

Abstract
Adult liver cells have been considered restricted regarding their fate and lineage potential. That is, hepatocytes have been thought able only to generate hepatocytes and duct cells, only duct cells. While this may be the case for the majority of scenarios in a state of quiescence or homeostasis, evidence suggests that liver cells are capable of interconverting between cellular states of distinct phenotypic traits. This interconversion or plasticity had been suggested by classical studies using cellular markers, but recently lineage tracing approaches have proven that cells are highly plastic and retain an extraordinary ability to respond differently to normal tissue homeostasis, to tissue repair, or when challenged to expand ex vivo or to differentiate upon transplantation. Stemness, as “self‐renewal and multipotency,” seems not to be limited to a particular cell type but rather to a cellular state in which cells exhibit a high degree of plasticity and can move back and forth in different phenotypic states. For instance, upon damage cells can dedifferentiate to acquire stem cell potential that allows them to self‐renew, repopulate a damaged tissue, and then undergo differentiation. In this review, we will discuss the evidence on cellular plasticity in the liver, focusing our attention on two markers, epithelial cell adhesion molecule and leucine‐rich repeat‐containing G protein‐coupled receptor 5, which identify cells with stem cell potential. (Hepatology 2016;64:652‐662)
Abbreviations
- EpCAM
epithelial cell adhesion molecule
- Lgr5
leucine‐rich repeat‐containing G protein‐coupled receptor 5
Stem Cell Fate and Stem Cell Potential: Different Sides of Cellular Plasticity
The stem cell state is defined by the ability of cells to fulfill the two following criteria: self‐renewal and multipotency.1 Several approaches have been used to identify cells that exhibit stem cell characteristics. In vivo, long‐term label‐retaining and genetic lineage tracing have been commonly used to identify both quiescent and actively cycling stem cells in several tissues.1 Alternatively, in vitro clonogenicity and multilineage differentiation as well as long‐term repopulation following transplantation have been regarded extensively as assays to demonstrate stem cell potential.1
Of note, stem cell fate and stem cell potential might have not always been adequately used. Stem cell fate indicates a cell that already fulfills the stem cell criteria, while stem cell potential represents a cell with the competence to acquire a stem cell state, depending on the environment or condition. Confusion might have been caused by the extensive plasticity of animal cells. Cellular plasticity is understood as the propensity of a cell to, under certain circumstances, acquire the biological properties of other cells.2 Because stem cell potential can be defined as the ability of cells (differentiated cells or progenitors) to acquire a stem cell state, stem cell potential would therefore be a specific manifestation of plasticity.2 On the other hand, one could also consider that this return to a more primitive state is a form of in vivo reprogramming. However, “reprograming” is associated with a complete reversion to a pluripotent state, as seen in Gurdon's tadpole experiments.3 In this review we use “plasticity” to mean the ability of cells to acquire other cellular fates, distinct from reprograming; and thus, acquisition of a tissue‐restricted stem cell fate or potential would be one form of plasticity.
Several authors have suggested the existence of plasticity in adult liver cells,4, 5, 6, 7 but advances in mouse genetic engineering, imaging tools, and the possibility of culturing cells in vitro have provided further evidence for cellular plasticity in the liver and other organs. Here, we review the evidence of liver cellular plasticity. We will use epithelial cell adhesion molecule (EpCAM) and leucine‐rich repeat‐containing G protein‐coupled receptor 5 (Lgr5) as examples of markers that identify cells with cellular plasticity and stem cell potential in the liver.
Cellular Plasticity: An Old Player in the New Viewpoint of Looking at Liver Repair
Increasing evidence of stem cell behavior in the intestine, hair follicle, and bone marrow suggests that cells often exist in two distinct states: an active stem cell state and a potential state that appears upon stem cell ablation. Studies on both intestinal and hair follicle cells show that when the stem cell pool is ablated, those cells which retain stem cell potential (usually early descendants of the stem cell) acquire properties of a stem cell (potential/plasticity), such as the ability to repair tissue and reinstate homeostasis (nicely reviewed by Blanpain and Fuchs2). Similarly to the intestine or skin, organs with slow physiological turnover, such as the lung, also possess a high degree of cellular plasticity. For instance, after ablation of airway stem cells, lineage tracing demonstrated that luminal secretory cells had dedifferentiated into multipotent basal stem cells.8 This capacity of cells to acquire a stem cell state may have a more general role in the regeneration of many tissues, including the liver.
The primary functional unit of the liver is the hepatic lobule or acinus, a structure resulting from the interaction between epithelial (hepatocytes and ductal cells), endothelial (sinusoidal cells), and mesenchymal (portal fibroblasts and stellate cells) cells.9
In the liver, during embryonic development, hepatoblasts behave like stem cells as they are capable of self‐duplicating while giving rise to hepatocytes and ductal cells (elegantly reviewed by Miyajima et al.9). During adulthood, cellular turnover is rather slow, with a period of more than several months.10 Extensive lineage tracing approaches in the mouse model indicate that if adult liver stem cells exist, their contribution to normal homeostasis is negligible, at least in the mouse model, with the exception of one report that demonstrated, using genetic lineage tracing based on Sox9CeER, that adult hepatocytes can also derive from specialized ductal progenitors.11 However, other studies did not find evidence for such liver progenitors.12, 13 Also, recently, a subset of centrilobular hepatocytes has been shown to contribute to normal homeostasis of the hepatocyte compartment.14, 15 On the other hand, using clonogenic assays, it has been reported that EpCAM+ human liver cells, isolated from healthy fetal, neonatal, pediatric, and adult16, 17 donors, display characteristics of liver stem/progenitors both in vitro and in vivo after transplantation. The latter could be understood as the ability of some resident cells to harbor stem cell potential during homeostasis. However, it is worth noting that clonogenic assays isolate the cells from their environment, which could trigger activation of a stem cell state as a result of damage to the tissue, as happens during a regenerative response. Therefore, as we will discuss below, the results from clonogenic assays could also be interpreted as the responses of cells to the external cue of being isolated from their tissue, which does not necessarily reflect what happens during in vivo homeostasis.
The liver excels in its extensive damage‐repair response (see Fig. 1A).18 The cells responsible for the facultative regenerative response of the liver have been the subject of extensive investigations. This has led to two schools of thought: the followers and the opponents of the existence/activation of a progenitor response that would contribute to the repair of the tissue after damage. On the one hand, mouse lineage‐tracing approaches in combination with specific cell markers have allowed the identification of cells that upon damage will differentiate into hepatocytes and/or ductal cells.11, 19, 20, 21, 22 However, in all of these studies, the lack of in vivo clonal analysis hampers the ability to conclude whether these cells are truly bipotential. Thus, until this is fully addressed, the existence of true bipotential cells induced after damage remains undetermined. Also, in vitro studies from several groups indicate that isolated progenitors from mouse injured livers display bipotentiality in vitro and in vivo following transplantation into FAH mutant mice.19, 20, 22, 23 Also, recently, Kaneko et al. showed that upon damage biliary cells expand toward the injured area, suggesting that the expanded biliary branches could contribute as a source or as a niche during the regeneration response.24 On the other hand, reports using virus‐mediated Cre lineage‐tracing approaches have recently ruled out the existence of progenitor‐driven regeneration in the mouse.25, 26 One explanation for this paradox could be that liver pathologies in these models are not sufficiently severe, so remaining “healthy” hepatocytes can still extensively proliferate and repair the lost tissue. Notably, in zebrafish, genetic ablation of the hepatocyte compartment followed by lineage tracing resulted in ductal cells dedifferentiating and acquiring a stem cell fate, where biliary tree stem/progenitors repair the damaged liver.27 Also, upon complete senescence of the hepatocyte compartment, Lu and colleagues observed a similar widespread ductular reaction in the mouse.28 Moreover, recent studies indicate that following transplantation and injury, mouse hepatocytes can acquire a ductal phenotype and stem cell state and can differentiate toward hepatocytes and ductal cells upon demand.29 Similarly, in human liver failure, ductal cells are detected close to clusters of hepatocytes that also express ductal markers.30 Whether in humans the ductal cells derive from hepatocytes or the inverse is true might be difficult to determine without the possibility of tracking the cells in vivo.
Figure 1.

Plasticity concept. (A) Diverse routes lead to liver regeneration. While in a quiescent, homeostatic state (1), tissue is maintained primarily by proliferation of the subpopulations of mature hepatic cells capable of cell division; upon damage (2) various cell sources have been described to be involved in the process of hepatic repair. Each of them is illustrated in one‐quarter of the gray hexagon; at the outer limits, the various cell sources are represented at their initial state, while the center of the hexagon represents their ultimate goal: to produce new hepatocytes (but also new duct cells or ducts). While each cell source has its color, their differentiated state is illustrated in green. Stromal cells are shaped as stars; bone marrow‐derived cells are represented as round cells; particular hepatocytes from the central vein are highlighted in blue. Biliary cells (and hepatic stem/progenitor cells) and transit‐amplifying cells are represented as small green and blue cells, respectively. (B) Diverse isolated cells are defined as liver progenitor cells. Using flow cytometry‐based cell separation methods in combination with cell surface markers or functions or genetic tracers, liver cells with stem cell potential have been isolated as viable cells. Black illustrates cell surface and genetic markers used in healthy livers, while red indicates markers used upon damage. Asterisk indicates function. Arrows indicate that whatever the nature of the isolated cells is, they all converge to the definition of “liver progenitor cells.” (C) Validation strategies of stem cell potential. Isolated cells are subjected to in vitro culture to evaluate their bidirectional differentiation, clonogenic potentials, and organoid formation and in vivo to repopulate the liver upon transplantation. Lately, genetic stability studies have been introduced. Until today, only EpCAM and Lgr5 have completed successfully this list. Abbreviations: HSPC, hepatic stem/progenitor cell; LPC, liver progenitor cell.
Overall, these studies suggest that the adult liver cellular state (either hepatocyte or duct) is not fixed but can be modulated upon request. Differentiated states can be dedifferentiated or pushed to a more “stem cell state” upon demand. In these reports, adult liver cells fulfill the stem cell criteria, whereby they will proliferate and differentiate depending on the type and extent of the damage and the model organism studied. The differences that are being observed might be due to the type of injury, the type of model (human, mouse, rat, zebrafish), or even the type of technique used to validate stem cell fate (lineage labeling) or stem cell potential (transplantation, clonogenicity, lineage tracing). Taking into account that cellular plasticity will enable cells that a priori do not exhibit stem cell properties to acquire stem cell potential if needed (self‐renew and differentiate), we therefore propose a more reconciled concept, whereby liver cells possess an extreme plasticity that allows the acquisition of different states (differentiation‐stemness) depending on the environment and tissue demand (Fig. 1A).
Isolation of Liver Cells With Clonogenic and Multilineage Potential
As mentioned, different experimental approaches have been used to identify stem cells or cells with stem cell potential: from lineage tracing to transplantation or colony formation (see Fig. 1).
Using antibodies and/or flow cytometry‐based cell separation methods, several groups have actually managed to isolate cell populations from the adult liver.31 Here, we will focus on the use of cellular markers that identify liver cells with clonogenic and multilineage potential.9, 32 EpCAM,16 Lgr5,22 CD133,33 MIC1‐1C3,33 Foxl1,19 OPN,12 Sox9,20 and CD2434 markers or antibodies, or a combination of them, have been mostly used to enrich for cells that, upon culture and/or transplantation, exhibit clonogenic and multilineage competency. Also, activities (functionality of the cell) that are enhanced in stem/progenitor cells can be used to isolate putative cells with stem cell potential, for instance, aldehyde dehydrogenase activity (Fig. 1B).35 Unfortunately, the aforementioned markers are usually expressed on regular biliary epithelial cells, which complicates their isolation. Similarly, expression of markers has been shown in a subpopulation of rat progenitors but is not found in the mouse counterpart. Conversely, OPN and MIC1‐1C3 are regarded as equivalent progenitor markers, at least in mice. Also, some markers appear only upon liver injury (like Lgr5 or Foxl1), while they are not present under homeostasis conditions. Together with the fact that stem/progenitor cell populations represent a spectrum of differentiation states, this makes the development of a unified isolation strategy difficult.
As a general view, assuming that liver stem cells are individual entities carrying specific markers is rather outdated. Perhaps our thinking on how liver stem cells work (and probably the parameters used to define stem cells) has been mistaken. For instance, it is becoming clear that the quiescent state is far from being a protected state, as used to be thought.36, 37, 38 Liver repair is also achieved by expansion of many cells, with plasticity of the stem cells and mature cells and dedifferentiation emerging as common themes (Fig. 1A). For instance, by switching on cellular and metabolic plasticity upon response to injury, the rates and types of cell production have to be rapidly adjusted to meet the tissue's cellular and metabolic requirements.38, 39 Could it be that the markers cited earlier are involved in these matters? In the future, it would be rewarding to examine whether such critical events may be correlated to the presence of the particular aforementioned markers. In this concise review, we focus on Lgr5 and EpCAM as markers that could potentially offer identification of such plasticity.
EpCAM as a Marker of Liver Cells During Homeostasis
EpCAM is a transmembrane glycoprotein that is frequently expressed in cancer.40 EpCAM is composed of a large N‐terminal extracellular domain (called EpEX) linked to a short C‐terminal fragment (named EpICD) by a single‐transmembrane domain (see Fig. 2A). Recently, EpCAM was recognized as a marker for pluripotent stem cells in humans and mice and for tissue stem cells (reviewed in Dollé et al.40). EpCAM can interact with proteins like E‐cadherin or claudins to modulate cell‐cell contact, regulate the activity of signaling pathways, or sequester molecules or receptors to prevent their biological effects (see Fig. 2A).40 EpCAM is a potent player in the maintenance of the polarized tissue and has been described to modulate the organization of the actin cytoskeleton41 and actomyosin contractility.42 While only its proliferative effect has formally been demonstrated,43 it is tempting to propose that EpCAM regulates the actomyosin network for functional purposes.
Figure 2.

EpCAM as a marker of liver cells during homeostasis. (A) Dynamics of EpCAM expression at the cellular level. The pleiotropic functions of EpCAM can be allocated to the full‐length protein as well as to EpCAM‐derived fragments generated upon intramembrane proteolysis.40 Some functions are illustrated. (B) Ex vivo, sorted and cultured EpCAM+ cells are able to form organoids, with a high degree of plasticity.22, 47 (C) Functional plasticity of EpCAM at the tissue scale is illustrated: EpCAM could be a player in a metabolic model of liver regeneration or as a molecular platform for cell recruitment. EpCAM expression on peribiliary hepatocytes (namely, canal of Hering‐associated hepatocytes found at the hepatocyte‐biliary interface) could allow an efficient hepatobiliary linkage to drain bile. Abbreviations: EpEX, EpCAM large N‐terminal extracellular domain; EpICD, EpCAM short C‐terminal fragment; ADAM, a disintegrin and metalloprotease; CTF, C‐terminal fragment; CV, central vein; PS2, presenilin‐2; PV, portal vein; TACE, tumor necrosis factor‐alpha‐converting enzyme.
Remarkably, EpCAM expression is not restricted to epithelial precursors but is also present in undifferentiated stem cells that are not yet assigned to a specific cell fate. During morphogenesis of pancreatic islets, EpCAM has been described as a morphoregulatory molecule44 whereby it is highly expressed in fetal endocrine pancreas, while the adult endocrine tissue exhibits low levels of expression. This developmentally regulated EpCAM expression has also been illustrated in other organs, such as kidney, lung, skin, and thymus (reviewed in Dollé et al.40). During liver development and homeostasis, EpCAM also demonstrates dynamic expression as it can be detected in immature cells, which gradually lose it along with their maturation into hepatocytes.16, 45, 46 So far, EpCAM is one of the most representative and successful markers used in isolating liver stem cells (Fig. 1B). Notably, long‐term culture of genome‐stable EpCAM+ bipotent stem cells from adult human liver has been developed (Fig. 2B).47
Still many questions regarding the role of EpCAM in liver regeneration remain unanswered. While in vitro these cells show bipotential competency, in vivo the reason for reexpression of EpCAM remains speculative (see Fig. 2C). Data by Yoon et al. clearly indicate the existence of a hierarchically structured regeneration of the liver based on differentiation processes that require the reexpression of EpCAM.48 Recently, a possible perspective on the role of EpCAM in the maturation of human hepatocyte buds has been elegantly shown. Briefly, the authors demonstrated that hepatocyte buds derived from progenitor cells (i.e., glutamine synthetase‐positive/EpCAM+ cells) and repopulate regions of extinct parenchyma in human cirrhosis by following a maturation process that involves dynamic expression of EpCAM and glutamine synthetase,49 allowing us to think that EpCAM might be required for stem cell maturation. Furthermore, the associated microvasculature develops in concert with the maturation of buds, resulting in a loss of CD34 expression in the bud center with the development of well‐defined sinusoids, while the periphery sustains a CD34 positivity matching the dynamics of EpCAM expression.49 This potentially exhibits EpCAM as a molecular platform permitting endothelial cell (CD34+) recruitment to ensure correct liver cell differentiation. A similar scenario has been speculated for explaining the hematopoietic cell migration from the fetal liver to the adult bone marrow (Fig. 2C).40 In response to injury, the plasticity of the hepatobiliary system has been recently unveiled.24 Interestingly, in this study EpCAM+ cell density matched the distance traveled by the emerging biliary branches. Consequently, one could consider that EpCAM has a role in this structural flexibility or possibly in the directionality of the biliary branches.
Several reports have demonstrated that immediately after an injury drastic changes in metabolism occur in the liver before the repair machineries are launched.50, 51, 52 Disturbance of the metabolic zonation upon injury led to the hypothesis of whether sensing of this metabolic insufficiency may in fact be the initiating trigger for the regenerative response. It is then tempting to speculate that de novo EpCAM expression on adult hepatocytes in the lobular parenchyma in response to injury could be an adaptive response to compensate for the hepatic insufficiency by creating a different metabolic zonation (see Fig. 2C). In this viewpoint, cellular plasticity of EpCAM, at cellular or tissue scale, is important because at one site (e.g., stem/progenitors) EpCAM might be dedicated for proliferation and at another site (e.g., hepatocytes) it can be required for response to hepatic insufficiency.
Overall, recent studies highlight the importance of the epithelial diversity that surrounds the bile ducts, which probably could partially explain the extraordinary plasticity of the biliary tree. Intriguingly enough, by (re)expressing EpCAM at cellular or tissue scale or by exposing a different integrity of the full‐length EpCAM molecule, the liver cells are champions of cellular plasticity. Whether EpCAM has a role in liver tissue plasticity remains an open question.
Lgr5 as a Marker of Liver Cells Following Damage
LGR5 is a G protein‐coupled receptor with a seven‐transmembrane domain. Together with its paralogue, LGR4, it is crucial for maintaining proliferating progenitors and stem cells in the intestine.53 Biochemical analyses have identified the LGRs as receptors for R‐spondins.54 Following association with R‐spondins, LGR4/5/6 strongly promote the activity of Wnt‐Frizzled mediated signaling (Fig. 3A). In fact, R‐spondin‐LGR binding results in removal of the E3 ubiquitin ligase RNF43, thus preventing the degradation of Frizzled, which results in a more robust and prolonged Wnt signal emanating from a “stabilized” Wnt/frizzled complex (reviewed in Koo and Clevers54). Lineage tracing studies have confirmed that Lgr5+ cells are fast‐dividing, long‐lived adult stem cells in the hair follicles, the antropyloric stomach, and the gut (reviewed in Koo and Clevers54). Also, the mammary epithelium, the developing kidney, the ovarian epithelium, and supporting cells in the inner ear possess LGR5+ cells (reviewed in Koo and Clevers54).
Figure 3.
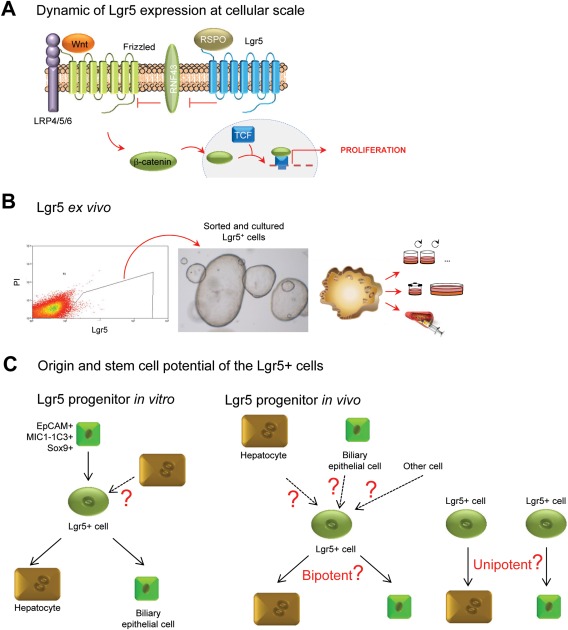
Lgr5 as a marker of liver cells following damage. (A) Dynamics of Lgr5 expression at the cellular level. (B) Ex vivo, sorted and cultured Lgr5+ cells are able to form organoids, with a high degree of plasticity.22, 47 (C) In vitro, Lgr5 cells derive from biliary epithelial cells.47, 60 Because medium to grow hepatocytes in culture has not been established yet, the origin of Lgr5+ cells from hepatocytes cannot be addressed. In vitro, Lgr5 cells are bipotential, generating the two epithelial liver lineages, hepatocytes and biliary epithelial cells.22, 47 The cell of origin of Lgr5+ cells in vivo is still unknown. In vivo, Lgr5 cells trace into hepatocytes and biliary epithelial cells.22 Whether in vivo these Lgr5+ cells can generate both lineages or there are Lgr5 committed progenitors to each lineage is still unknown. Abbreviations: RSPO, R‐spondin; PI, propidium iodide; TCF, T‐cell factor.
In the liver, Wnt signaling is active in perivenous hepatocytes55 and has been shown to induce metabolic zonation of the liver lobule.56 Upon damage, either by hepatectomy,57 oval cell response,58 or central vein damage,22 Wnt signaling is highly activated (the role of Wnt and its effector, beta‐catenin, is elegantly reviewed in Nejak‐Bowen and Monga55 and is not the focus of this review). While classical canonical Wnt target genes, such as Axin 2, are detected in homeostasis in centrilobular hepatocytes, reporter mice have failed to show expression of Lgr5 under normal physiological conditions,22 although RNA analysis indicates basal expression of Lgr5 in this area.59 However, following liver damage, Lgr5, similarly to Foxl1,19 marks a population of cells that proliferates and, as shown by lineage tracing, upon damage caused by carbon tetrachloride, 3,5‐diethoxycarbonyl‐1,4‐dihydrocollidine, or methionine and choline‐deficient diet supplemented with ethionine, differentiate into hepatocytes and/or ductal cells.22 In vitro (Fig. 3B), these damage‐induced Lgr5+ cells exhibit stem cell potential; they can be expanded from single cells (clonogenic) into self‐sustaining liver organoids, while at the same time being able to differentiate toward cholangiocytes and hepatocytes (bipotentiality) in vitro and in vivo, after transplantation in the FAH‐/‐ mouse model.22 Unfortunately, Lgr5+ cells have not been transplanted in other liver disease models, such as following partial hepatectomy or injury from liver toxins. These models have proven very useful for the analysis of bipotentiality and stem cell behavior of neonatal and adult EpCAM+ cells derived from human donors.16, 17 Future studies are expected to answer this question.
Still, many questions regarding the role of Lgr5 in liver regeneration remain unresolved. While in vitro these cells show bipotential competency, their behavior in vivo is still unknown (Fig. 3C). In fact, a drawback of the lineage tracing experiments using an Lgr5Cre driver is that these experiments were not performed at the clonal level (as discussed above). Therefore, whether in vivo these Lgr5+ cells that appear after damage are bipotential or indeed there are two types of Lgr5 progenitors for the hepatocyte and ductal lineages (Fig. 3C) remains unresolved. Also, because this marker only appears after damage, the cell of origin from which these Lgr5+ cells arise in vivo is still unknown. Of note, in vitro mouse ductal MIC1‐1C3+ cells60 or human EpCAM+ liver cells generate liver organoids that express LGR5.47 Whether in vivo EpCAM+ cells are the cells of origin of Lgr5 damage‐induced cells is unknown.
Because of the essential role of Lgr5 in enhancing Wnt signaling, it is tempting to hypothesize that LGR5 could be sensing higher levels of Wnt upon damage, which in turn could be inducing an active proliferative response on those specific cells to repair the tissue and reinstate homeostasis. It is worth mentioning that the dynamics of Lgr5 expression following injury indicate that LGR5 should be expressed early after the onset of damage and should be switched off again once the tissue is regenerated.22 Thus, it is plausible to speculate that Lgr5 could be acting as a switch between on and off states that instructs the cells whether to proliferate or not depending on the levels of Wnt in the environment. If that is the case, then Lgr5 would be marking cells that exhibit high plasticity and can move back and forth between different stem and differentiation states. If so, it is feasible to hypothesize that perturbations in the system could break the fine line between proliferation and differentiation and result in disastrous consequences such as tissue hyperproliferation (cancer) or degeneration (cirrhosis). With respect to that option, it has been recently shown that murine liver cancer cells have a similar expression pattern to Lgr5 liver progenitors induced after damage,61 suggesting that deregulation of a Wnt‐driven regenerative response could be a factor contributing to liver cancer. Of note, hepatocellular carcinomas harbor mutations in beta‐catenin or other Wnt pathway components, which could be reflecting a mechanism of the tissue to activate proliferation by enhancing Wnt signaling.
Overall, we are just beginning to understand the role of Lgr5 in stem cell maintenance and repair. In the liver, future studies will be required to identify the cells from which Lgr5+ damaged cells arise and the role of Lgr5 during regeneration. Whether in the liver Lgr5 is implicated in tissue plasticity remains an open question.
Conclusions
It is well established that high proliferative tissues such as the gut and the skin have evolved mechanisms to prevent tissue degeneration in the event of damage to their bona fide stem cell compartments. Thus, ablation of the stem cell pool results in activation of “reserve” populations or, also on the dedifferentiation of mature cell types toward a more stem cell state (potential/plasticity), to allow the repair of the tissue and reinstate homeostasis.2 Similarly, increasing evidence suggests that activation of a “stem cell state” in a priori non‐stem cell pools is not unique to the gut or the skin but occurs across many tissues. Thus, stomach62 and lung8 differentiated cells have also demonstrated the acquisition of stem cell properties (stem cell potential) upon damage to the tissue, in what are examples of cellular plasticity. Here, we have discussed the evidence on stem cell plasticity in the liver. The remarkable regenerative capacity of the liver under many different types of liver injury makes it a champion of cellular plasticity. Liver differentiated cells, potential resident stem cells, and even bone marrow stem cells can be dedifferentiated, activated, or recruited, respectively, to recover the damaged liver. This capacity of cells to acquire a stem cell state may highlight a more universal phenomenon. Whether this plasticity is relevant to disease states is yet to be determined, but recent evidence suggests that, at least in the intestine, dedifferentiation of non‐stem cells results in acquisition of a tumor‐initiating stem cell competency,63 thus highlighting the concept of bidirectional conversion and cellular plasticity as potentially relevant not only to tissue repair but also to tumorigenesis.
Author names in bold designate shared co‐first authorship.
Conflict of interest: Nothing to report.
Supported by the Wellcome Trust and the Royal Society (104151/Z/14/Z, to M.H., who is a Wellcome Trust Sir Henry Dale Fellow) and by the Interuniversity Attraction Poles (phase VII, contract P7/47, Federal Science Policy, to L.D.).
REFERENCES
- 1. Li L, Clevers H. Coexistence of quiescent and active adult stem cells in mammals. Science 2010;327:542‐545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Blanpain C, Fuchs E. Stem cell plasticity. Plasticity of epithelial stem cells in tissue regeneration. Science 2014;344:1242281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gurdon JB. Adult frogs derived from the nuclei of single somatic cells. Dev Biol 1962;4:256‐273. [DOI] [PubMed] [Google Scholar]
- 4. Sell S. Heterogeneity and plasticity of hepatocyte lineage cells. Hepatology 2001;33:738‐750. [DOI] [PubMed] [Google Scholar]
- 5. Theise ND, Wilmut I. Cell plasticity: flexible arrangement. Nature 2003;425:21. [DOI] [PubMed] [Google Scholar]
- 6. Sell S. The hepatocyte: heterogeneity and plasticity of liver cells. Int J Biochem Cell Biol 2003;35:267‐271. [DOI] [PubMed] [Google Scholar]
- 7. Theise ND. Gastrointestinal stem cells. III. Emergent themes of liver stem cell biology: niche, quiescence, self‐renewal, and plasticity. Am J Physiol Gastrointest Liver Physiol 2006;290:G189‐G193. [DOI] [PubMed] [Google Scholar]
- 8. Tata PR, Mou H, Pardo‐Saganta A, Zhao R, Prabhu M, Law BM, et al. Dedifferentiation of committed epithelial cells into stem cells in vivo . Nature 2013;503:218‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Miyajima A, Tanaka M, Itoh T. Stem/progenitor cells in liver development, homeostasis, regeneration, and reprogramming. Cell Stem Cell 2014;14:561‐574. [DOI] [PubMed] [Google Scholar]
- 10. Macdonald RA. “Lifespan” of liver cells. Autoradiographic study using tritiated thymidine in normal, cirrhotic, and partially hepatectomized rats. Arch Intern Med 1961;107:335‐343. [DOI] [PubMed] [Google Scholar]
- 11. Furuyama K, Kawaguchi Y, Akiyama H, Horiguchi M, Kodama S, Kuhara T, et al. Continuous cell supply from a Sox9‐expressing progenitor zone in adult liver, exocrine pancreas and intestine. Nat Genet 2011;43:34‐41. [DOI] [PubMed] [Google Scholar]
- 12. Carpentier R, Suner RE, van Hul N, Kopp JL, Beaudry JB, Cordi S, et al. Embryonic ductal plate cells give rise to cholangiocytes, periportal hepatocytes, and adult liver progenitor cells. Gastroenterology 2011;141:1432‐1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tarlow BD, Finegold MJ, Grompe M. Clonal tracing of Sox9+ liver progenitors in mouse oval cell injury. Hepatology 2014;60:278‐289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang B, Zhao L, Fish M, Logan CY, Nusse R. Self‐renewing diploid Axin2+ cells fuel homeostatic renewal of the liver. Nature 2015;524:180‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dollé L, Gilgenkrantz H. The puzzle of liver homeostasis: the centrilobular hepatocyte, a novel master piece on the chessboard? International Journal of Stem Cell Research 2015;1.2. [Google Scholar]
- 16. Schmelzer E, Zhang L, Bruce A, Wauthier E, Ludlow J, Yao HL, et al. Human hepatic stem cells from fetal and postnatal donors. J Exp Med 2007;204:1973‐1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cardinale V, Wang Y, Carpino G, Cui CB, Gatto M, Rossi M, et al. Multipotent stem/progenitor cells in human biliary tree give rise to hepatocytes, cholangiocytes, and pancreatic islets. Hepatology 2011;54:2159‐2172. [DOI] [PubMed] [Google Scholar]
- 18. Michalopoulos GK. Liver regeneration. J Cell Physiol 2007;213:286‐300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shin S, Walton G, Aoki R, Brondell K, Schug J, Fox A, et al. Foxl1‐Cre‐marked adult hepatic progenitors have clonogenic and bilineage differentiation potential. Genes Dev 2011;25:1185‐1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dorrell C, Erker L, Schug J, Kopp JL, Canaday PS, Fox AJ, et al. Prospective isolation of a bipotential clonogenic liver progenitor cell in adult mice. Genes Dev 2011;25:1193‐1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Espanol‐Suner R, Carpentier R, Van Hul N, Legry V, Achouri Y, Cordi S, et al. Liver progenitor cells yield functional hepatocytes in response to chronic liver injury in mice. Gastroenterology 2012;143:1564‐1575. [DOI] [PubMed] [Google Scholar]
- 22. Huch M, Dorrell C, Boj SF, van Es JH, Li VS, van de Wetering M, et al. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt‐driven regeneration. Nature 2013;494:247‐250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Suzuki A, Sekiya S, Onishi M, Oshima N, Kiyonari H, Nakauchi H, et al. Flow cytometric isolation and clonal identification of self‐renewing bipotent hepatic progenitor cells in adult mouse liver. Hepatology 2008;48:1964‐1978. [DOI] [PubMed] [Google Scholar]
- 24. Kaneko K, Kamimoto K, Miyajima A, Itoh T. Adaptive remodeling of the biliary architecture underlies liver homeostasis. Hepatology 2015;61:2056‐2066. [DOI] [PubMed] [Google Scholar]
- 25. Schaub JR, Malato Y, Gormond C, Willenbring H. Evidence against a stem cell origin of new hepatocytes in a common mouse model of chronic liver injury. Cell Rep 2014;8:933‐939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yanger K, Knigin D, Zong Y, Maggs L, Gu G, Akiyama H, et al. Adult hepatocytes are generated by self‐duplication rather than stem cell differentiation. Cell Stem Cell 2014;15:340‐349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Choi TY, Ninov N, Stainier DY, Shin D. Extensive conversion of hepatic biliary epithelial cells to hepatocytes after near total loss of hepatocytes in zebrafish. Gastroenterology 2014;146:776‐788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lu WY, Bird TG, Boulter L, Tsuchiya A, Cole AM, Hay T, et al. Hepatic progenitor cells of biliary origin with liver repopulation capacity. Nat Cell Biol 2015;17:971‐983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tarlow BD, Pelz C, Naugler WE, Wakefield L, Wilson EM, Finegold MJ, et al. Bipotential adult liver progenitors are derived from chronically injured mature hepatocytes. Cell Stem Cell 2014;15:605‐618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hattoum A, Rubin E, Orr A, Michalopoulos GK. Expression of hepatocyte epidermal growth factor receptor, FAS and glypican 3 in EpCAM‐positive regenerative clusters of hepatocytes, cholangiocytes, and progenitor cells in human liver failure. Hum Pathol 2013;44:743‐749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dollé L, Best J, Mei J, Al Battah F, Reynaert H, van Grunsven LA, et al. The quest for liver progenitor cells: a practical point of view. J Hepatol 2010;52:117‐129. [DOI] [PubMed] [Google Scholar]
- 32. Itoh T, Miyajima A. Liver regeneration by stem/progenitor cells. Hepatology 2014;59:1617‐1626. [DOI] [PubMed] [Google Scholar]
- 33. Rountree CB, Barsky L, Ge S, Zhu J, Senadheera S, Crooks GM. A CD133‐expressing murine liver oval cell population with bilineage potential. Stem Cells 2007;25:2419‐2429. [DOI] [PubMed] [Google Scholar]
- 34. Qiu Q, Hernandez JC, Dean AM, Rao PH, Darlington GJ. CD24‐positive cells from normal adult mouse liver are hepatocyte progenitor cells. Stem Cells Dev 2011;20:2177‐2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dollé L, Best J, Empsen C, Mei J, Van Rossen E, Roelandt P, et al. Successful isolation of liver progenitor cells by aldehyde dehydrogenase activity in naive mice. Hepatology 2012;55:540‐552. [DOI] [PubMed] [Google Scholar]
- 36. Dollé L, Streetz KL, van Grunsven LA. Sharpen your look on liver progenitor cells. Hepatology 2012;55:319‐321. [DOI] [PubMed] [Google Scholar]
- 37. Cheung TH, Rando TA. Molecular regulation of stem cell quiescence. Nat Rev Mol Cell Biol 2013;14:329‐340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wabik A, Jones PH. Switching roles: the functional plasticity of adult tissue stem cells. EMBO J 2015;34:1164‐1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Folmes CD, Dzeja PP, Nelson TJ, Terzic A. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell 2012;11:596‐606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dollé L, Theise ND, Schmelzer E, Boulter L, Gires O, van Grunsven LA. EpCAM and the biology of hepatic stem/progenitor cells. Am J Physiol Gastrointest Liver Physiol 2015;308:G233‐G250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Guillemot JC, Naspetti M, Malergue F, Montcourrier P, Galland F, Naquet P. Ep‐CAM transfection in thymic epithelial cell lines triggers the formation of dynamic actin‐rich protrusions involved in the organization of epithelial cell layers. Histochem Cell Biol 2001;116:371‐378. [DOI] [PubMed] [Google Scholar]
- 42. Maghzal N, Kayali HA, Rohani N, Kajava AV, Fagotto F. EpCAM controls actomyosin contractility and cell adhesion by direct inhibition of PKC. Dev Cell 2013;27:263‐277. [DOI] [PubMed] [Google Scholar]
- 43. Maetzel D, Denzel S, Mack B, Canis M, Went P, Benk M, et al. Nuclear signalling by tumour‐associated antigen EpCAM. Nat Cell Biol 2009;11:162‐171. [DOI] [PubMed] [Google Scholar]
- 44. Cirulli V, Crisa L, Beattie GM, Mally MI, Lopez AD, Fannon A, et al. KSA antigen Ep‐CAM mediates cell‐cell adhesion of pancreatic epithelial cells: morphoregulatory roles in pancreatic islet development. J Cell Biol 1998;140:1519‐1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Balzar M, Winter MJ, de Boer CJ, Litvinov SV. The biology of the 17‐1A antigen (Ep‐CAM). J Mol Med (Berl) 1999;77:699‐712. [DOI] [PubMed] [Google Scholar]
- 46. Schmelzer E, Reid LM. EpCAM expression in normal, non‐pathological tissues. Front Biosci 2008;13:3096‐3100. [DOI] [PubMed] [Google Scholar]
- 47. Huch M, Gehart H, van Boxtel R, Hamer K, Blokzijl F, Verstegen MM, et al. Long‐term culture of genome‐stable bipotent stem cells from adult human liver. Cell 2015;160:299‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yoon SM, Gerasimidou D, Kuwahara R, Hytiroglou P, Yoo JE, Park YN, et al. Epithelial cell adhesion molecule (EpCAM) marks hepatocytes newly derived from stem/progenitor cells in humans. Hepatology 2011;53:964‐973. [DOI] [PubMed] [Google Scholar]
- 49. Stueck AE, Wanless IR. Hepatocyte buds derived from progenitor cells repopulate regions of parenchymal extinction in human cirrhosis. Hepatology 2015;61:1696‐1707. [DOI] [PubMed] [Google Scholar]
- 50. Rudnick DA, Davidson NO. Functional relationships between lipid metabolism and liver regeneration. Int J Hepatol 2012;2012:549241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gebhardt R, Matz‐Soja M. Liver zonation: novel aspects of its regulation and its impact on homeostasis. World J Gastroenterol 2014;20:8491‐8504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Huang J, Rudnick DA. Elucidating the metabolic regulation of liver regeneration. Am J Pathol 2014;184:309‐321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. de Lau W, Barker N, Low TY, Koo BK, Li VS, Teunissen H, et al. Lgr5 homologues associate with Wnt receptors and mediate R‐spondin signalling. Nature 2011;476:293‐297. [DOI] [PubMed] [Google Scholar]
- 54. Koo BK, Clevers H. Stem cells marked by the R‐spondin receptor LGR5. Gastroenterology 2014;147:289‐302. [DOI] [PubMed] [Google Scholar]
- 55. Nejak‐Bowen KN, Monga SP. Beta‐catenin signaling, liver regeneration and hepatocellular cancer: sorting the good from the bad. Semin Cancer Biol 2011;21:44‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Benhamouche S, Decaens T, Godard C, Chambrey R, Rickman DS, Moinard C, et al. Apc tumor suppressor gene is the “zonation‐keeper” of mouse liver. Dev Cell 2006;10:759‐770. [DOI] [PubMed] [Google Scholar]
- 57. Tan X, Behari J, Cieply B, Michalopoulos GK, Monga SP. Conditional deletion of beta‐catenin reveals its role in liver growth and regeneration. Gastroenterology 2006;131:1561‐1572. [DOI] [PubMed] [Google Scholar]
- 58. Hu M, Kurobe M, Jeong YJ, Fuerer C, Ghole S, Nusse R, et al. Wnt/beta‐catenin signaling in murine hepatic transit amplifying progenitor cells. Gastroenterology 2007;133:1579‐1591. [DOI] [PubMed] [Google Scholar]
- 59. Yamamoto Y, Sakamoto M, Fujii G, Tsuiji H, Kenetaka K, Asaka M, et al. Overexpression of orphan G‐protein‐coupled receptor, Gpr49, in human hepatocellular carcinomas with beta‐catenin mutations. Hepatology 2003;37:528‐533. [DOI] [PubMed] [Google Scholar]
- 60. Dorrell C, Tarlow B, Wang Y, Canaday PS, Haft A, Schug J, et al. The organoid‐initiating cells in mouse pancreas and liver are phenotypically and functionally similar. Stem Cell Res 2014;13:275‐283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nikolaou KC, Moulos P, Chalepakis G, Hatzis P, Oda H, Reinberg D, et al. Spontaneous development of hepatocellular carcinoma with cancer stem cell properties in PR‐SET7‐deficient livers. EMBO J 2015;34:430‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Stange DE, Koo BK, Huch M, Sibbel G, Basak O, Lyubimova A, et al. Differentiated Troy+ chief cells act as reserve stem cells to generate all lineages of the stomach epithelium. Cell 2013;155:357‐368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Goktuna SI, Ziegler PK, et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem‐cell‐like properties. Cell 2013;152:25‐38. [DOI] [PubMed] [Google Scholar]