Native BAG3 has been implicated in apoptosis, development, cytoskeletal organization, and autophagy, allowing cells to adapt to external stimuli.1 Mechanical stimulation itself induces BAG3 expression,2, 3, 4 which may indicate its role in the maintenance of the skeletal muscle sarcomere in disease.
Myofibrillar myopathies are a heterogeneous group of skeletal and cardiac muscle diseases caused by mutations in eight sarcomere proteins, including BAG3.5 At least 10 mutations have been reported in BAG3, nine resulting in a dilated cardiomyopathy.6, 7, 8 One of the most commonly reported mutations is the P209L missense mutation, resulting in particularly affected cardiac phenotype, constituting a subset of myofibrillar myopathies leading to both skeletal muscle and cardiac complications.6, 9, 10, 11 Because the P209L mutation is found at amino acid 209, it is worth mentioning that recent studies have found that the Bag3 Ile-Pro-Val (IPV) motif found at amino acids 200 to 213 in Bag3 bind to the heat shock protein HSPB6.12 In patient biopsies, the Bag3 P209L mutant leads to increased levels of HSP27 (HSPB1), CRYAB, and Bcl-2 by immunohistochemistry.10 The mutant BAG3 complex migrates farther than the wild-type BAG3 complex in denaturing gel electrophoresis, suggesting that the P209L mutant Bag3 interacts less with partner proteins.10
Although little is known about the Bag3 P209L mutation, it is distinctly not a loss-of-function mutation. Neonatal cardiomyocytes transfected with Bag3 P209L have a greater susceptibility to apoptosis when stressed,6 consistent with Bag3−/− mice that have this same pro-apoptotic phenotype.11 Reducing BAG3 expression in zebrafish results in a severe cardiac defect,8 likely related to its structural role facilitating Hsc70 and CapZ, a protein linking actin filaments to the Z-disk.2 In zebrafish, expression of Bag3 P209L induces protein aggregates and gradually reduce the pool of BAG3.13 However, the underlying pathological mechanisms with which the Bag3 P209L mutation causes disease have not been identified, nor has it been previously associated with autophagy regulation. The slowly progressive loss of cardiac function, a key feature in BAG3 P209L patients, has similarly not previously been identified in animal models.
To elucidate the underlying mechanisms of disease and identify potentially targetable intermediates driving the Bag3 P209L pathology in patients, we generated cardiomyocyte-specific (αMHC-) Bag3 P209L transgenic mice and characterized their progressive cardiac phenotype in vivo over time. Given the increasing appreciation of the role of misfolded proteins in neurodegenerative diseases in heart failure,14 we focused on characterizing the misfolded proteins in the Bag3 P209L Tg+ hearts, and their effects on altering mitochondrial dynamics, effects on native BAG3 expression, and activation of mitogen-activated protein kinase signaling paralleling that found in the pathogenesis of neurodegenerative disease. Unexpected proliferation of cardiac fibroblasts (lacking Bag3 P209L only found in cardiomyocytes) and activation of p38 were identified at 1 year of age, where a progressive systolic and diastolic heart failure was identified by echocardiography. Together, these findings point to a previously undescribed therapeutic target that may have application to mutation-induced myopathies and other common causes of heart failure that harbor misfolded proteins, such as idiopathic dilated cardiomyopathy.15, 16, 17, 18
Materials and Methods
Generation of Bag3 P209L Mouse Strain and Experimental Design
The human Bag3 cDNA with P209L (http://www.ncbi.nlm.nih.gov; GenBank accession number NM_004281) was PCR amplified by the following primers: forward: XhoI-ATG-Flag-Bag3 (5′-GGCTCGAGATGGACTACAAAGACGATGACGACAAGGGTAGCGCCGCCACCCACTCG-3′) and reverse: HindIII-Bag3 with stop codon (5′-CCATTGGGTCGTCGTGGCATCTTCGAAGG-3′) and cloned into the SalI and HindIII site of the αMHC promoter-pBSIISK+ construct (gifted by Dr. Jeff Robbins, Cincinnati Children's Hospital, Cincinnati, OH). Positive clones were digested with NotI, generating a 9-kb fragment, which was injected into the pronuclei of BDF1 embryos and implanted into pseudo-pregnant recipient females. Transgene-positive founders were then bred onto C57BL/6 mice (Charles River Laboratories, Inc., Wilmington, MA). Mice were genotyped from isolated tail DNA (DNeasy Tissue Kit; Qiagen Inc., Valencia, CA) at both weaning and post-mortem. The Bag3 transgene was identified using standard PCR conditions (forward primer: 5′-CATGCCAGAAACCACTCAGC-3′; reverse primer: 5′-GTAGACAGGTCCCACGTAGC-3′ or forward primer/F2: 5′-AGAGACGGTGTCAGGAAGGT-3′; reverse primer/R2: 5′-GAACTACCCACCGTAGGGA-3′). The Bag3 transgene expression levels were approximately twofold wild-type levels. All experiments were approved by the Institutional Animal Care and Use Committee review boards at the University of North Carolina and were performed in accordance with federal guidelines.
Experimental Design
All experiments described used age-matched sibling mice (approximately 50% male/approximately 50% female). Conscious echocardiographic analysis of cardiac function began at 4 months of age and repeated until cardiac dysfunction was identified (8 months) through 12 months of age. At 12 months of life, mice were anesthetized with isoflurane, euthanized with cervical spine dislocation, and heart muscle was flash frozen (stored at −80°C) or perfused by gravity. After abdominal aorta transection, cardiac perfusion was performed using a 23 g butterfly needle in the apex, first perfusing 10 mL sterile phosphate-buffered saline, followed by 20 mL 4% paraformaldehyde at a constant rate of 1 mL/minute. Cardiac tissue was then fixed in 4% paraformaldehyde for exactly 24 hours, followed by storage in 70% ethanol, and processed for routine paraffin-embedded histology (5 mm thick sections) and stained with hematoxylin and eosin, Mason's trichrome, or fixed in 2% paraformaldehyde and 2.5% glutaraldehyde in 0.15 mol/L sodium phosphate buffer (pH 7.4) overnight and then post-fixed with 1% osmium tetroxide/0.15 mol/L sodium phosphate buffer and processed for transmission electron microscopy (TEM). Samples were dehydrated with increasing concentrations of ethanol, infiltrated, and embedded in Poly/Bed812 epoxy resin (Polysciences, Inc., Warrington, PA). Sections (1 μm thick) were prepared to select representative areas by light microscopy, and sections (70 nm ultrathin) were cut with a diamond knife. Sections were mounted on 200 mesh copper grids and stained with 4% aqueous uranyl acetate and Reynolds' lead citrate.
Quantitative Measure of Mitochondria by qPCR Analysis
Mitochondrial number was quantified by real-time quantitative PCR (qPCR), and DNA was isolated from 50 μL whole-heart homogenates using the DNAeasy Blood and Tissue Kit (Qiagen; catalogue number 69506). Isolated DNA and oligomer primers for mitochondrial cytochrome c oxidase subunit 1 (CO1; mt-CO1), cytochrome b (Cyt-b; mt-Cyb), and NADH dehydrogenase 1 (ND1; mt-nd1) DNA normalized to nuclear H19 (imprinted maternally expressed transcript, nonprotein coding) DNA were run in SYBR Green mastermix by qPCR, including melting curves as previously detailed.19
Mouse Echocardiography
Conscious cardiac transthoracic echocardiography was performed on mice at monthly time points using a VisualSonics Vevo 2100 ultrasound biomicroscopy system (VisualSonics, Inc., Toronto, ON, Canada), as previously described.20 Briefly, two-dimensional M-mode echocardiography was performed in the parasternal long-axis view at the level of the papillary muscle on loosely restrained mice by investigators (M.T.Q., M.S.W.) blinded to mouse genotype throughout the process of collection and analysis. Anterior and posterior wall thickness was measured as distance from epicardial to endocardial leading edges. Left ventricular internal diameters were also measured. Left ventricular systolic function was assessed by ejection fraction, left ventrical (LV) ejection fraction % = [(LV Volume; diastole-LV Volume; systole/LV Vol; diastole) × 100], and fractional shortening % = [(LV end-diastolic dimension − LV end-systolic diameter)/LV end-diastolic diameter] × 100.
Measurements represent the average of three cardiac cycles from each mouse. To measure diastolic function, Doppler measurement of the mitral valve flow was taken in an apical view into the LV outflow tract under isoflurane anesthesia, as previously described.21
Measurement of Autophagic Flux
Autophagic flux was determined in vivo from the titration of bafilomycin A1 to optimize animal survival without eliciting other major dysfunction, while inhibiting autophagy in the heart, as previously described.22, 23 Briefly, stock bafilomycin A1 (number 201550A; Santa Cruz Biotechnology, Dallas, TX; 3 mmol/L stock in dimethyl sulfoxide) diluted in phosphate-buffered saline (40% dimethyl sulfoxide final) was injected i.p. to a final concentration of 3 micromol per kilogram in 200 μL and repeated 1 hour later. One hour after the second injection, mice were euthanized and heart tissues were collected for analysis by Western immunoblot for LC3B isoforms (Sigma-Aldrich number L7543).
Histological and TEM Morphometric Analysis
Cross-Sectional Areas
Imaging of hematoxylin and eosin- and Masson's trichrome–stained sections was obtained using Aperio Scanscope and Aperio Imagescope software version 10.0.36.1805 (Aperio Technologies, Inc., Vista, CA). Masson's trichrome–stained images were taken using Aperio Imagescope (TIFF) and analyzed using NIH ImageJ software version 1.47 (NIH, Bethesda, MD) using Aperio exported image ruler. Cardiomyocyte cross-sectional area was measured using ImageJ. A minimum of 100 random cardiomyocytes was imaged from at least three different sections from four biological replicates per experimental group.
Fibrosis Analysis
Using ImageScope, positive pixel countv9 algorithm was used on Masson's trichrome–stained histological section (hue value, 0.66; hue width, 0.1) analyzing tissue sections isolated using the pen tool. After analysis, the calculated N positive/N total was multiplied by 100 to give a percentage collagen content (identifying the blue as a percentage of the tissue present).
Nuclei Counts
The ImageScope positive pixel countv9 algorithm was used to analyze hematoxylin and eosin-stained slides for nuclei (hue value, 0.1; hue width, 0.45; color saturation 0.3), similarly isolated for fibrosis. The nuclei per area was identified per mm squared.
TEM Morphometric Analysis
Cardiac sections were observed with a LEO EM910 TEM operating at 80 kV (LEO Electron Microscopy, Thornwood, NY) and photographed with a Gatan Orius SC1000 CCD Digital Camera and Digital Micrograph 3.11.0 (Gatan, Pleasanton, CA). Mitochondria cross-sectional areas were measured using NIH ImageJ at magnifications of ×5000 to ×10,000. The global scale was set according to the image-specific scale generated by Gatan camera output. An average of 2500 mitochondria were analyzed from 30 fields from multiple levels from three hearts per mouse cohort.
Immunofluorescence Staining of Cardiac Histological Sections for Vimentin, AIF, FLAG (Bag3 P209L), and TUNEL
Immunostaining was performed as described previously.24, 25 Cardiac sections were stained with antibodies against the following: i) vimentin (1:100, catalogue number SC-6260; Santa Cruz Biotechnology, Dallas, TX), or an unrelated isotype mouse, rabbit, or goat IgG (as a negative control) at 4°C overnight; ii) apoptosis-inducing factor (AIF; 1:250; Abcam, Cambridge, MA) or an unrelated isotype mouse, rabbit, or goat IgG (as a negative control) at 4°C overnight; iii) FLAG (DYKDDDK Tag) (1:100; Cell Signaling Technology, Danvers, MA) or an unrelated isotype mouse, rabbit, or goat IgG (as a negative control) at 4°C overnight. Slides were then incubated with Alexa Fluor 488–conjugated secondary antibodies and counterstained with DAPI (Vector Laboratories, Burlingame, CA). Sections were then treated with Alexa Fluor 488–conjugated secondary antibodies and counterstained with DAPI. Total positive vimentin stain cells were identified and counted in each of the four cross-sectional defined areas using Meta-Morph software version 7.8 (Molecular Devices). Images were taken using EVOS XL Core cell imaging system (Life Technologies).
Apoptosis was determined in histological sections by identifying the presence of fragmented DNA by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) using the Roche TUNEL in situ staining kit (Roche Molecular Biochemicals, Basel, Switzerland), according to the manufacturer's instructions. To detect DNA fragmentation associated with apoptosis, we used a fluorescence-based TUNEL followed by counterstaining with DAPI. Histological sections were treated with a recombinant DNase I to allow TUNEL labeling of all nuclei, which were used as positive controls.
Western Blot Analysis
For Western blot analysis, 24- to 36-day-old hearts were homogenized in radioimmunoprecipitation assay buffer and loaded 10 μg/lane and blotted for using mouse anti-FLAG M2 (Sigma number A8592, 1:1000) to detect the Flag-tagged Bag3 P209L protein, anti-serum raised against purified GST-BAG3 recognizing both human/mouse BAG3, and anti-actin (Calbiochem Corp., catalogue number CP01, Ab-1/JLA20). Western analysis of ventricular tissue was performed on lysates generated from approximately 25 mg tissue placed in 8 mol/L Urea Lysis Buffer (8 mol/L Urea Sigma, catalogue number U0631, 5 mol/L NaH2PO4 Sigma, catalogue number S3139, 1 mol/L Tris-Cl, pH 8.0) at a ratio of 15 μL lysis buffer/mg of tissue and was homogenized at 4°C (TissueLyser LT, Qiagen, catalogue number 85600) for 2 minutes. The homogenate was centrifuged at 4°C, 16,000 × g for 15 minutes, and the supernatant stored at −80°C. Protein concentration was determined using the Bio-Rad DC Protein Assay Reagent Package (Bio-Rad Laboratories, Inc., Hercules, CA, catalogue number 500-0116). Proteins (30 to 50 μg/lane) were resolved on NuPAGE Bis-Tris or Tris-Acetate 10- and 15-well gels. Rabbit anti-ubiquitin (Sigma L7543, 1:500) was used to measure changes in ubiquitination. Mouse anti–NF-κB p65 (total) and rabbit anti–phospho-NF-κB p65 (Ser536) were used to measure NF-κB signaling (Cell Signaling Technologies, catalogue number 4767, each 1:500). Rabbit anti-LC3 (Sigma L7543, 1:500) was used to measure autophagic flux. Rabbit anti-p38 and phospho-p38 (Cell Signaling, 9212, 9211, each 1:1000) was used to measure p38 activation. Rabbit anti-PRDX1 and anti-SSADH (Sigma SAB2101877 and PRS4807, each 1:1000) were used to detect periredoxin and succinic semialdehyde dehydrogenase. Primary antibodies were diluted in 1× Tris-buffered saline and Tween 20 or 4% bovine serum albumin and incubated at 4°C overnight. Horseradish peroxidase–labeled secondary antibodies against mouse (Sigma A9917, 1:10,000), goat (Sigma A4174, 1:10,000), and rabbit (Sigma A9169, 1:5000) were used to detect the primary antibodies diluted in 1× Tris-buffered saline and Tween 20 and incubated 1 hour at room temperature. Mouse anti–β-actin (Sigma, Inc., catalogue number A2228, 1:4000) or mouse anti–glyceraldehyde-3-phosphate dehydrogenase (Sigma, Inc., catalogue numberG8795, 1:5000) were used as a loading control. Secondary antibody horseradish peroxidase was detected using ECL Select (GE Healthcare, catalogue number RPN2235) and imaged using the MultiDoc-it Imaging System (UVP, LLC, Upland, CA).
RNA Isolation and Quantitative PCR Analysis of Gene Expression, Mitochondrial Dynamics
Total RNA was isolated using TRIzol reagent, according to the manufacturer's protocols (Life Technologies, Inc., catalogue number 15596-026). Approximately 25 mg of cardiac ventricular tissue was put into TRIzol reagent and homogenized at 4°C (TissueLyser LT, Qiagen, catalogue number 85600). Total mRNA expression was determined using a two-step reaction. cDNA was made from total RNA using the iScript Reverse Transcription Supermix for RT-qPCR kit (catalogue number 170-8841, Bio-Rad), with a total volume of 20 μL per reaction. The complete reaction mix was incubated in an Eppendorf Cycler (Hamburg, Germany) using the following protocol: priming 5 minutes at 25°C, reverse transcription 30 minutes at 42°C, room temperature inactivation 5 minutes at 85°C. PCR products were amplified on a Roche Lightcycler 480II system using cDNA, Taqman Probes (Applied Biosciences), and Lightcycler 480 Probe Master Mix 2X (catalogue number 04 707 494 001). The TaqMan probes used in this study were Hs99999901_s1 (18S), Mm00600555_m1 (β-MHC, Myh7), Mm01255747_g1 (ANF), Mm00435304_g1 (BNP), Mm00808218_g1 (Acta1, skeletal muscle α1-actin), Mm01278171_m1 (mouse Bag3, spanning exons 2 to 3), Hs00188713_m1 (human Bag3, spanning exons 1 to 2), Opa1 (Mm00453879_m1), Drp1 (Mm01342903_m1), Fis1 (Mm00481580_m1), and Mfn1 (Mm00612599_m1) (Applied Biosystems, Inc., Foster City, CA). Samples were run in triplicate, and relative mRNA expression was determined using 18S as an internal endogenous control. RNase-free water, 2X Master Mix, Taqman Probe, or Roche UPL primer and probe, and cDNA were used for each reaction.
Protein Aggregation Assay and Preamyloid Oligomer Staining
Protein aggregation was assessed using an Enzo Proteostat Protein Aggregation Assay (ENZ-51023, Farmingdale, NY) in conjunction with their preformulated aggregation standards (ENZ-51039). Briefly, protein lysates were prepared from flash-frozen apical cardiac tissues using 8 mol/L urea lysis buffer, protein concentrations were determined by Bradford assays, and assays were performed in 96-well, black-walled, clear-bottom plates read on a Clariostar microplate reader at 550-nm excitation and 600-nm emission with gain setting between 1000 and 2000. Preamyloid oligomer staining was performed as previously described using the A11 and MF20 antibodies.26
Nontargeted Metabolomics Determination by Gas Chromatography–Mass Spectrometry Instrumentation
Cardiac tissue was flash frozen with liquid nitrogen cooled in a biopress, a fraction weighed (approximately 25 to 30 mg weight), finely ground, and added to fresh 50% acetylnitrile, 50% water, and 0.3% formic acid at a standard concentration of 25 mg/475 mcl buffer, then fully homogenized on ice for 10 to 25 seconds and placed on dry ice/stored at −80°C. Samples were crash deprotonized by methanol precipitation and spiked with D27-deuterated myristic acid (D27-C14:0) as an internal standard for retention-time locking and dried. The trimethylsilyl (TMS)-D27-C14:0 standard retention time was set at *16.727 minutes. Reactive carbonyls were stabilized at 50°C with methoxyamine hydrochloride in dry pyridine. Metabolites were made volatile with TMS groups using N-methyl-N-(trimethylsilyl) trifluoroacetamide or MSTFA with catalytic trimethylchlorosilane at 50°C. Gas chromatography–mass spectrometry methods were adopted from Roessner et al,27 Fiehn et al,28 and Kind et al,29 which used a 6890 N gas chromatograph connected to a 5975 Inert single quadrupole mass spectrometer (Agilent Technologies, Santa Clara, CA). The two wall-coated, open-tubular gas chromatography columns connected in series are both from J&W/Agilent (part 122–5512), DB5-MS, 15 m in length, 0.25 mm in diameter, with an 0.25-lm luminal film. Positive ions generated with conventional electron-ionization (EI) at 70 eV are scanned broadly from 600 to 50 m/z in the detector throughout the 45-minute cycle time. Data were acquired and analyzed as previously described.30, 31 All raw data and transformed data included in the analysis are included in Supplemental Tables S1 and S2, respectively.
Thioredoxin Reductase Assay
Flash-frozen cardiac tissue (approximately 20 mg) was homogenized at a ratio of 10 μL lysis buffer/mg of tissue according to the manufacturer's protocols to prepare assay homogenate (TissueLyser LT, Qiagen, catalogue number 85600). Protein concentration was determined using the Pierce 660 nm Protein Assay Reagent (Thermo Fisher Scientific, catalogue number 22660) and assayed for thioredoxin reductase activity using a Thioredoxin Reductase Assay (Sigma, catalogue number CS0170) according to the manufacturer's protocol. Protein (100 μg) was incubated on a U bottom 96-well plate with DTNB containing reaction mix in the presence or absence of a thioredoxin reductase–specific inhibitor and read at 412 nm using a Clariostar microplate reader.
Statistical Analysis
Prism 6 for Mac OS X (Version 6.0f, August 5, 2014) or SigmaPlot for Windows (Version 11.0 Build 11.0.0.77) was used to plot and statistically analyze data using a t-test with significance defined as P < 0.05. Statistical analysis on metabolomics data was performed as previously described.30, 31 Briefly, Metaboanalyst (v2.0) run on the statistical package R (v2.14.0) used metabolite peak areas (as representative of concentration).32, 33 These data were first analyzed by an unsupervised principal component analysis. To sharpen the separation between groups, data were next analyzed using a partial least squares discriminant analysis to further determine which metabolites were responsible for separating these two groups. The specific metabolites contributing most significantly to the differences identified by partial least squares discriminant analysis between Bag3 P209L and wild-type control group hearts were determined using the variable importance in projection (VIP) analysis in the Metaboanalyst environment. The metabolites that best differentiated the groups were then individually tested using the t-test (Microsoft Excel 2011, Seattle, WA). The VIP and t-test significant metabolites were matched to metabolomics pathways using the Pathway Analysis feature in Metaboanalyst 2.0. Heat maps of the metabolite data (individual and grouped) were generated using the GENE E software version 3.0.2014 (http://www.broadinstitute.org/cancer/software/GENE-E/index.html; last accessed February 20, 2016).
Results
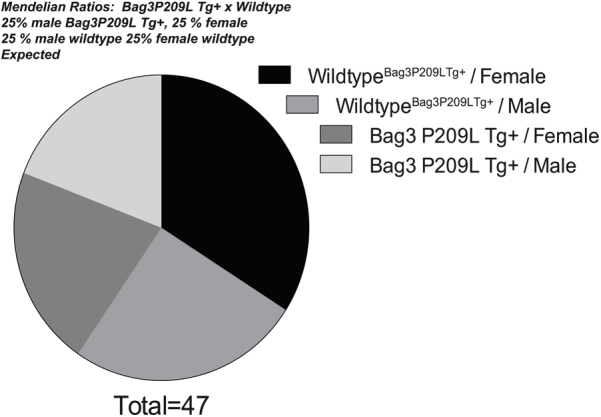
The BAG3 P209L missense mutation in exon 3 is one of the most recently identified causes of myofibrillar myopathy resulting in cardiac complications.10 In contrast to other myofibrillar myopathies, the BAG3 cardiomyopathy manifests as hypertrophic cardiomyopathy in the first to second decades of life.5, 10 Bag3 constitutive expression is restricted to few cell types, including skeletal muscle and cardiomyocytes.2 However, it is induced in a subset of cells (retinal, glial, epithelial, leukocytes) by a variety of stressors to induce resistance.34, 35, 36, 37 To generate a cardiac model that paralleled human disease without the indirect effects of a skeletal phenotype, including respiratory distress and limited mobility, we generated an αMHC-driven BAG3 P209L transgenic mouse. The resulting mouse exhibited a modest constitutive increase in total BAG3 expression (approximately twofold wild-type levels) (Figure 1A), localized to the heart (cardiomyocyte). Because we crossed Bag3 P209L Tg+ with wild-type mice, we looked for evidence of prenatal death by comparing the expected 50% ratios with the observed. There was no evidence that the Bag3 P209L Tg+ offspring were underrepresented, with >50% Bag3 P209L Tg+ by genotyping (Figure 1B and Table 1). The distribution of offspring with Bag3 P209L Tg+ and sex similarly did not differ between groups (Supplemental Figure S1 and Supplemental Table S3), demonstrating that the Bag3 P209L Tg+ did not alter viability during development. Body weight from 4 months of age through 12 months was not significantly different between wild-type Bag3 P209L Tg+ mice, as shown in Table 2 (row 1).
Figure 1.

Cardiac function and natural history of αMHC-Bag3 P209L transgenic mouse cardiac phenotype up to 12 months of age. A: Western blot analysis of Bag3 P209L expression by anti-FLAG (FLAG-Bag3 P209L) demonstrating specificity and by anti-BAG3 (human and mouse) to compare relative levels of total BAG3 in cardiac and gastrocnemius tissue from αMHC-Bag3 P209L and littermate wild-type mice. B: Mendelian ratios of offspring at the time of genotyping at 14 to 21 days post-partum. C: Kaplan-Meier survival curves of Bag3 P209L Tg+ and sibling wild-type mice. D–G: Temporal changes in fractional shortening %, calculated as (LVEDD-LVESD)/LVEDD × 100, see Table 2 (D), left ventricular end diameter; diameter (E), anterior wall thickness in diastole (AWTD) (F), and left ventricular mass (G), corresponding to the complementary morphometrics in Table 2. H: Measured total heart weight (normalized to tibia length) at 1 year of age. I: Analysis of diastolic function made from Doppler measures of mitral valve at 1 year of age. A t-test was used to compare genotypes at each time point. Data are expressed as means ± SEM (D–I). N = 47 (B); N = 68 (C); N = 3 (D–G, baseline Bag3 P209LTg+ and 5 months Bag3 P209LTg+); N = 4 (D–G, 8 months Bag3 P209LTg+); N = 5 (D–G, baseline wild-typeBag3 P209L Tg+ and 5 months wild-typeBag3 P209L Tg+); N = 7 (D–G, 8 months wild-typeBag3 P209L Tg+); N = 10 (D–G,12 months wild-typeBag3 P209L Tg+ and BAG3 P209LTg+); N = 4 (H, Bag3 P209LTg+); N = 7 (H, wild-typeBag3 P209L Tg+); N = 3 per group (I). ∗P < 0.05, ∗∗P < 0.005 versus age-matched sibling wild-type mice. IB, immunoblot; LV, left ventricle; LVEDD, left ventricular end-diastolic diameter.
Table 1.
Analysis of Mendelian Ratios Breeding Bag3 P209L Tg+ with Strain-Matched Wild-typeBag3 P209L Tg+ Mice (50% Expected Tg+)
| Outcome | Expected (no.) | Observed (no.) | Expected (%) | Observed (%) | 95% CI of observed % |
|---|---|---|---|---|---|
| Wild-typeBag3 P209L Tg+ | 23.5 | 28 | 50 | 59.57 | 44.27–73.63 |
| Bag3 P209L Tg+ | 23.5 | 19 | 50 | 40.43 | 26.37–55.73 |
| Total | 47 | 47 | 100 | 100 |
A binomial test was performed and a one-tailed and two-tailed P was determined to be not significant (0.1215 and 0.2430, respectively). See Figure 1B for pie chart of distribution.
Table 2.
High-Resolution Transthoracic Echocardiography Performed on Conscious Bag3 P209L Tg+ and Strain-Matched Sibling Controls over 1 Year
| Variable | Baseline wild-typeBag3 P209L Tg+ control | Baseline Bag3 P209L Tg+ | 5 Months wild-typeBag3 P209L Tg+ control | 5 Months Bag3 P209L Tg+ | 8 Months wild-typeBag3 P209L Tg+ control | 8 Months Bag3 P209L Tg+ | 12 Months wild-typeBag3 P209L Tg+ control | 12 Months Bag3 P209L Tg+ |
|---|---|---|---|---|---|---|---|---|
| N | 5 | 3 | 5 | 3 | 7 | 4 | 10 | 10 |
| Body weight (g) | 27.2 ± 1.8 | 23.4 ± 1.8 | 28.5 ± 1.7 | 25.5 ± 2.8 | 35.6 ± 2.4 | 28.6 ± 2.6 | 37.2 ± 2.1 | 38.8 ± 3.2 |
| AWTS (mm) | 1.74 ± 0.05 | 1.68 ± 0.03 | 1.77 ± 0.05 | 1.73 ± 0.06 | 1.82 ± 0.04 | 1.64 ± 0.05∗ | 1.81 ± 0.04 | 1.74 ± 0.04 |
| LVESD (mm) | 1.83 ± 0.06 | 1.85 ± 0.15 | 1.9 ± 0.11 | 1.87 ± 0.16 | 1.76 ± 0.06 | 2.33 ± 0.12∗ | 1.83 ± 0.03 | 2.28 ± 0.05∗ |
| PWTD | 0.89 ± 0.01 | 0.97 ± 0.02∗ | 0.96 ± 0.03 | 1.03 ± 0.02 | 1.06 ± 0.02 | 0.99 ± 0.04 | 1.06 ± 0.02 | 0.99 ± 0.04∗ |
| PWTS | 1.45 ± 0.09 | 1.40 ± 0.06 | 1.47 ± 0.05 | 1.43 ± 0.09 | 1.65 ± 0.04 | 1.50 ± 0.06 | 1.65 ± 0.04 | 1.50 ± 0.06 |
| LV %EF | 81.3 ± 0.9 | 78.6 ± 0.9 | 80.1 ± 1.6 | 77.5 ± 2.9 | 80.8 ± 0.8 | 69.2 ± 1.1∗ | 80.5 ± 0.6 | 64.2 ± 1.1∗ |
| LV Mass (mg) | 121.8 ± 5.0 | 124.0 ± 16.1 | 127.2 ± 11.3 | 129.8 ± 7.3 | 137.3 ± 8.2 | 145.1 ± 10.2 | 137.3 ± 8.1 | 145.1 ± 10.2 |
| LV Vol; d (mL) | 54.4 ± 4.1 | 49.2 ± 8.3 | 57.0 ± 6.5 | 47.7 ± 4.5 | 49.0 ± 4.9 | 61.8 ± 8.3 | 51.9 ± 1.7 | 64.8 ± 2.3∗ |
| LV Vol; s (mL) | 10.2 ± 0.9 | 10.7 ± 2.1 | 11.4 ± 1.6 | 11.0 ± 2.5 | 9.3 ± 0.9 | 19.0 ± 2.6∗ | 10.1 ± 0.4 | 17.8 ± 1.1∗ |
| PWTH† (%) | 63.6 ± 10.0 | 44.7 ± 5.6 | 53.8 ± 3.8 | 39.2 ± 6.2 | 55.9 ± 4.2 | 52.8 ± 9.4 | 59.7 ± 4.5 | 33.6 ± 2.0∗ |
| RWT; d‡ | 0.52 ± 0.02 | 0.58 ± 0.04 | 0.52 ± 0.03 | 0.61 ± 0.03 | 0.62 ± 0.03 | 0.53 ± 0.04 | 0.60 ± 0.01 | 0.66 ± 0.02∗ |
| RWT; s§ | 1.75 ± 0.09 | 1.69 ± 0.11 | 1.73 ± 0.14 | 1.72 ± 0.16 | 1.98 ± 0.09 | 1.35 ± 0.06∗ | 1.91 ± 0.04 | 1.44 ± 0.04∗ |
| HR (bpm) | 645 ± 14 | 713 ± 12 | 677 ± 17 | 663 ± 2 | 683 ± 9 | 679 ± 20 | 671 ± 12 | 672 ± 10 |
Baseline echos taken at 4 months of age. Approximately 50% female and approximately 50% male mice were assayed throughout the study. Data represent means ± SEM. A t-test was performed comparing Bag3 P209L Tg+ to sibling wild-type controls.
∗P < 0.05.
AWTD, anterior wall thickness in diastole; AWTS, anterior wall thickness in systole; bpm, heart beats per minute; d, in diastole; EF%, ejection fraction calculated as (end Simpson's diastolic volume – end Simpson's systolic volume)/end Simpson's diastolic volume * 100; ExLVD, external left ventricular diameter; FS, fractional shortening, calculated as (LVEDD − LVESD)/LVEDD × 100; HR, heart rate; LVEDD, left ventricular end-diastolic dimension; LVESD, left ventricular end-systolic dimension; ND, not determined; PWTD, posterior wall thickness in diastole; PWTH, posterior wall thickness; PWTS, posterior wall thickness in systole; RWT; relative wall thickness; s, in systole; vol, volume.
PWTH = [(LVPW; s − LVPW; d)*100%/LVPW; d].
RWT; d = LVPW; d + LVAntW; d/LVEDDd.
RWT; s = LVPW; s + LVAntW; s/LVEDDs; LVPW; d.
Because the Bag3 P209L Tg+ was expressed specifically in cardiomyocytes, we focused our analysis on cardiac function, performing conscious echocardiography over time, starting at 4 months of age and continuing through 1 year, along with mechanistic studies. Postnatally, we identified only one mouse that died prematurely, which was a Bag3 P209L Tg+ mouse on day 243 (Figure 1C). Unfortunately, we were unable to perform a necropsy to determine any contributions of the mutation to its death, but did confirm we had identified systolic dysfunction by echocardiography prior. By 8 months of age, significant systolic dysfunction was seen, evidenced by decreases in fractional shortening (Figure 1D) and ejection fraction (Table 2). Although Bag3 P209L Tg+ hearts demonstrated significant dilation at 8 months (Figure 1E) and had significantly increased anterior wall thickness (Figure 1F), there was no difference in LV mass determined by echocardiographic measurements (Figure 1G) or measured heart weight at 12 months of age (Figure 1H).
Of the six genes found to be mutated and leading to myofibrillar myopathies [desmin, aB-crystallin, myotilin, LDB3 (ZASP), FLNC, and BAG3], only 15% to 30% of patients with these mutations present with a cardiomyopathy.5 As the disease progresses, up to 60% develop cardiomyopathies, including dilation (17%), restrictions (12%), and/or hypertrophic (6%) cardiomyopathy.38 The first case of a Bag3 P209L mutation reported in 2009 identified a childhood onset with severe progressive muscle weakness with atrophy, along with a restrictive cardiomyopathy pattern.5, 10 Because of the restrictive cardiomyopathy pattern, we hypothesized that the Bag3 P209L Tg+ mice had exhibited diastolic dysfunction, being unable to relax. We performed Doppler analysis on the mitral valve in Bag3 P209L Tg+ mice at 12 months of age and determined an E/a waveform ratio (Figure 1I). Wild-type mice exhibited a dynamic relation during the cardiac cycle, exhibited an E/a ratio of nearly 2.0, consistent with healthy human heart function generally around 1.5 to 2.0 E/a ratio. In contrast, the Bag3 P209L Tg+ hearts had a reduced diastolic function, with an E/a ratio of approximately 1.0 (Figure 1I).
The severity of the heart failure was limited in the Bag3 P209L Tg+ hearts, because there was no evidence of congestive heart failure, such as fluid accumulation in the lungs (Figure 2A). Blinded cross-sectional analysis (M.T.Q.) of Masson's trichrome–stained histology revealed that the Bag3 P209L Tg+ hearts exhibited a significant increase in cardiomyocyte size (Figure 2B), with no evidence of increased fibrosis, quantified by algorithmic analysis of collagen (Figure 2C). Transmission electron microscopy revealed no alterations or abnormalities in the Bag3 P209L sarcomere structures throughout the heart (Figure 2D). Taken together, the Bag3 P209L Tg+ hearts were normal at birth, and progressively developed both systolic and diastolic heart failure at 1 year of age, with no evidence of prenatal lethality and minimal mortality (3.7% = 1/27).
Figure 2.

Morphometric analysis of αMHC-Bag3 P209L transgenic hearts. A: Heart weight and lung weights at 12 months of age. B: Cross-sectional analysis of cardiomyocytes from Masson's trichrome–stained histological sections. C: Analysis of collagen in αMHC-Bag3 P209L and littermate wild-type hearts by nonbias computer logarithm analysis of collagen as a measure of fibrosis (N = 6 wild-typeBag3 P209L Tg+, N = 4 Bag3 P209L Tg+). D: Transmission electron microscopy analysis of αMHC-Bag3 P209L and littermate wild-type mice at ×5 and ×20,000 (N = 3/group). A t-test was used to compare genotypes at each time point. Measurements represent means ± SEM (A–C). N = 3 per group (A–C). ∗∗∗P < 0.001. Scale bars: 0.5 μm (D, top row); 1 μm (D, bottom row). Original magnification: ×20 (B); ×10 (C).
To better understand the mechanistic cause of the observed cardiac dysfunction, we next investigated how alterations in autophagy, also recognized as the lysosomal degradation pathway, may be related to the cardiac dysfunction seen in the Bag3 P209L Tg+ hearts. The primary reason autophagy was of interest in this model was the fact that wild-type Bag3 has recently been implicated in the regulation of chaperone-assisted selective autophagy in muscle.14, 39 In these studies, the protein Starvin and Bag3 coordinated machinery were required for the maintenance of the Z disk while Bag3 coordinated the connection between HSC70, the small heat shock protein HspB8, and the ubiquitin ligase CHIP with the autophagic ubiquitin adaptor p62.40 Second, mutations in other myofibrillar proteins such as αB crystallin (eg, CryAB R120G) have been shown to induce endoplasmic reticulum stress41 and increase autophagy to enhance the removal of proteotoxic misfolded proteins.42 We first investigated if amyloid fibrils were present by measuring amyloid using a dye-based Proteostat assay to measure protein aggregation. In Bag3 P209L hearts expressing the transgenic protein at 12 months of age (Figure 3A), no increase in protein aggregation was detected in heart lysates compared with wild-typeBag3 P209L Tg+ mice in vivo (Figure 3B). We next investigated if alterations in autophagic flux were present by quantifying the amount of LC3II levels in the heart. No significant differences were seen between Bag3 P209L Tg+ and age-matched wild-type hearts at 12 months of age (Figure 3C) when treated with and without bafilomycin A1 treatment, using doses previously described to accumulate stop lysosomal degradation23, 42, 43 (Figure 3D). Quantitative measurement of genes involved in the autophagic process (mRNA) was then analyzed for evidence of altered autophagy (Figure 3E). No differences in Atg5, Atg7, Atg12, Bnip3, LC3, or Vps34 mRNA were identified compared to wild-type hearts (Figure 3E). Although mutations in other myofibrillar proteins causing cardiomyopathy enhance autophagy, the Bag3 P209L mutation did not alter this lysosomal degradation pathway significantly. Alterations in cardiac function do not appear to be related to changes in autophagic flux in this model.
Figure 3.

Analysis of protein aggregate and autophagic flux in αMHC-Bag3 P209L transgenic hearts at 12 months. A: Immunofluorescence detection of FLAG-Bag3 P209L in transgenic, but not wild-type, hearts. B: Colorimetric staining of protein aggregation. C: Western blot analysis of cardiac autophagic flux from αMHC-Bag3 P209L and littermate wild-type mice at 12 months of age. D: Western blot analysis demonstrating that bafilomycin induction is adequate. E: Real-time quantitative RT-PCR analysis of Atg5, Atg7, Atg12, Bnip3, LC3, and Vps34, mediating autophagy. A t-test was used to compare genotypes at each time point. Measurements represent means ± SEM (B, C, and E). N = 6 per group (B and E); N = 3 per group (C). IB, immunoblot; IF, immunofluorescence.
An essential underlying mechanism of heart failure is the loss of myocytes by apoptosis.44, 45 In tumor cell lines, silencing of Bag3 enhances apoptosis, whereas increasing Bag3 inhibits spontaneous or drug-induced apoptosis.37, 46, 47, 48, 49, 50, 51, 52, 53 Different mechanisms have been identified in cancer, including Bag3's protection of IKKγ from proteasome delivery promoting cell survival.37 We next investigated the potential role of apoptosis in the Bag3 P209L Tg+ hearts by measuring cardiac apoptosis by TUNEL and AIF immunofluorescence (Figure 4A). TUNEL positivity was investigated in both fibroblasts (TUNEL+, vimentin+) and in nonfibroblast cells (TUNEL+, vimentin negative) made up primarily of cardiomyocytes. Significantly less apoptosis (TUNEL+) was seen in the Bag3 P209L Tg+ fibroblasts (Figure 4A), whereas Bag3 P209L Tg+ cardiomyocytes (TUNEL+ and vimentin negative cells) trended higher compared to sibling wild-type controls at 1 year of age (Figure 4B). No differences in AIF staining were seen in between Bag3 P209L Tg+ hearts and sibling-matched wild-type controls (Supplemental Figure S2). This led us to further analyze the fibroblast (Vimentin+) characteristics histologically (Figure 4B). Unexpectedly, we identified that the Bag3 P209L Tg+ hearts exhibited more vimentin+ cells compared to wild-type hearts (Figure 4B). Hearts were analyzed in quadrants as outlined in Figure 4C, with quadrant 2 and quadrant 3 (Q2 and Q3) including primarily the left ventricle and quadrant 1 and quadrant 4 (Q1 and Q4) making up the right and upper left ventricle and septum. Blinded quantitative analysis (C.C.Y.) of vimentin (+) cells from multiple animals identified a consistent and significant increase in the left ventricle (Figure 4A), which was distinctly different from that found in Q1 and Q4 (Supplemental Figure S3). Identification of vimentin in parallel studies of cardiac ventricles in Bag3 P209L Tg+ hearts at 12 months illustrates that approximately fivefold more vimentin is present (Figure 4D). Qualitatively, these vimentin (+) cells in the Bag3 P209L Tg+ hearts were distributed around the vasculature, which may represent smooth muscle or fibroblast pericytes (Figure 4B). This vascular distribution of vimentin (+) cells was not seen in any of the age-matched wild-type controls (Figure 4B). Together, these findings indicate that the Bag3 P209L Tg+ hearts do not have increases in cardiomyocyte apoptosis at 12 months of age, but have an enhanced fibroblast presence in the left ventricle and an increase in pericytes. With the ability to inhibit vessel formation and induce vessel dissociation,54 their role in the heart has been attributed to their contributions to tissue repair.55, 56
Figure 4.

αMHC-Bag3 P209L transgenic hearts have increased left ventrical (LV) fibroblast areas in a perivenular distribution and decreased fibroblast apoptosis. A: Costaining of TUNEL positive nonfibroblast (left) and fibroblast (vimentin positive) cells by immunohistochemistry. B: Representative confocal immunofluorescence images of cardiac fibroblasts from histological sections of αMHC-Bag3 P209L and littermate wild-type mice at 12 months of age. Shown are quadrant 2 and quadrant 3, tracking with the LV (quadrant 1 and quadrant 1 primarily right ventricle). C: Nonbias quantitative logarithmic analysis of vimentin (+) cell staining in quadrants 1 to 4. D: Western immunoblot analysis of cardiac vimentin in Bag3 P209L hearts compared to wild-type hearts at 12 months of age. A t-test was used to compare genotypes at each time point. Measurements represent means ± SEM (A). N = 12 (A, wild-typeBag3 P209L Tg+); N = 6 (A, Bag3 P209L Tg+ cardiac sections). ∗P < 0.05 versus age-matched sibling wild-type mice. Scale bars = 200 μm (B). IHC, immunohistochemistry; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick-end labelling. AU, arbitrary units; IB, immunoblot; IF, immunofluorescence.
Recent studies have linked the presence of pre-amyloid oligomers to cardiac dysfunction and heart failure. Mutations in the cardiac myofibrillar protein CryAB, another small heat shock protein, have been shown to result in the accumulation of proteotoxic misfolded protein, characterized by reactivity to an amyloid recognizing antibody.17, 18, 57 Interestingly, antibodies recognizing amyloid conformations have been shown to react similarly in multiple causes of human heart failure.17 Subsequently, cardiac expression of a misfold prone protein [polyglutamate 83 (PQ83) repeat] was compared to the cardiac expression of a protein not prone to misfold (PQ19 repeat) to demonstrate the proteotoxicity of these proteins in cardiomyocytes.16 The resulting PQ83 mice progressed rapidly into heart failure and death, whereas the PQ19 were entirely unaffected.16 The PQ83 hearts had cardiomyocyte accumulation of the preamyloid oligomers (PAOs), whereas the PQ19 did not, illustrating the causative role of the misfolded proteins in heart failure that are identified as PAOs.16 This led us to hypothesize that the Bag3 P209L Tg+ heart failure may be mediated by the accumulation of PAO. Immunohistochemistry of PAO (recognized by the A11 antibody) colocalizing with cardiomyocytes (recognized by the MF20 antibody) was performed on Bag3 P209L Tg+ heart sections (Figure 5A). Although gross histological analysis did not reveal overt differences in the Bag3 P029L Tg+ hearts (Figure 5B), differences in infiltrating immune cells were detected on higher-power evaluation (Figure 5C). When quantified by nuclei counts using an algorithm recognizing nuclei as a function of area, significantly more nuclei were present in the Bag3 P029L Tg+ hearts (Figure 5D), consistent with an increase in infiltrates, likely immune cells. Several hypotheses have emerged concerning the cellular mechanisms by which misfolded proteins trigger inflammation, including the direct activation of the inflammasome by misfolded proteins (eg, amyloid-β, amylin, serum amyloid A, and prions) that could trigger downstream IL-1, caspase activation, and heart failure.58
Figure 5.

Analysis nuclei in αMHC-Bag3 P209L transgenic hearts at 12 months. A: Immunohistochemistry analysis of preamyloid oligomers (PAO; anti-A11 antibody), cardiomyocytes (anti-MF20 antibody), and IgG controls showing representative green (PAO) as a percentage of red (cardiomyocytes) in cardiac sections. B: Gross comparison of αMHC-Bag3 P209L and littermate wild-type mice [hematoxylin and eosin (H&E) stained). C: Algorithmic analysis of nuclei quantitated per area of cross-sectional area. D: Representative Bag3 P209L Tg+ hearts and age-matched sibling wild-type hearts with cellular infiltrates. Representative histological section of wild-typeBag3 P209L Tg+ heart stained with H&E (D) and parallel representative cross sections of sibling Bag3 P209L Tg+ hearts with inflammatory cell infiltrates (arrows) at 12 months of age (E). A t-test was used to compare genotypes at each time point. N = 3 Bag3 P209L Tg+ (A); N = 4 Bag3 P209L Tg+ biological replicates, 3 cross-sectional areas each (A and B); N = 5 wild-typeBag3 P209L Tg+ (A); N = 3 wild-typeBag3 P209L Tg+ (C). ∗P < 0.05 versus age-matched sibling wild-type mice. Original magnification: ×20 (A, D, and E); ×0.7 (B).
In neurodegenerative diseases where amyloidogenic proteins, including PAOs, mediate proteotoxicity, abnormal mitochondrial dynamics composed of fission and fusion events have been reported.59, 60 One of the distinctive findings by TEM of the Bag3 P209L Tg+ hearts was the apparent dysregulation of the mitochondrial sizes, briefly illustrated in Figure 2D. We next analyzed the TEM images in more detail, assaying sections throughout the ventricles of the Bag3 P209L Tg+ and wild-type hearts (Figure 6). We identified an apparent increase in mitochondria by manual counting of TEM images (Figure 6A), but only mild decreases in area (Figure 6B). These manual counts were complemented by quantitative analysis of mitochondrial genes normalized to a nuclear intron (H19) (Figure 6C). Although mitochondrial NADH dehydrogenase 1 (ND1), cytochrome-b (Cytb), and cytochrome-c oxidase 1 (CO1) were increased in the Bag3 P209L hearts, significant increases were found only in ND1, consistent with a nearly doubling of the mitochondrial number (Figure 6C). Mitochondrial dynamics are regulated by a number of key proteins encoded by mitofusins (Mfn1, Mfn2) and optic atrophy (Opa1) to regulate mitochondrial fusion and dystrophin-related protein 1 (Drp1) and fission 1 homolog (Fis1) that regulate mitochondrial fission.61 We identified that the Bag3 P209L Tg+ hearts had significant decreases in the mitochondrial fusion Opa1 mRNA (Figure 6D) and the mitochondrial fission Drp1 mRNA (Figure 6E). No differences in Bag3 P209L cardiac Mfn1 or Fis1 were present compared to wild-type hearts (Figure 6, D and E). With an increased number of smaller mitochondria, Bag3 P209L Tg+ hearts exhibited significant alterations in Opa1 and Drp1 involved in regulating fusion and fission, respectively.
Figure 6.

αMHC-Bag3 P209L transgenic hearts exhibit alterations in mitochondria number and dynamics. Quantitative analysis of mitochondrial number (A) and mitochondrial area (B) in ×5000 TEM sections. C: Quantitative analysis of the mean cytochrome c oxidase subunit 1 (CO1, aka mt-CO1)/cytochrome b (Cyt-b, aka mt-Cyb)/NADH dehydrogenase 1 (ND1, aka mt-nd1) normalized to nuclear H19 (imprinted maternally expressed transcript, non-protein coding) of αMHC-Bag3 P209L and littermate wild-type mice at 12 months of age. Real-time quantitative RT-PCR (RT-qPCR) analysis of genes regulating mitochondrial fusion (mitofusin 1/Mfn1 and optic atrophy1/opa1) (D) and mitochondrial fission (mitochondrial fission 1 protein/Fis1 and dynamic-related protein 1/Drp1) (E). A t-test was used to compare genotypes at each time point. Data are expressed as means ± SEM. N = 4 (A and B, Bag3 P209L Tg+); N = 5 per group (C); N = 6 per group (A and B, wild-typeBag3 P209L Tg+, and D and E). ∗P < 0.05 versus age-matched sibling wild-type mice. nu-DNA, nuclear DNA; qPCR, real-time quantitative PCR.
Alterations in mitochondria led to a nontargeted metabolomics analysis to determine any gross metabolic changes in the Bag3 P029L Tg+ hearts at 12 months of age (Figure 7A). By principal components analysis, the Bag3 P029L Tg+ hearts had a distinct signature from the wild-type hearts (Figure 7B), with 11 metabolites significantly different by t-test (Figure 7C and Table 3). A partial least squares discriminant analysis was also run to differentiate the two groups (Figure 7D) by identifying unique metabolites of interest, on the basis of their known relationships indicated by their VIP score (Figure 7E). Together, these studies found that Bag3 P209L Tg+ hearts had elevated aspartic acid and dehydroascorbic acid and decreased taurine, O-methylphosphate, and adenosine-5-monophosphate (AMP). Pathway enrichment analysis of both t-test and VIP identified metabolites found pathways involved in d-glutamine metabolism, taurine metabolism, and alanine, aspartate, and glutamate metabolism had the highest impact (Supplemental Figure S4A and Supplemental Table S4). Disease association enrichments identified that these metabolites were most closely associated (lowest P value) with the heart failure metabolomics reference set, continuous ambulatory peritoneal dialysis, and tumor-bearing mice (Supplemental Figure S4B). These metabolites also tracked with locations within the cell involved in mitochondria, peroxisome, and skeletal muscle (lowest P value) (Supplemental Figure S4C).
Figure 7.

Nontargeted metabolomics analysis of αMHC-Bag3 P209L transgenic hearts at 12 months of age. A: Heat map representation of metabolites identified in αMHC-Bag3 P209L and littermate wild-type hearts at 12 months of age. B: Principal components (PC) analysis of αMHC-Bag3 P209L and littermate wild-type hearts. C:t-Test analysis of metabolites, including significant metabolites (above horizontal line), listed below with P-value and false discovery rate (FDR) estimate. D: Partial least squares discriminant analysis (PLS-DA) score plot demonstrating significant separation between groups. E: The variable influence on the projection (VIP) parameter to select variables that have the most significant contribution in discriminating between metabolomics profiles in a PLS-DA model. Statistical analyses were performed as described in Materials and Methods. Double asterisks denote fold-change inaccurate because of imputing a non-zero value resulting from the metabolite not being detected in one group. N = 3 biological replicates per group (A–E). WT, wild-type.
Table 3.
t-Test Significant Metabolites Identified by Nontargeted Metabolomics Analysis of Bag3 P029L Tg+ Heart versus Sibling-Matched Wild-TypeBag3 P209L Tg Controls
| t-Test significant | P-value | FDR |
|---|---|---|
| No ID 11 | 4.33 × 10−6 | 0.00037239 |
| Aspartic acid 1 | 0.0001159 | 0.0049839 |
| No ID 21 | 0.00081798 | 0.023449 |
| O-Methylphosphate | 0.0014924 | 0.032087 |
| Dehydroascorbic acid | 0.0043074 | 0.074088 |
| C13 Hydrocarbon | 0.0072195 | 0.10348 |
| Taurine | 0.010598 | 0.12587 |
| Aspartic acid 2 | 0.011709 | 0.12587 |
| No ID 14 | 0.021674 | 0.18684 |
| No ID 10 | 0.021726 | 0.18684 |
| Adenosine-5′-monophosphate | 0.045195 | 0.35334 |
Identification of metabolites indicated in Figure 7C. N = 3 hearts per group. FDR, false discovery rate.
Recent studies have identified that Bag3 mutations in dilated cardiomyopathy are predicted to result from loss-of-function mutations, in which knock-down of Bag3 in a zebrafish model recapitulated the observed heart failure.8 Because these studies implied that the loss of function, also known as haploinsufficiency, was because of the Bag3 mutations directly, we hypothesized that the Bag3 P209L transgene would have no effect on the endogenous wild-type Bag3 levels. Because the Bag3 P029L Tg+ mouse model described herein used a human Bag3 P209L transgene, we next assayed the endogenous levels of (mouse) Bag3 at 1 year of age. To our surprise, the endogenous Bag3 P209L mRNA levels (measured by qPCR of mouse Bag3 mRNA spanning exons 2 to 3) were suppressed to <20% of wild-type heart levels (Figure 8A) with evidence of the human Bag3 P209L present (Figure 8B). This paralleled decreased levels of total Bag3 protein recognizing both mouse and human P209L Bag3 (Figure 8C). There is limited information on how Bag3 is regulated transcriptionally, but involvement of the transcription factors cJun/cFos (AP1), NF-κB, and HSF1 has been reported and may be related to the observed findings.50, 62, 63, 64, 65
Figure 8.

αMHC-Bag3 P209L down-regulates wild-type BAG3 and activates P38 at 12 months of age. Cardiac expression of endogenous mouse Bag3 (A) and human Bag3 (B) mRNA expression analysis by real-time quantitative RT-PCR. Western blot analysis of total (mouse and human) Bag3 protein (C), phospho-p38/p38 (D), and ubiquitinated proteins from αMHC-BAG3 P209L and littermate wild-type mice (E) at 12 months of age. A t-test was used to compare genotypes at each time point. Measurements represent means ± SEM (A–E). N = 4 (A, wild-typeBag3 P209L Tg+); N = 5 (A and B, Bag3 P209L Tg+); N = 3 per group (C and D); N = 6 per group (B, wild-typeBag3 P209L Tg+, and E). ∗P < 0.05 versus age-matched sibling wild-type mice. IB, immunoblot; ND, not detected.
The Bag3 P209L transgenic hearts had several notable parallels with neurodegenerative disease, including an increase in pre-amyloid oligomers and a dysregulation of mitochondrial dynamics.59, 60 We hypothesized further that the Bag3 P209L Tg+ hearts exhibited p38 activation and an increase in ubiquitinated proteins, on the basis of recent discoveries in models of neurodegenerative diseases. Misfolded protein aggregates have been reported in aged Drosophila to activate p38 signaling66 and in neurons to activate both the unfolded protein response and p38 signaling.67, 68 Analysis of phospho-p38 protein identified that Bag3 P209L Tg+ hearts exhibited significant increases in activated p38 compared to wild-type hearts at 1 year (Figure 8D), with modest increases in ubiquitinated proteins (Figure 8E). Targeted activation of p38 mitogen-activated protein kinase in cardiomyocytes has been shown to induce heart failure, having negative inotropic and restrictive diastolic effects.69 Activation of p38 in cardiomyocytes has also been implicated in the inflammatory response, including IL-1b–induced COX-2 expression, PGE2 biosynthesis.70 Together, these findings suggest that the Bag3 P209L drives an increase in cardiomyocyte PAOs, which leads to p38 activation and increase protein ubiquitination, as is seen in neurodegenerative diseases in vivo.
Discussion
The 575 amino acid BAG3 protein is expressed predominantly in the heart and skeletal muscle and functions as a cochaperone with members of the heat shock protein family to facilitate multiple important cellular processes, including the removal of misfolded and degraded proteins, apoptosis by interacting with Bcl2, the structural integrity of the Z-disk by binding CapZ, and chaperone-associated selective autophagy.71 Single allele mutations in BAG3 have been associated with adult onset hereditary dilated cardiomyopathy. Significant decreases in BAG3 have been identified in end-stage failing human heart and in animal models of heart failure secondary to myocardial infarction and pressure overload.71 Herein, we report the first mammalian model of a single amino acid mutation of BAG3 (P209L) in exon 3 of Bag3 associated with the development of muscle disease with left ventricular dysfunction and heart failure first reported in 2009.10 We identified previously unreported underlying mechanisms, including altered mitochondrial dynamics (increased fragmented mitochondria), the presence of pre-amyloid oligomers, the development of wild-type BAG3 haploinsufficiency, and activation of p38 signaling, paralleling the emerging pathogenesis in other diseases of misfolded proteins, such as Alzheimer and Huntington diseases.
Many of the findings in the present study may be related to domain P209L mutation, as the domain is linked to the cell's general ability to clear misfolded proteins. BAG3 has five domains: i) the WW domain (with two tryptophan motifs = WW) (amino acids 21 to 55); ii) the IPV motif 1 (amino acids 87 to 101); iii) the IPV motif 2 (amino acids 200 to 213, where P209L is located); iv) the PXXP motif (amino acids 302 to 412); and v) the BAG motif (amino acids 420 to 499). Each of these domains binds specific proteins, each with distinct cellular processes. In the N-terminal end, the WW domain is involved in the induction of autophagy, IPV motifs (1 and 2) bind HSPB8 (and to a lesser extent, HSPB6), the Bcl2 binds the region around amino acid 440, and HSC70, HSP70, and CapZ bind the c-terminal BAG domain.12, 71, 72 Because the P209L mutation affects the IPV motif (amino acids 200 to 213), our findings that the Bag3 P209L hearts did not have altered autophagy (Figure 3) or apoptosis (Figure 4) mediated by the WW (amino acids 21 to 55) and Bcl2 binding region (amino acids 440), respectively, were not surprising because they are not related to this region. Similarly, a recent report identified that Bag3 P209L was as effective as wild-type BAG3 in enhancing LC3 lipidation and maintaining autophagic flux.73 Similarly, the lack of any alterations in the Bag3 P209L Tg+ heart sarcomeres (Figure 2D) is also consistent with the recent finding that the Bag3 P209L is able to rescue the myofibrillar disintegration found when Bag3 is knocked down in zebrafish models,13 demonstrating that the mutation does not have dominant negative activity and maintains structural integrity. More important, the IPV domains (where P209L is located) associates with critical HSPB proteins found necessary to degrade the Huntington disease–causing Htt43Q mutation, caused by its misfolding (ie, PAO) and proteotoxicity.72 Taken together, these findings implicate the Bag3 P209L mutation in producing a protein more prone to forming PAOs, or by altering the cells' ability to clear PAOs, or possibly both. Either way, the altered physiology leading to heart failure identified in the current study is mechanistically consistent with a proteotoxic mechanism in vivo.
Although the formation of pre-amyloid oligomers (Figure 5A) in the αMHC-Bag3 P209L parallel those seen in other small heat shock protein mutations, notably the CryAB R120G, many differences exist in the cardiac phenotypes that these mutant and native proteins have in vivo. At 12 months, the Bag3 P029L Tg+ heart does not have any increase in fibrosis, relatively mild heart failure with minimal death (only one recorded), and no classic fibril deposition by TEM. In contrast, the αMHC-CryAB R120G have 100% mortality by early adulthood, considerable fibril deposition, and impaired myofibril alignment.74 These differences may have only to do with the amount of protein present, secondary to its solubility and relationship to interacting proteins they chaperone, because the native small heat shock proteins Bag3 and CryAB have considerably different roles in the adult heart. This issue with the concentration of the mutant protein is significant, as recent studies overexpressing the Bag3 P209L mutant protein in cells result in dramatic increases in aggregated protein in cell culture,75 in contrast to the present study in vivo having only onefold to twofold increase in Bag3 P209L and a slower clinical course as seen in the human disease. Recent studies have found that Bag3 knockdown in adult zebrafish results in myofibrillar disintegration without forming protein aggregates,13 whereas CryAB−/− (and CryAB/HSPB2 double knockout) mice do not have any ultrastructural abnormalities.76 The ability of the Bag3 P209L mutant protein to rescue the BAG3 knockdown myofibillar disarray indicates that its structural functions in the sarcomere are still intact.13
The importance of maintaining BAG3 levels is highlighted by the Bag3 knockdown studies in zebrafish, making the transcriptional down-regulation of wild-type BAG3 a critical finding in the present study (Figure 8A). How BAG3 is transcriptionally regulated in the cardiomyocyte is unknown, but multiple studies have identified that Jun proteins (c-Jun, JunB, and JunD), which form AP-1 homodimers (Jun/Jun) or heterodimers with Fos family members, ATF/CREB family members, or JDP family members regulate BAG3 expression in noncardiac cells.77 In cancer cells, c-Jun can be found to interact with the BAG3 promoter, with less c-Jun occupancy driving a suppression of BAG3.77
Misfolded tau proteins caused by truncation mutations have been linked to the activation of mitogen-activated protein kinase signaling.78 The p38 activation in Alzheimer disease animal models (ie, Aβ and tau toxicities) can be attenuated with vitamin E79 and by medications that are used to successfully reduce loss of memory impairment in Alzheimer disease (Donepezil, an inhibitor of acetylcholinesterase, attenuates amyloid-β–induced iNOS, cyclooxygenase-2, and the phosphorylation of p38 as well as NF-κB80). Taken together, these studies illustrate the functional role of PAO-prone amyloid-β and tau proteins in activating inflammation via p38 activation, which is a therapeutic target to attenuate disease progression. Similarly, Bag3 Tg+ hearts have both an increase in inflammatory infiltrates (Figure 5, C–E) and trending increases in NF-κB activity (increased phospho-p65/p65 by Western blot) at 12 months of age (Supplemental Figure S5).
These finding have broader implications in treating PAO-mediated cardiac diseases, such as the Bag3 P029L mutation, which currently have no therapies available beyond traditional RAAS-targeted heart failure therapies. Similar to the link between misfolded proteins and p38 activation in the brain,81 the cardiac literature suggests a parallel role of p38 in mediating heart failure and may be partially responsible for the cardiac phenotype of the current study. Targeted activation of cardiomyocyte p38 potently induces cardiac hypertrophy and heart failure in vivo.69, 82, 83 In the current study, a clear progressive heart failure ensues in the Bag3 P029L Tg+ mice by 8 months of age (Figure 1, D–I), along with cardiac hypertrophy (Figure 2B). Also like the amyloid-β and tau protein–induced p38 mediated–inflammation, preliminary evidence of inflammation was seen in the 12-month Bag3 P029L Tg+ hearts (Figure 5, B and C).70 Evidence is accumulating that p38 inhibitors are effective in protecting in cardiac ischemia, arrhythmias, myocardial contractile dysfunction, myocardial infarction, LV remodeling, atherosclerosis, and endothelial cell dysfunction in both preclinical models and in human clinical trials (eg, losmapimod/NCT02145468 in phase 3 for acute MI and BMS-582949/NCT00570752 in phase 1 trials for atherosclerosis) with potential benefit identified in patients with early and late atherosclerosis.84 To our knowledge, p38 inhibitors have not been reported in hereditary myopathies to date, but their use may provide a therapy that has shown promise in other cardiac diseases, for which a specific mechanistically rationale therapy is currently unavailable.
Acknowledgments
We thank Robert Bagnell and Vicky Madden (Microscopy Services Laboratory, UNC Department of Pathology and Laboratory Medicine) for their assistance with the electron microscopy experiments and Carolyn Spaniel (Hillhouse), for her management of the Bag3 P209L colony.
M.S.W. is the guarantor of this work and, as such, had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
Supported by the Jefferson-Pilot Corporation Fellowship in Academic Medicine (M.S.W.), the NIH National Heart, Lung, and Blood Institute (R01HL104129), and the Leducq Foundation (M.S.W.).
Disclosures: None declared.
Supplemental material for this article can be found at http://dx.doi.org/10.1016/j.ajpath.2016.03.017.
Supplemental Data
Supplemental Figure S1.
Experimental design and analysis of mendelian ratios of αMHC-Bag3 P209L transgenic offspring. Experimental design of studies. Mendelian ratios of offspring further analyzed by sex at the time of genotyping at 14 to 21 days post-partum with χ2 analysis. N = 47 total, with 9 to 16 per group as illustrated.
Supplemental Figure S2.
Quantification of Bag3 P209L apoptosis-inducing factor (AIF) expression by immunofluorescence. Representative confocal immunofluorescence images of AIF positive cells in histological cardiac cross sections of αMHC-Bag3 P209L and littermate wild-type mice at 12 months of age. N = 3 biological controls per group. Scale bar = 400 μm. AIF, apoptosis-inducing factor; IF, immunofluorescence.
Supplemental Figure S3.

Immunofluorescence analysis of left ventricular fibroblasts in quadrant 1 (Q1) and quadrant 4 (Q4). Representative confocal immunofluorescence images of cardiac fibroblasts from histological sections of αMHC-Bag3 P209L and littermate wild-type mice at 12 months of age. A: Quadrant 1, B: Quadrant 4. Two representative biological replicates shown for each group. Scale bar = 200 μm (A and B).
Supplemental Figure S4.
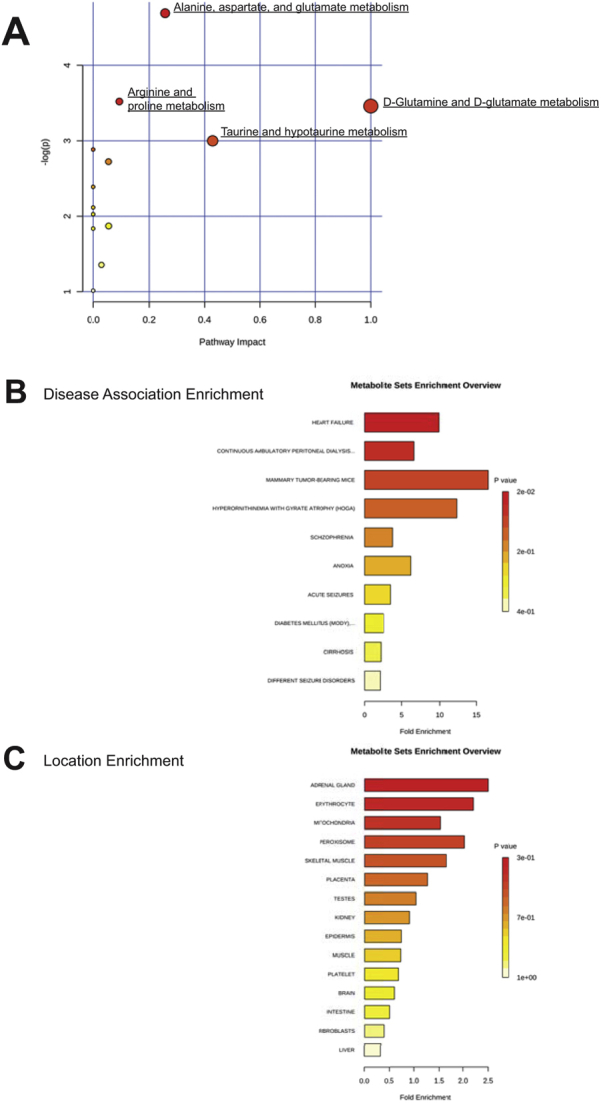
Higher-order enrichment analysis of metabolites is significantly altered in αMHC-Bag3 P209L transgenic hearts at 12 months. Enrichment analysis of t-test/variable importance in projection (VIP) significant metabolites by pathway (A), disease association (B), and subcellular location (C) using Metaboanalyst data sets.
Supplemental Figure S5.

Analysis of thioredoxin reductase activity, NF-κB, and fetal gene expression in Bag3 P209L Tg+ hearts. A: Thioredoxin reductase activity of ventricular tissue from αMHC-BAG3 P209L and littermate wild-type mice at 12 months of age. Western blot analysis of the thioredoxin enzymes succinic semialdehyde dehydrogenase (SSAHD) (B) and periredoxin 1 (Prdx1) (C). D: Immunoblot analysis of activated NF-κB subunit p65 (phospho-p65 subunit) normalized to total p65. E: Quantitative RT-PCR analysis of BNP, αMHC, and skeletal muscle actin mRNA in Bag3 P209L Tg+ hearts. A t-test was used to compare genotypes at each time point. Data are expressed as means ± SEM. N = 5 mice per group (A, Bag3 P209L Tg+ and wildtypeBag3 P209L Tg+, and D, wild-typeBag3 P209L Tg+); N = 3 per group (B, C, and E, Bag3 P209L Tg+ and B and C, wild-typeBag3 P209L Tg+); N = 6 (D, Bag3 P209L Tg+ and E, wild-typeBag3 P209L Tg+). ∗P < 0.05 versus age-matched sibling wild-type mice. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IB, immunoblot.
References
- 1.Rosati A., Graziano V., De Laurenzi V., Pascale M., Turco M.C. BAG3: a multifaceted protein that regulates major cell pathways. Cell Death Dis. 2011;2:e141. doi: 10.1038/cddis.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hishiya A., Kitazawa T., Takayama S. BAG3 and Hsc70 interact with actin capping protein CapZ to maintain myofibrillar integrity under mechanical stress. Circ Res. 2010;107:1220–1231. doi: 10.1161/CIRCRESAHA.110.225649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sanoudou D., Corbett M.A., Han M., Ghoddusi M., Nguyen M.A., Vlahovich N., Hardeman E.C., Beggs A.H. Skeletal muscle repair in a mouse model of nemaline myopathy. Hum Mol Genet. 2006;15:2603–2612. doi: 10.1093/hmg/ddl186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Warren G.L., Summan M., Gao X., Chapman R., Hulderman T., Simeonova P.P. Mechanisms of skeletal muscle injury and repair revealed by gene expression studies in mouse models. J Physiol. 2007;582:825–841. doi: 10.1113/jphysiol.2007.132373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olive M., Kley R.A., Goldfarb L.G. Myofibrillar myopathies: new developments. Curr Opin Neurol. 2013;26:527–535. doi: 10.1097/WCO.0b013e328364d6b1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arimura T., Ishikawa T., Nunoda S., Kawai S., Kimura A. Dilated cardiomyopathy-associated BAG3 mutations impair Z-disc assembly and enhance sensitivity to apoptosis in cardiomyocytes. Hum Mutat. 2011;32:1481–1491. doi: 10.1002/humu.21603. [DOI] [PubMed] [Google Scholar]
- 7.Lee H.C., Cherk S.W., Chan S.K., Wong S., Tong T.W., Ho W.S., Chan A.Y., Lee K.C., Mak C.M. BAG3-related myofibrillar myopathy in a Chinese family. Clin Genet. 2012;81:394–398. doi: 10.1111/j.1399-0004.2011.01659.x. [DOI] [PubMed] [Google Scholar]
- 8.Norton N., Li D., Rieder M.J., Siegfried J.D., Rampersaud E., Zuchner S., Mangos S., Gonzalez-Quintana J., Wang L., McGee S., Reiser J., Martin E., Nickerson D.A., Hershberger R.E. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am J Hum Genet. 2011;88:273–282. doi: 10.1016/j.ajhg.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Odgerel Z., Sarkozy A., Lee H.S., McKenna C., Rankin J., Straub V., Lochmuller H., Paola F., D'Amico A., Bertini E., Bushby K., Goldfarb L.G. Inheritance patterns and phenotypic features of myofibrillar myopathy associated with a BAG3 mutation. Neuromuscul Disord. 2010;20:438–442. doi: 10.1016/j.nmd.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Selcen D., Muntoni F., Burton B.K., Pegoraro E., Sewry C., Bite A.V., Engel A.G. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Ann Neurol. 2009;65:83–89. doi: 10.1002/ana.21553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Homma S., Iwasaki M., Shelton G.D., Engvall E., Reed J.C., Takayama S. BAG3 deficiency results in fulminant myopathy and early lethality. Am J Pathol. 2006;169:761–773. doi: 10.2353/ajpath.2006.060250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCollum A.K., Casagrande G., Kohn E.C. Caught in the middle: the role of Bag3 in disease. Biochem J. 2010;425:e1–e3. doi: 10.1042/BJ20091739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruparelia A.A., Oorschot V., Vaz R., Ramm G., Bryson-Richardson R.J. Zebrafish models of BAG3 myofibrillar myopathy suggest a toxic gain of function leading to BAG3 insufficiency. Acta Neuropathol. 2014;128:821–833. doi: 10.1007/s00401-014-1344-5. [DOI] [PubMed] [Google Scholar]
- 14.Willis M.S., Patterson C. Proteotoxicity and cardiac dysfunction: Alzheimer's disease of the heart? N Engl J Med. 2013;368:455–464. doi: 10.1056/NEJMra1106180. [DOI] [PubMed] [Google Scholar]
- 15.Subramanian K., Gianni D., Balla C., Assenza G.E., Joshi M., Semigran M.J., Macgillivray T.E., Van Eyk J.E., Agnetti G., Paolocci N., Bamburg J.R., Agrawal P.B., Del Monte F. Cofilin-2 phosphorylation and sequestration in myocardial aggregates: novel pathogenetic mechanisms for idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 2015;65:1199–1214. doi: 10.1016/j.jacc.2015.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pattison J.S., Sanbe A., Maloyan A., Osinska H., Klevitsky R., Robbins J. Cardiomyocyte expression of a polyglutamine preamyloid oligomer causes heart failure. Circulation. 2008;117:2743–2751. doi: 10.1161/CIRCULATIONAHA.107.750232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sanbe A., Osinska H., Saffitz J.E., Glabe C.G., Kayed R., Maloyan A., Robbins J. Desmin-related cardiomyopathy in transgenic mice: a cardiac amyloidosis. Proc Natl Acad Sci U S A. 2004;101:10132–10136. doi: 10.1073/pnas.0401900101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X., Klevitsky R., Huang W., Glasford J., Li F., Robbins J. AlphaB-crystallin modulates protein aggregation of abnormal desmin. Circ Res. 2003;93:998–1005. doi: 10.1161/01.RES.0000102401.77712.ED. [DOI] [PubMed] [Google Scholar]
- 19.Hayashi M., Imanaka-Yoshida K., Yoshida T., Wood M., Fearns C., Tatake R.J., Lee J.D. A crucial role of mitochondrial Hsp40 in preventing dilated cardiomyopathy. Nat Med. 2006;12:128–132. doi: 10.1038/nm1327. [DOI] [PubMed] [Google Scholar]
- 20.Willis M.S., Wadosky K.M., Rodriguez J.E., Schisler J.C., Lockyer P., Hilliard E.G., Glass D.J., Patterson C. Muscle ring finger 1 and muscle ring finger 2 are necessary but functionally redundant during developmental cardiac growth and regulate E2F1-mediated gene expression in vivo. Cell Biochem Funct. 2014;32:39–50. doi: 10.1002/cbf.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Willis M.S., Dyer L.A., Ren R., Lockyer P., Moreno-Miralles I., Schisler J.C., Patterson C. BMPER regulates cardiomyocyte size and vessel density in vivo. Cardiovasc Pathol. 2013;22:228–240. doi: 10.1016/j.carpath.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Su H., Wang X. Autophagy and p62 in cardiac protein quality control. Autophagy. 2011;7:1382–1383. doi: 10.4161/auto.7.11.17339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Su H., Li F., Ranek M.J., Wei N., Wang X. COP9 signalosome regulates autophagosome maturation. Circulation. 2011;124:2117–2128. doi: 10.1161/CIRCULATIONAHA.111.048934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Santiago J.J., Dangerfield A.L., Rattan S.G., Bathe K.L., Cunnington R.H., Raizman J.E., Bedosky K.M., Freed D.H., Kardami E., Dixon I.M. Cardiac fibroblast to myofibroblast differentiation in vivo and in vitro: expression of focal adhesion components in neonatal and adult rat ventricular myofibroblasts. Dev Dyn. 2010;239:1573–1584. doi: 10.1002/dvdy.22280. [DOI] [PubMed] [Google Scholar]
- 25.Yates C.C., Krishna P., Whaley D., Bodnar R., Turner T., Wells A. Lack of CXC chemokine receptor 3 signaling leads to hypertrophic and hypercellular scarring. Am J Pathol. 2010;176:1743–1755. doi: 10.2353/ajpath.2010.090564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sidorova T.N., Mace L.C., Wells K.S., Yermalitskaya L.V., Su P.F., Shyr Y., Byrne J.G., Petracek M.R., Greelish J.P., Hoff S.J., Ball S.K., Glabe C.G., Brown N.J., Barnett J.V., Murray K.T. Quantitative imaging of preamyloid oligomers, a novel structural abnormality, in human atrial samples. J Histochem Cytochem. 2014;62:479–487. doi: 10.1369/0022155414535782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roessner U., Wagner C., Kopka J., Trethewey R.N., Willmitzer L. Technical advance: simultaneous analysis of metabolites in potato tuber by gas chromatography-mass spectrometry. Plant J. 2000;23:131–142. doi: 10.1046/j.1365-313x.2000.00774.x. [DOI] [PubMed] [Google Scholar]
- 28.Fiehn O., Wohlgemuth G., Scholz M., Kind T., Lee do Y., Lu Y., Moon S., Nikolau B. Quality control for plant metabolomics: reporting MSI-compliant studies. Plant J. 2008;53:691–704. doi: 10.1111/j.1365-313X.2007.03387.x. [DOI] [PubMed] [Google Scholar]
- 29.Kind T., Wohlgemuth G., Lee do Y., Lu Y., Palazoglu M., Shahbaz S., Fiehn O. FiehnLib: mass spectral and retention index libraries for metabolomics based on quadrupole and time-of-flight gas chromatography/mass spectrometry. Anal Chem. 2009;81:10038–10048. doi: 10.1021/ac9019522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Banerjee R., Bultman S.J., Holley D., Hillhouse C., Bain J.R., Newgard C.B., Muehlbauer M.J., Willis M.S. Non-targeted metabolomics of double-mutant cardiomyocytes reveals a novel role for SWI/SNF complexes in metabolic homeostasis. Metabolomics. 2015;11:1287–1301. doi: 10.1007/s11306-015-0786-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Banerjee R., He J., Spaniel C., Quintana M.T., Wang Z.J., Bain J.R., Newgard C.B., Muehlbauer M.J., Willis M.S. Non-targeted metabolomics analysis of cardiac Muscle Ring Finger-1 (MuRF1), MuRF2, and MuRF3 in vivo reveals novel and redundant metabolic changes. Metabolomics. 2015;11:312–322. doi: 10.1007/s11306-014-0695-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xia J., Mandal R., Sinelnikov I.V., Broadhurst D., Wishart D.S. MetaboAnalyst 2.0: a comprehensive server for metabolomic data analysis. Nucleic Acids Res. 2012;40:W127–W133. doi: 10.1093/nar/gks374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xia J., Psychogios N., Young N., Wishart D.S. MetaboAnalyst: a web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 2009;37:W652–W660. doi: 10.1093/nar/gkp356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosati A., Ammirante M., Gentilella A., Basile A., Festa M., Pascale M., Marzullo L., Belisario M.A., Tosco A., Franceschelli S., Moltedo O., Pagliuca G., Lerose R., Turco M.C. Apoptosis inhibition in cancer cells: a novel molecular pathway that involves BAG3 protein. Int J Biochem Cell Biol. 2007;39:1337–1342. doi: 10.1016/j.biocel.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 35.Pagliuca M.G., Lerose R., Cigliano S., Leone A. Regulation by heavy metals and temperature of the human BAG-3 gene, a modulator of Hsp70 activity. FEBS Lett. 2003;541:11–15. doi: 10.1016/s0014-5793(03)00274-6. [DOI] [PubMed] [Google Scholar]
- 36.Rosati A., Di Salle E., Luberto L., Quinto I., Scala G., Turco M.C., Pascale M. Identification of a Btk-BAG3 complex induced by oxidative stress. Leukemia. 2009;23:823–824. doi: 10.1038/leu.2009.23. [DOI] [PubMed] [Google Scholar]
- 37.Ammirante M., Rosati A., Arra C., Basile A., Falco A., Festa M., Pascale M., d'Avenia M., Marzullo L., Belisario M.A., De Marco M., Barbieri A., Giudice A., Chiappetta G., Vuttariello E., Monaco M., Bonelli P., Salvatore G., Di Benedetto M., Deshmane S.L., Khalili K., Turco M.C., Leone A. IKK{gamma} protein is a target of BAG3 regulatory activity in human tumor growth. Proc Natl Acad Sci U S A. 2010;107:7497–7502. doi: 10.1073/pnas.0907696107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Spaendonck-Zwarts K.Y., van Hessem L., Jongbloed J.D., de Walle H.E., Capetanaki Y., van der Kooi A.J., van Langen I.M., van den Berg M.P., van Tintelen J.P. Desmin-related myopathy. Clin Genet. 2011;80:354–366. doi: 10.1111/j.1399-0004.2010.01512.x. [DOI] [PubMed] [Google Scholar]
- 39.Ulbricht A., Gehlert S., Leciejewski B., Schiffer T., Bloch W., Hohfeld J. Induction and adaptation of chaperone-assisted selective autophagy CASA in response to resistance exercise in human skeletal muscle. Autophagy. 2015;11:538–546. doi: 10.1080/15548627.2015.1017186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arndt V., Dick N., Tawo R., Dreiseidler M., Wenzel D., Hesse M., Furst D.O., Saftig P., Saint R., Fleischmann B.K., Hoch M., Hohfeld J. Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr Biol. 2010;20:143–148. doi: 10.1016/j.cub.2009.11.022. [DOI] [PubMed] [Google Scholar]
- 41.Jiao Q., Sanbe A., Zhang X., Liu J.P., Minamisawa S. alphaB-Crystallin R120G variant causes cardiac arrhythmias and alterations in the expression of Ca(2+) -handling proteins and endoplasmic reticulum stress in mice. Clin Exp Pharmacol Physiol. 2014;41:589–599. doi: 10.1111/1440-1681.12253. [DOI] [PubMed] [Google Scholar]
- 42.Zheng Q., Su H., Ranek M.J., Wang X. Autophagy and p62 in cardiac proteinopathy. Circ Res. 2011;109:296–308. doi: 10.1161/CIRCRESAHA.111.244707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Willis M.S., Min J.N., Wang S., McDonough H., Lockyer P., Wadosky K.M., Patterson C. Carboxyl terminus of Hsp70-interacting protein (CHIP) is required to modulate cardiac hypertrophy and attenuate autophagy during exercise. Cell Biochem Funct. 2013;31:724–735. doi: 10.1002/cbf.2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marunouchi T., Tanonaka K. Cell death in the cardiac myocyte. Biol Pharm Bull. 2015;38:1094–1097. doi: 10.1248/bpb.b15-00288. [DOI] [PubMed] [Google Scholar]
- 45.van Empel V.P., Bertrand A.T., Hofstra L., Crijns H.J., Doevendans P.A., De Windt L.J. Myocyte apoptosis in heart failure. Cardiovasc Res. 2005;67:21–29. doi: 10.1016/j.cardiores.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 46.Doong H., Vrailas A., Kohn E.C. What's in the “BAG”?–A functional domain analysis of the BAG-family proteins. Cancer Lett. 2002;188:25–32. doi: 10.1016/s0304-3835(02)00456-1. [DOI] [PubMed] [Google Scholar]
- 47.Chiappetta G., Ammirante M., Basile A., Rosati A., Festa M., Monaco M., Vuttariello E., Pasquinelli R., Arra C., Zerilli M., Todaro M., Stassi G., Pezzullo L., Gentilella A., Tosco A., Pascale M., Marzullo L., Belisario M.A., Turco M.C., Leone A. The antiapoptotic protein BAG3 is expressed in thyroid carcinomas and modulates apoptosis mediated by tumor necrosis factor-related apoptosis-inducing ligand. J Clin Endocrinol Metab. 2007;92:1159–1163. doi: 10.1210/jc.2006-1712. [DOI] [PubMed] [Google Scholar]
- 48.Liu P., Xu B., Li J., Lu H. BAG3 gene silencing sensitizes leukemic cells to Bortezomib-induced apoptosis. FEBS Lett. 2009;583:401–406. doi: 10.1016/j.febslet.2008.12.032. [DOI] [PubMed] [Google Scholar]
- 49.Jacobs A.T., Marnett L.J. HSF1-mediated BAG3 expression attenuates apoptosis in 4-hydroxynonenal-treated colon cancer cells via stabilization of anti-apoptotic Bcl-2 proteins. J Biol Chem. 2009;284:9176–9183. doi: 10.1074/jbc.M808656200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang H.Q., Liu B.Q., Gao Y.Y., Meng X., Guan Y., Zhang H.Y., Du Z.X. Inhibition of the JNK signalling pathway enhances proteasome inhibitor-induced apoptosis of kidney cancer cells by suppression of BAG3 expression. Br J Pharmacol. 2009;158:1405–1412. doi: 10.1111/j.1476-5381.2009.00455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Festa M., Del Valle L., Khalili K., Franco R., Scognamiglio G., Graziano V., De Laurenzi V., Turco M.C., Rosati A. BAG3 protein is overexpressed in human glioblastoma and is a potential target for therapy. Am J Pathol. 2011;178:2504–2512. doi: 10.1016/j.ajpath.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jung S.E., Kim Y.K., Youn D.Y., Lim M.H., Ko J.H., Ahn Y.S., Lee J.H. Down-modulation of Bis sensitizes cell death in C6 glioma cells induced by oxygen-glucose deprivation. Brain Res. 2010;1349:1–10. doi: 10.1016/j.brainres.2010.06.043. [DOI] [PubMed] [Google Scholar]
- 53.Cesaro E., Montano G., Rosati A., Crescitelli R., Izzo P., Turco M.C., Costanzo P. WT1 protein is a transcriptional activator of the antiapoptotic bag3 gene. Leukemia. 2010;24:1204–1206. doi: 10.1038/leu.2010.68. [DOI] [PubMed] [Google Scholar]
- 54.Bodnar R.J., Rodgers M.E., Chen W.C., Wells A. Pericyte regulation of vascular remodeling through the CXC receptor 3. Arterioscler Thromb Vasc Biol. 2013;33:2818–2829. doi: 10.1161/ATVBAHA.113.302012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Avolio E., Rodriguez-Arabaolaza I., Spencer H.L., Riu F., Mangialardi G., Slater S.C., Rowlinson J., Alvino V.V., Idowu O.O., Soyombo S., Oikawa A., Swim M.M., Kong C.H., Cheng H., Jia H., Ghorbel M.T., Hancox J.C., Orchard C.H., Angelini G., Emanueli C., Caputo M., Madeddu P. Expansion and characterization of neonatal cardiac pericytes provides a novel cellular option for tissue engineering in congenital heart disease. J Am Heart Assoc. 2015;4:e002043. doi: 10.1161/JAHA.115.002043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wong S.P., Rowley J.E., Redpath A.N., Tilman J.D., Fellous T.G., Johnson J.R. Pericytes, mesenchymal stem cells and their contributions to tissue repair. Pharmacol Ther. 2015;151:107–120. doi: 10.1016/j.pharmthera.2015.03.006. [DOI] [PubMed] [Google Scholar]
- 57.Vicart P., Caron A., Guicheney P., Li Z., Prevost M.C., Faure A., Chateau D., Chapon F., Tome F., Dupret J.M., Paulin D., Fardeau M. A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat Genet. 1998;20:92–95. doi: 10.1038/1765. [DOI] [PubMed] [Google Scholar]
- 58.Parry T.L., Melehani J.H., Ranek M.J., Willis M.S. Functional amyloid signaling via the inflammasome, necrosome, and signalosome: new therapeutic targets in heart failure. Front Cardiovasc Med. 2015;2:25. doi: 10.3389/fcvm.2015.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nakamura T., Lipton S.A. S-nitrosylation of critical protein thiols mediates protein misfolding and mitochondrial dysfunction in neurodegenerative diseases. Antioxid Redox Signal. 2011;14:1479–1492. doi: 10.1089/ars.2010.3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nakamura T., Lipton S.A. Redox regulation of mitochondrial fission, protein misfolding, synaptic damage, and neuronal cell death: potential implications for Alzheimer's and Parkinson's diseases. Apoptosis. 2010;15:1354–1363. doi: 10.1007/s10495-010-0476-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zungu M., Schisler J., Willis M.S. All the little pieces: regulation of mitochondrial fusion and fission by ubiquitin and small ubiquitin-like modifer and their potential relevance in the heart. Circ J. 2011;75:2513–2521. doi: 10.1253/circj.cj-11-0967. [DOI] [PubMed] [Google Scholar]
- 62.Du Z.X., Zhang H.Y., Meng X., Gao Y.Y., Zou R.L., Liu B.Q., Guan Y., Wang H.Q. Proteasome inhibitor MG132 induces BAG3 expression through activation of heat shock factor 1. J Cell Physiol. 2009;218:631–637. doi: 10.1002/jcp.21634. [DOI] [PubMed] [Google Scholar]
- 63.Franceschelli S., Rosati A., Lerose R., De Nicola S., Turco M.C., Pascale M. Bag3 gene expression is regulated by heat shock factor 1. J Cell Physiol. 2008;215:575–577. doi: 10.1002/jcp.21397. [DOI] [PubMed] [Google Scholar]
- 64.Song S., Kole S., Precht P., Pazin M.J., Bernier M. Activation of heat shock factor 1 plays a role in pyrrolidine dithiocarbamate-mediated expression of the co-chaperone BAG3. Int J Biochem Cell Biol. 2010;42:1856–1863. doi: 10.1016/j.biocel.2010.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang H.Q., Meng X., Liu B.Q., Li C., Gao Y.Y., Niu X.F., Li N., Guan Y., Du Z.X. Involvement of JNK and NF-kappaB pathways in lipopolysaccharide (LPS)-induced BAG3 expression in human monocytic cells. Exp Cell Res. 2012;318:16–24. doi: 10.1016/j.yexcr.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 66.Belozerov V.E., Ratkovic S., McNeill H., Hilliker A.J., McDermott J.C. In vivo interaction proteomics reveal a novel p38 mitogen-activated protein kinase/Rack1 pathway regulating proteostasis in Drosophila muscle. Mol Cell Biol. 2014;34:474–484. doi: 10.1128/MCB.00824-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kang E.B., Kwon I.S., Koo J.H., Kim E.J., Kim C.H., Lee J., Yang C.H., Lee Y.I., Cho I.H., Cho J.Y. Treadmill exercise represses neuronal cell death and inflammation during Abeta-induced ER stress by regulating unfolded protein response in aged presenilin 2 mutant mice. Apoptosis. 2013;18:1332–1347. doi: 10.1007/s10495-013-0884-9. [DOI] [PubMed] [Google Scholar]
- 68.Huang Y., Li X., Wang Y., Wang H., Huang C., Li J. Endoplasmic reticulum stress-induced hepatic stellate cell apoptosis through calcium-mediated JNK/P38 MAPK and Calpain/Caspase-12 pathways. Mol Cell Biochem. 2014;394:1–12. doi: 10.1007/s11010-014-2073-8. [DOI] [PubMed] [Google Scholar]
- 69.Liao P., Georgakopoulos D., Kovacs A., Zheng M., Lerner D., Pu H., Saffitz J., Chien K., Xiao R.P., Kass D.A., Wang Y. The in vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proc Natl Acad Sci U S A. 2001;98:12283–12288. doi: 10.1073/pnas.211086598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Degousee N., Martindale J., Stefanski E., Cieslak M., Lindsay T.F., Fish J.E., Marsden P.A., Thuerauf D.J., Glembotski C.C., Rubin B.B. MAP kinase kinase 6-p38 MAP kinase signaling cascade regulates cyclooxygenase-2 expression in cardiac myocytes in vitro and in vivo. Circ Res. 2003;92:757–764. doi: 10.1161/01.RES.0000067929.01404.03. [DOI] [PubMed] [Google Scholar]
- 71.Knezevic T., Myers V.D., Gordon J., Tilley D.G., Sharp T.E., 3rd, Wang J., Khalili K., Cheung J.Y., Feldman A.M. BAG3: a new player in the heart failure paradigm. Heart Fail Rev. 2015;20:423–434. doi: 10.1007/s10741-015-9487-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fuchs M., Poirier D.J., Seguin S.J., Lambert H., Carra S., Charette S.J., Landry J. Identification of the key structural motifs involved in HspB8/HspB6-Bag3 interaction. Biochem J. 2010;425:245–255. doi: 10.1042/BJ20090907. [DOI] [PubMed] [Google Scholar]
- 73.Meister M, Minoia M, Kanon B, Carra S, Kampinga H: Characterization of the myopathy associated BAG3 P209L mutation. The biology of molecular chaperones: from molecules, organelles and cells to risfolding diseases. European Molecular Biology Organization Conference, May 17–22, 2013, Santa Margherita di Pula, Italy. Poster 275.
- 74.Wang X., Osinska H., Klevitsky R., Gerdes A.M., Nieman M., Lorenz J., Hewett T., Robbins J. Expression of R120G-alphaB-crystallin causes aberrant desmin and alphaB-crystallin aggregation and cardiomyopathy in mice. Circ Res. 2001;89:84–91. doi: 10.1161/hh1301.092688. [DOI] [PubMed] [Google Scholar]
- 75.Minoia M. University Medical Center Groningen and University of Groningen; Netherlands: 2014. Chaperones, Protein Homeostasis & Protein Aggregation Diseases. [Google Scholar]
- 76.Benjamin I.J., Guo Y., Srinivasan S., Boudina S., Taylor R.P., Rajasekaran N.S., Gottlieb R., Wawrousek E.F., Abel E.D., Bolli R. CRYAB and HSPB2 deficiency alters cardiac metabolism and paradoxically confers protection against myocardial ischemia in aging mice. Am J Physiol Heart Circ Physiol. 2007;293:H3201–H3209. doi: 10.1152/ajpheart.01363.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li C., Li S., Kong D.H., Meng X., Zong Z.H., Liu B.Q., Guan Y., Du Z.X., Wang H.Q. BAG3 is upregulated by c-Jun and stabilizes JunD. Biochim Biophys Acta. 2013;1833:3346–3354. doi: 10.1016/j.bbamcr.2013.10.007. [DOI] [PubMed] [Google Scholar]
- 78.Kovac A., Zilka N., Kazmerova Z., Cente M., Zilkova M., Novak M. Misfolded truncated protein tau induces innate immune response via MAPK pathway. J Immunol. 2011;187:2732–2739. doi: 10.4049/jimmunol.1100216. [DOI] [PubMed] [Google Scholar]
- 79.Giraldo E., Lloret A., Fuchsberger T., Vina J. Abeta and tau toxicities in Alzheimer's are linked via oxidative stress-induced p38 activation: protective role of vitamin E. Redox Biol. 2014;2:873–877. doi: 10.1016/j.redox.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim H.G., Moon M., Choi J.G., Park G., Kim A.J., Hur J., Lee K.T., Oh M.S. Donepezil inhibits the amyloid-beta oligomer-induced microglial activation in vitro and in vivo. Neurotoxicology. 2014;40:23–32. doi: 10.1016/j.neuro.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 81.Zilka N., Kazmerova Z., Jadhav S., Neradil P., Madari A., Obetkova D., Bugos O., Novak M. Who fans the flames of Alzheimer's disease brains? misfolded tau on the crossroad of neurodegenerative and inflammatory pathways. J Neuroinflammation. 2012;9:47. doi: 10.1186/1742-2094-9-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang Y., Huang S., Sah V.P., Ross J., Jr., Brown J.H., Han J., Chien K.R. Cardiac muscle cell hypertrophy and apoptosis induced by distinct members of the p38 mitogen-activated protein kinase family. J Biol Chem. 1998;273:2161–2168. doi: 10.1074/jbc.273.4.2161. [DOI] [PubMed] [Google Scholar]
- 83.Braz J.C., Bueno O.F., Liang Q., Wilkins B.J., Dai Y.S., Parsons S., Braunwart J., Glascock B.J., Klevitsky R., Kimball T.F., Hewett T.E., Molkentin J.D. Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopathy through upregulation of calcineurin-NFAT signaling. J Clin Invest. 2003;111:1475–1486. doi: 10.1172/JCI17295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Martin E.D., Bassi R., Marber M.S. p38 MAPK in cardioprotection: are we there yet? Br J Pharmacol. 2015;172:2101–2113. doi: 10.1111/bph.12901. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.