

Abstract
Although the complement system is primarily perceived as a host defence system, a more versatile, yet potentially more harmful side of this innate immune pathway as an inflammatory mediator also exists. The activities that define the ability of the complement system to control microbial threats and eliminate cellular debris — such as sensing molecular danger patterns, generating immediate effectors, and extensively coordinating with other defence pathways — can quickly turn complement from a defence system to an aggressor that drives immune and inflammatory diseases. These host-offensive actions become more pronounced with age and are exacerbated by a variety of genetic factors and autoimmune responses. Complement can also be activated inappropriately, for example in response to biomaterials or transplants. A wealth of research over the past two decades has led to an increasingly finely tuned understanding of complement activation, identified tipping points between physiological and pathological behaviour, and revealed avenues for therapeutic intervention. This Review summarizes our current view of the key activating, regulatory, and effector mechanisms of the complement system, highlighting important crosstalk connections, and, with an emphasis on kidney disease and transplantation, discusses the involvement of complement in clinical conditions and promising therapeutic approaches.
Clinical observations, results from genome-wide association studies (GWAS), and insights from improved disease models have renewed interest in the human complement system. The complement system is now understood to contribute to a growing list of immune, inflammatory and age-related conditions, with kidney disorders assuming a particularly prominent place1–3. The strong association between the complement system and disease might at first seem unexpected, given the beneficial role of complement as a first line of defence against microbial threats. Protection from infection is certainly a defining function of this ancient innate immune pathway, especially during the early stages of life when adaptive immunity is still developing. Rapid recognition and opsonic tagging of microbial intruders by complement components not only facilitates elimination of pathogens but also supports the development of adaptive immune responses1,4,5.
In the past few decades, however, seminal discoveries have led to the realization that such an intricate system with unique surface recognition capabilities would not be evolutionarily maintained for the sole purpose of killing microorganisms. Rather, complement exerts much broader functions in immune surveillance and homeostasis1. For example, complement assists in the clearance of immune complexes, cellular debris, and apoptotic cells and has been associated with early development and tissue repair1,6–9. Such a broad involvement in physiological processes can only be achieved through close communication of the complement system with other regulatory systems. Indeed, numerous relationships involving crosstalk between complement and other biological systems have meanwhile been described, underscoring a role for complement as an immunological mediator, rather than merely an antimicrobial effector1,5. Newer studies also suggest that complement is not a strictly intravascular system; instead, local secretion of complement components by tissue and infiltrating cells, and potentially even intracellular complement turnover, contribute to the overall complement response in many circumstances10. Yet the prime surface-recognition capabilities, functional versatility, and tight interconnectivity of the complement system also provide a basis for its undesirable effects. The emerging picture of complement is one of a powerful surveillance system that can be inadvertently triggered by injured, damaged, or altered cells and by biosurfaces used in modern medicine. Autoimmune reactions, age-related modifications, deficiencies, and genetic alterations in complement proteins often exacerbate complement-mediated damage and tip the balance from protection to destruction2,11. This Review provides an overview of the complement network and its crosstalk with other systems. We discuss the mechanisms by which complement contributes to disease pathogenesis with relevant clinical examples, and highlight therapeutic opportunities for complement-targeted drugs.
Key points.
The complement system is a critical modulator of immune responses that triages microbial and other threats through pattern recognition, tailored effector functions, and intensive crosstalk with other systems
When disrupted by dysregulation or age-related effects, or excessively triggered by acute and chronic tissue damage, biomaterials or transplants, complement can attack host cells and contribute to inflammatory conditions
The kidney is particularly sensitive to complement-mediated damage, exemplified by the involvement of complement in several kidney diseases (such as atypical haemolytic uraemic syndrome (aHUS) and C3 glomerulopathies) and in complications of kidney transplantation and haemodialysis
Therapeutic complement inhibition is successfully used in paroxysmal nocturnal haemoglobinuria and aHUS, and has shown promise in other clinical conditions, raising hope for improved treatment options for other disorders
Complement in immune surveillance
Despite its diverse functions, activation of the complement system typically follows a simple yet effective sequence that involves surface recognition and opsonic tagging of a target cell, self-amplification, generation of effector molecules, and induction of immune signalling. The involvement of individual recognition molecules and modulators determines the breadth and severity of the complement response1,4 to ensure that the reaction to an apoptotic cell is more tempered than that toward an invading pathogen, for example, and to spare healthy host cells from complement activation. Studies over the past decade have revealed insights into the molecular machinery that facilitates this immune surveillance4. The below section summarizes the major mechanisms by which complement is activated and modulated under physiological conditions, focusing on overarching concepts rather than the traditional distinction of classical, lectin, and alternative pathways (BOX 1).
Box 1. The complement ‘pathways’.
Complement activation is typically described to occur through three distinct ’pathways’1,4: the ‘classical pathway’ involves antibody-mediated activation via the C1 complex comprising C1q, C1r and C1s; the ‘lectin pathway’ is triggered by carbohydrate signatures on cell surfaces; and the ‘alternative pathway’ involves direct activation of C3 through surface-binding or tick-over, and properdin (FP)-mediated binding of C3b. However, improved understanding of the mechanisms of complement activation and the recognition of complement functions beyond host defence has led to a move away from this strict separation of initiation routes. For example, antibodies can also initiate the lectin pathway via their carbohydrate portions, and proteases of the MASP family that are typically attributed to lectin pathway activation might well have a role in the alternative pathway175–177. Of note, however, some of the activities of MASP-1/3, such as the cleavage of pro-factor D (FD) to mature FD, might not depend on lectin pathway activation but rather be mediated in a ‘homeostatic’ manner independent of prior pattern recognition175,176. Depending on the cellular target or disease state, several initiation mechanisms might be triggered simultaneously. The traditional pathway terminology can itself cause confusion, since the ‘alternative’ pathway is often the dominant contributor to the overall complement response through the amplification loop14,15. Moreover, the alternative pathway can be divided into at least two arms: one that initiates complement activation and invokes pattern recognition (that is, by properdin) or hydrolysis of C3 (that is, by tick-over), and one that mediates amplification independent of the initiating mechanism. Finally, ‘extrinsic’ proteases that are not traditionally associated with the complement system, in particular those from the coagulation cascade (for example thrombin and plasmin) can directly activate C3 and/or C5, thereby providing an additional, context-specific pathway of complement activation31,178. Although the differentiation of the classical, lectin and alternative pathways remains helpful in many circumstances, this distinction should be used with care.
Danger sensing and complement activation
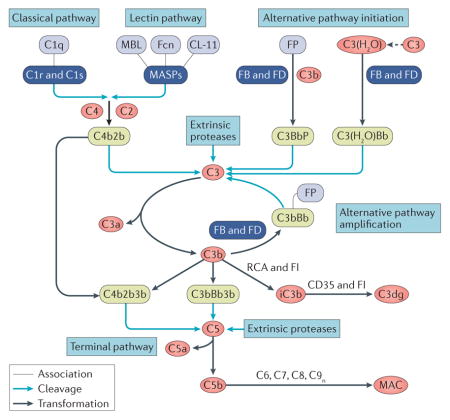
A key function of complement is the sensing of potential insults, which is achieved through the detection of pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) by specialized recognition molecules (FIG. 1). C1q predominantly binds to IgG and IgM when arranged in antigen–antibody complexes12 but also detects other molecules such as lipopolysaccharides and phosphatidylserine4,13. In contrast, mannose-binding lectin (MBL) mainly recognizes carbohydrate patches that are rich in D-mannose and L-fucose. Although MBL has long been considered the prime initiator of the lectin pathway (BOX 1), it is now clear that other recognition molecules such as ficolins and collectin-11 are also involved in sensing carbohydrates and/or acetylated moieties on microbial and apoptotic cells4. In the circulation, the recognition molecules are associated with serine proteases: C1q associates with C1r and C1s, and MBL, ficolins, and collectin-11 form complexes with MBL-associated serine proteases (MASPs). Binding of these complexes to target surfaces leads to activation of the associated serine proteases, which then cleave the plasma proteins C2 and C4. Activation of C4 produces the opsonin C4b, which is deposited on the target surface via a thioester moiety. The cleavage fragment of C2, in turn, binds to C4b to generate a C3 convertase complex (termed either C4b2b or C4b2a in the literature), which activates the abundant complement component C3. Convertase-mediated cleavage of C3 liberates the anaphylatoxin C3a and generates the versatile opsonin C3b that, like C4b, covalently binds to activating surfaces. The high plasma concentration of C3 (~1 mg/ml), which is relatively inert in its native form, is a critical factor in the rapid reactivity of the complement system. Conversely, the ultra-short reaction time of the activated thioester moiety of C3b and C4b restricts opsonization to the immediate site of activation and reduces bystander opsonization of other cells4.
Figure 1. Mechanisms of differential complement activation and regulation under physiological conditions.

When complement encounters a foreign cell, the recognition molecules C1q, mannose-binding lectin (MBL), ficolins, and collectin-11 sense antibody clusters or pathogen-associated molecular patterns (PAMPs) and activate associated serine proteases (C1r, C1s or MBL-associated serine proteases (MASPs)) that cleave C4 and C2 to form the C3 convertase (C4b2b) of the classical pathway (CP) and lectin pathway (LP) on the triggering cell. In addition, tick-over or surface interactions of C3 with certain cell surfaces leads to C3 hydrolysis and generation of initial alternative pathway (AP) C3 convertases. All C3 convertases cleave C3 into the anaphylatoxin C3a and C3b, the latter of which is deposited on activating surfaces (opsonization). Factor B (FB) binds to C3b and gets activated by Factor D (FD) to form the final AP convertases (C3bBb), which activate more C3 to C3b and fuel an amplification loop. Increasing densities of C3b deposition favour the generation of C5 convertases, which activate C5 to release the anaphylatoxin C5a and C5b, which, after tiered interactions with C6–C9 and membrane insertion, in turn forms membrane attack complexes (MACs), which leads to lysis, damage, or activation of target cells. The released anaphylatoxins act as immune mediators and, particularly in the case of C5a, attract and prime immune cells. Interaction of the C3b opsonin and its degradation products iC3b and C3dg with complement receptors (CRs) mediates cell adhesion (via CR1 (also known as CD35)) and/or phagocytic uptake (via CR3, CR4, and CRIg). Concurrently, iC3b and C3dg modulate adaptive immune responses by binding to CR2 on the B cell co receptor, thereby lowering the threshold of B cell stimulation. Properdin (FP) stabilizes the C3 and C5 convertases and increases complement-mediated opsonization and effector generation. On healthy cells, complement activation is tightly controlled. C1 esterase inhibitor (C1-INH), MAP1 and sMAP regulate the activity of recognition complexes, whereas soluble C4b-binding protein (C4BP) and factor H (FH) — which recognize self-surface patterns such as heparin and sialic acid, and bind to host cells — or membrane-bound regulators of complement activation (RCA) proteins (CD35, CD46 and CD55) control convertase activity by accelerating convertase decay and/or act as cofactors for the factor I (FI)-mediated degradation of C3b and C4b. Finally, CD59, clusterin (clu) and vitronectin (vtn) prevent formation of MAC. ‘Silent’ removal of damaged and apoptotic cells, debris, and immune complexes is achieved through the sensing of damage-associated molecular patterns (DAMPs), either directly by complement recognition complexes or mediated by modulators such as pentraxins (for example, PTX3). Controlled activation of complement occurs with limited deposition of opsonins that facilitate phagocytic clearance. In addition to C3b/iC3b/C3dg, C1q also contributes to clearance by binding to C1q receptors (gC1qR and cC1qR). CA, cofactor activity; CRIg, CR of the immunoglobulin superfamily; DAA, decay acceleration activity; FcγR, Fcγ receptor.
The amplification step that follows initial opsonization often determines the fate of the target cell (FIG. 1). Surface-deposited C3b binds the protease factor B (FB), which is subsequently cleaved by factor D (FD) to form another C3 convertase, C3bBb. Importantly, this convertase is able to activate additional C3 to form C3b and produce more C3 convertases, thereby creating a self-amplification loop that leads to efficient opsonization of unprotected cells and surfaces. Under most circumstances, C3-mediated amplification by the ‘alternative pathway’ (BOX 1) contributes most of the overall complement response14,15. The increasing density of C3b opsonins on the target surface leads to the formation of convertases that preferentially cleave complement component C5. Upon activation, with release of the anaphylatoxin C5a, the resulting C5b associates with complement components C6 and C7, inserts into membranes and forms a membrane-attack complex (MAC) through binding to C8 and multiple copies of C9. Of note, although C4b can generate a C3 convertase, the presence of C3b is required for C5 convertase formation. Furthermore, C3 is continuously hydrolysed at a low rate, producing a C3b-like conformational state that can form alternative pathway-initiating C3 convertases with FB and FD. This ‘tick-over mechanism’ provides a state of complement alertness in the circulation but can also be actively induced when C3 is adsorbed on certain interfaces (for example, on platelet surfaces or biomaterials), and changes its conformation16.
Effector functions of complement
Activation and amplification of complement creates a variety of potent effectors (FIG. 1). The MAC with its lytic pore is probably the best-recognized of these effectors. However, not all cells are equally susceptible to MAC, and swift MAC-induced lysis mainly occurs in aged erythrocytes and certain Gram-negative bacteria. On nucleated cells, MAC formation is tightly regulated, assembled MAC complexes are actively removed from the membrane, and the lytic effects of MAC are counteracted by ion pumps17. These protective effects can, however, be overwhelmed — a principle that is used to achieve complement-dependent cytotoxicity in cancer therapy18. Of note, cell lysis is not the only effector function of MAC; even when formed at sublytic levels, MAC can induce damage or activation of host cells and act as a proinflammatory mediator on various cells ranging from neutrophils to platelets17.
The dominant effector in signalling danger and in the induction of immune-modulatory responses by complement is typically the other C5-derived activation product, C5a8,19. This anaphylatoxin is a potent chemoattractant, recruiting neutrophils and other cells to the activation site and priming them by binding to C5a receptor 1 (C5aR1, also known as CD88). The role of a second C5a receptor, C5aR2 (also known as C5L2), is not fully resolved and might involve decoy or modulatory functions19. C5a also exerts a variety of other effects on various cell types, including contraction of smooth muscle cells, and is an important orchestrator of immuno–inflammatory crosstalk (see below). Although C3a and C5a were once thought to have analogous functions because of their structural similarities, the two anaphylatoxins are now considered to have distinct activity spectra. Compared to the proinflammatory C5a, the activity of C3a, which signals through the C3a receptor (C3aR), seems to be more diverse and context-specific. For example, C3a does not attract neutrophils but might even counteract the neutrophil-attractant effects of C5a, and is reported to have direct antimicrobial activity20,21. As such, C3a must be regarded as a unique inflammatory modulator, not just a less-potent homologue of C5a20.
Anaphylatoxin-mediated attraction and priming of immune cells are important prerequisites for phagocytosis, which contributes to the removal of microbial threats and cellular debris through antibody and complement-assisted mechanisms. Although C1q can contribute to phagocytosis by binding to its receptors gC1qR and cC1qR (also known as calreticulin)22, complement-mediated phagocytosis is mainly driven by the interactions of opsonins on target surfaces with complement receptors on phagocytes. Complement receptor 1 (CR1, also known as CD35), which is ubiquitously expressed on erythrocytes and many nucleated cells, binds both C3b and C4b. This adhesion receptor can facilitate Fcγ-receptor-mediated uptake of antibody-tagged cells, and erythrocyte-bound CR1 is instrumental in transporting opsonized particles to the spleen and liver5. Importantly, CR1 acts as a regulator of complement activation by mediating the degradation of C3b to iC3b and C3dg, thereby shifting the signalling profile of the opsonins. Although the term iC3b had been introduced to imply an ‘inactive’ form in terms of convertase formation, iC3b is in fact among the most versatile effectors. It is a major ligand for the integrin receptors CR3 (also known as CD11b/CD18) and CR4 (also known as CD11c/CD18), which are found on most phagocytic cells. Moreover, C3b and iC3b bind to the complement receptor of the immunoglobulin family (CRIg), a phagocytic receptor on tissue-resident macrophages such as Kupffer cells23. Finally, iC3b and C3dg modulate adaptive responses by binding to CR2 (also known as CD21) on B-cells and follicular dendritic cells (as discussed below). Thus, C3-derived opsonins fulfil various immune-modulatory functions, and their density and dynamic conversion on distinct cell surfaces shape the effector profile of the complement system.
Regulating the complement response
Modulators of complement activity in the circulation and on the surface of cells are essential to prevent complement from attacking healthy host cells and to adjust the response of the complement system to microbial and homeostatic insults (FIG. 1). Regulation of the complement system starts at the initiation steps through the control of recognition molecules and their associated proteases. The circulating C1 esterase inhibitor (C1-INH) inactivates C1r, C1s, and MASP-1/2 but also acts on non-complement serine proteases4,24. Splice products of MASPs (MAP-1 and sMAP), which lack the protease domains of MASPs, compete with MASPs for binding to MBL, ficolins, and collectins25.
The assembly of C3 and C5 convertases is particularly well-controlled by proteins from the ‘regulators of complement activation’ (RCA) family. These modular proteins, which are composed of complement control protein (CCP) domains, prevent the formation of convertase complexes and destabilize existing ones. They also act as cofactors for the serine protease factor I (FI) to enable the degradation of C3b and/or C4b. Among RCA members, the membrane-bound CR1 is particularly potent, since it acts on all C3 and C5 convertases and mediates full cleavage of both C4b and C3b. The other membrane-bound RCA family members confer either cofactor activity (membrane cofactor protein; also known as CD46) or convertase decay activity (decay accelerating factor; also known as CD55). The soluble RCAs C4b-binding protein (C4BP) and factor H (FH) control complement activation in the circulation and assist in modulating the response on host cell surfaces. C4BP acts on C4b-containing convertases and mediates the degradation of C4b. FH is a 20-domain protein, the regulatory activity of which is mediated by its binding to hydrolysed C3 and C3b, thereby controlling both tick-over activation and the amplification loop. The N-terminus of FH provides convertase decay activity for C3bBb and enables FI-mediated degradation of C3b to iC3b. FH also has surface-recognition capabilities that discriminate between healthy and apoptotic host cells and microbial particles. The C-terminus of FH harbours an additional binding site for C3-derived opsonins and areas that recognize polyanionic patches found in glycosaminoglycans and sialic acids on host surfaces. FH-like protein 1 (FHL-1), a splice product that consists of the first seven FH domains, has the same regulatory capacity as FH but lacks some recognition capabilities; its smaller size, however, might allow better tissue penetration with potential implications for local convertase regulation26.
By tightly controlling opsonization and convertase formation, RCAs impair C3 and C5 activation and largely prevent effector generation on host cells. However, given the potentially devastating effects of C5-derived effectors, additional mechanisms exist to keep their activity in check. The membrane-bound protein CD59 binds to active forms of C8 and C9 and prevents MAC formation. In addition, clusterin and vitronectin act as MAC regulators and scavenge partially formed MAC in solution. Finally, many host cells swiftly remove C5b–9 complexes via exocytosis or endocytosis17.
Anaphylatoxin activity is mainly regulated by carboxypeptidases that rapidly convert C3a and C5a to their desarginated forms (des-Arg), which profoundly reduces their effector functions. Residual and sometimes distinct activities of C3a-desArg and C5a-desArg when compared to their parent anaphylatoxins have been reported, although the physiological consequences of these activities remain unclear19,27. Nevertheless, these findings indicate that conversion steps do not lead to complete inactivation of complement effectors, but rather to a shift in their activity and/or signalling profiles.
Whereas the above-mentioned regulators tame the complement response to protect host cells, the plasma protein properdin (also known as factor P) acts as a positive modulator. Properdin binds to and stabilizes the C3 convertase of the amplification loop, enhancing the overall complement response. Although properdin is considered to be an initiating recognition molecule because of its ability to bind to surface structures such as lipopolysaccharide and heparin and its ability to provide an interaction platform for C3b or hydrolysed C3, the implications of this function are still debated28,29. Properdin had long been thought to be the only positive regulator of complement activation; however, several molecules that tailor complement activity on specific cell surfaces have now been described, giving rise to the concept of context-specific complement modulation (BOX 2).
Box 2. Context-specific complement modulation.
In addition to traditional activators and regulators of complement, a new set of complement modulators have emerged in the past decade. These physiological proteins bind to altered surfaces and enhance or compete with the binding of recognition molecules, activators and regulators. Among those, the factor H-related (FHR) proteins have gained particular attention. FHRs are composed of 4–9 complement control protein domains, some of which share homology with the surface-recognizing C-terminus of factor H (FH); this recognition activity can be increased by forming dimers and tetramers. Current experimental evidence points to a major role for FHR proteins in competing with FH for binding to C3b and self-surface patterns, thereby causing ‘de-regulation’ of complement on certain surfaces113. Conversely, certain members of the pentraxin family modulate complement responses by acting as a molecular bridge between non-self or altered cells and complement components. For example, the acute-phase marker C-reactive protein is reported to bind the recognition molecules C1q and ficolins but also the regulators FH and C4b-binding protein (C4BP)179. Similarly, pentraxin-3 interacts with C1q, mannose-binding lectin, and ficolins as well as C4BP, FH and the FH-competitor FHR-5.179,180 Although this dual binding of activators and regulators might seem counterintuitive, an emerging hypothesis suggests that these complex molecular interactions temper and finely tune the response of complement to homeostatic insults such as damaged or apoptotic cells. The modulator-mediated binding of recognition molecules directs the complement response to target cells and provides opsonic tagging, whereas regulator recruitment prevents amplification and excessive effector generation and rapidly converts opsonins to forms that facilitate clearance by phagocytosis181. Several aspects of this complex interplay remain to be explored, particularly regarding surface discrimination and (patho)physiological implications. However, these new discoveries demonstrate that timing, localization, and collaboration between complement proteins and modulators discriminate between a forceful inflammatory response to microbial intruders, a tempered reaction to immune complexes and cellular debris, and a non-consequential probing of healthy host cells.
Although complement is mainly regarded as an intra-vascular system, increasing evidence supports an important role for complement components that are produced locally by specific cells or tissues30. Indeed, complement components are not only synthesized by the liver but by many other cells. For example, adipocytes are the primary source of FD production, and platelets and neutrophils store several complement proteins in their granules. Differential activation of complement in particular tissues is an important but not yet fully appreciated phenomenon that might help the immune system to distinguish between beneficial and pathogenic triggers.
Complement beyond the cascade
The effector mechanisms of complement contribute directly to the elimination of undesired particles; however, full capacity of the complement system is only achieved through its extensive collaboration with other defence systems1,5. Indeed, a plethora of crosstalk mechanisms have been described and extensively reviewed over the past few years1,5,10. Although the extent of its molecular and cellular connections could suggest an indispensable role for complement in the coordination of immunological responses, it is important to remember its primary role as a first-in-line threat detector. The crosstalk between effectors generated after upstream complement-mediated sensing of PAMPs and DAMPs with other defence systems is important for translating the threat message into cellular signals that modulate a downstream response1,8. Whereas a reduction in complement crosstalk, for example due to deficiencies in complement components, could influence the responsiveness of the system to certain insults, the overall effect on the functioning of the connected systems might often be small. Conversely, as discussed later, inadvertent triggering of the complement system will often have downstream inflammatory consequences. The following section describes important examples and emerging concepts related to complement-mediated crosstalk. Some of these mechanisms might be highly context-specific and many mechanisms that have been described in animal models await confirmation in human systems; however, these examples illustrate how tightly embedded the complement system is in many aspects of immune surveillance and homeostasis.
Crosstalk with coagulation systems
Cooperation between the complement and coagulation systems has many potential implications for health and disease31. Both cascades are driven by tiered, serine protease-mediated activation steps, and examples of each pathway activating the other have been reported. For instance, coagulation enzymes such as thrombin and kallikrein can activate C3 and/or C5, whereas certain MASPs can cleave fibrinogen, prothrombin and factor XIII, among others. However, these cross-activation activities are typically low when compared to their normal routes of activation, and the (patho) physiological implications of this crosstalk remain to be explored. Independent of their mode of generation, anaphylatoxins influence coagulation through direct effects on platelets, neutrophils and endothelial cells or by stimulating pro-coagulant cytokines. For example, C5aR1 can induce expression of tissue factor (TF), thereby triggering the extrinsic coagulation pathway32. Conversely, thrombin-activatable fibrinolysis inhibitor (TAFI) is a carboxypeptidase (carboxypeptidase B2), the active form of which desarginates anaphylatoxins and tames their effects. von Willebrand factor (vWF) has a complex modulatory role in complement–coagulation interplay. Ultra-large multimeric forms of vWF, as observed after tissue injury, can provide a binding platform for C3b to trigger complement activation. Concurrently, vWF interacts with FH and enhances its cofactor activity for the FI-mediated degradation of C3b. Conversely, FH seems to interfere with the hydrolysis of vWF multimers by ADAMTS13. This bidirectional modulation has been proposed to strengthen platelet aggregation while keeping complement activation in control33.
The connection between complement and platelet activation has garnered considerable interest in the past few decades, but remains obscure34. Platelets seem largely unaffected by complement when in their quiescent form; upon activation, however, platelets engage with complement through a complex interplay. For example, chondroitin sulfate exposed on the platelet surface is recognized by both the activator C1q and the regulators FH and C4BP. Exposed gC1qR can trigger additional C1q binding to platelets, and P-selectin, acting as a ligand for C3b, can initiate convertase formation on the surface of activated platelets. A 2015 study demonstrated that C3 can adhere to activated platelets whereupon it is transformed to a hydrolysed-like state that is capable of forming convertases and binding to CR3 (REF. 35). The release of complement components such as FD from the α-granules of activated platelets might fuel convertase-mediated complement turnover. Platelets themselves are well-protected from complement damage through the expression and engagement of complement regulators31,34; however, the generation of complement effectors could be important in enhancing platelet activation. Soluble MAC (that is, sC5b–9) can trigger the secretion of the platelets’ α-granules, and C1q induces the expression of P-selectin, among other effects34. Anaphylatoxins have also been described as platelet activators. Although C3aR and C5aR1 are barely detectable on resting platelets, both are strongly upregulated upon platelet activation. The physiological effects of complement–platelet crosstalk on the defensive actions of complement or on platelet clearance remains debated, but current observations suggest that tempered complement activation on activated platelets by effector-mediated stimulation serves to sustain or amplify platelet activation31.
Coordination with other immune pathways
Complement activation products, particularly C3a and C5a, can modulate the activation state of most immune cell types, including neutrophils, monocytes, macrophages, and dendritic cells19,36,37 (FIG. 2). For example, a positive feedback mechanism between complement and neutrophil activation has been described in vitro: C5a can activate neutrophils to secrete properdin that, in turn, activates complement on released neutrophil extracellular traps to generate more C5a. This perpetuation of neutrophil-derived inflammation signals might enhance defence mechanisms but also have pathological consequences in diseases such as antineutrophil cytoplasmic autoantibody (ANCA)-associated vasculitis (see below)38,39. By contrast, C3a-mediated signalling demonstrated a protective effect in a mouse model of ischaemia–reperfusion injury (IRI), constraining neutrophil mobilization in response to injury40. Complement has also been linked to the development and recruitment of myeloid-derived suppressor cells (MDSCs), which are increasingly recognized as important players in the immune response to cancer or transplants. For example, the induction of MDSC development by hepatic stellate cells in cultured murine dendritic cells was strongly dependent on C3, and addition of exogenous iC3b to dendritic cell cultures influenced MDSC differentiation41. In the tumour microenvironment, complement activation leads to the recruitment and/or activation of MDSCs in a C5a-dependent manner, as shown in a syngeneic cervical cancer model42.
Figure 2. Examples of complement crosstalk with immune cells and defence pathways.

The generation of complement effectors stimulates a broad spectrum of downstream immune, inflammatory, and procoagulative responses. The anaphylatoxin C5a, for example, exerts strong proinflammatory effects by acting as a chemoattractant and stimulator of various immune cells via C5aR1-mediated signalling, thereby influencing priming and activation with release of mediators (for example, cytokines, neutrophil extracellular traps (NETs)), differentiation, and functional activity. C3a has a distinct spectrum from C5a and has, for example, been shown to activate mast cells. Activation of professional phagocytes induces the expression of complement receptors, which enable complement-mediated phagocytosis, whereas crosstalk between C5aR, FcγR, and dectin-1 also affects antibody-mediated uptake. Adherence of opsonins to CR1 on erythrocytes is an important mechanism that directs immune complexes to the liver and spleen. Complement activation also modulates adaptive immune responses by lowering the threshold of B-cell stimulation (via iC3b or C3dg interaction with CD21) or by influencing T-cell activation (for example, by binding of C3b to CD46), differentiation, and homeostasis. Complement effectors such as C5a, sublytic membrane attack complex (MAC), and MASP-1, can directly activate endothelial cells and, for example, increase expression of tissue factor (TF) as an inducer of coagulation. Serine proteases of the complement and coagulation systems might cross-activate under certain circumstances to contribute to thrombo-inflammation. Concomitantly, the release of complement proteins and binding of both complement activators and regulators to platelets might amplify the platelet response and contribute to clearance of platelets and pathogens alike. BCR, B-cell receptor; NK, natural killer; TLR, Toll-like receptor.
Interactions with antigen presenting cells
Complement has a dual role in the activation of antigen presenting cells (APCs), depending on the target receptor and cell type involved. Activation of macrophages by C1q or iC3b induces the production of IL-10 and their participation in the clearance of apoptotic cells and damaged molecules — physiologic mechanisms that are not associated with an inflammatory process. Conversely, macrophage stimulation with C3a, C5a or C5b–9 usually induces a proinflammatory phenotype with the production of iNOS, TNF, and IL-1β driving pathogen elimination37. Similarly, engagement of C3aR or C5aR1 on dendritic cells is associated with their activation via PI3K/AKT, ERK, and NF-κB signalling, whereas C1q supports the differentiation of monocytes toward dendritic cells by engaging the leukocyte-associated immunoglobulin-like receptor 1 (REFS 43,44).
Given the broad effects of complement on APC responses, it is reasonable to assume that APCs primed in the presence or absence of complement-derived signals will differentially modulate T-cell responses. A role for C5a in APC activation, with consequent polarization of CD4+ T cells, has long been appreciated45. C3a and C5a modulate T-cell responses by regulating the expression levels of major histocompatibility complex class II and co-stimulatory molecules, and the production of cytokines by APCs46.
Modulation of T cell activity
In the past 5 years, new concepts relating to the direct modulation of T-cell responses by complement have emerged. Local activation of T cell-derived autocrine C3 and C5 has been suggested to upregulate C3aR and C5aR1 expression on both APCs and T cells, resulting in direct activation of CD4+ T cells46. However contradictory findings, such as a reported inability to detect C5aR1 on T cells in C5aR-GFP reporter mice47, suggest further research is needed to determine the conditions under which anaphylatoxins might affect T cells47,48. Signalling via CD46, which serves as a regulator of and a receptor for C3b and other ligands, represents another mechanism by which complement interacts with immune pathways. Activated T cells produce C3, which, when locally activated to C3b, can trigger CD46-derived signals that are critical for the induction of type 1 T helper (TH1) cell-mediated immunity via the Notch signalling pathway and IL-10 production49,50. Consistent with these findings, individuals who are deficient in CD46 or C3 have suboptimal TH1 responses in vitro50,51. A 2013 study demonstrated that intracellular C3 can be processed into C3a and C3b by the T cell-produced protease cathepsin L; the resulting CD46 and C3aR-mediated signalling regulated the induction of TH1 and TH17 cell responses52.
Interactions with pattern recognition receptors
In addition to complement, DAMPs and PAMPs are sensed by several classes of pattern recognition receptors (PRRs), including Toll-like receptors (TLRs), leucine-rich repeat-containing receptors (NLRPs), nucleotide-binding oligomerization domain (NOD)-like receptors, RIG-I-like receptors, and C-type lectin receptors53. Although some physiological roles of these receptors are still under investigation, evidence indicates that crosstalk between complement and PRRs determines the quality and magnitude of innate responses and the polarization of adaptive immunity1,5,46,53,54. Notably, while PRRs are characterized by their tissue-specific expression, complement components are expressed by virtually all tissues but at differing levels55. The distribution of and interplay between different PRRs and complement is essential for host defence, particularly in exposed organs such as the kidneys, but might also drive adverse events. Indeed, PRRs and complement are involved in the pathology of the same kidney diseases and are critical factors that determine the outcome of kidney transplantation56–60.
A considerable body of work has demonstrated intense collaboration between responses mediated by complement and TLRs1,5,54,61. Complement effector signalling via C3aR and C5aR1, in particular, but also CR3 and gC1qR modulate TLR responses with effects on the production of proinflammatory cytokines by mouse and/or human APCs54,62. Of note, seemingly contrasting effects are observed depending on whether dendritic cells, monocytes, or macrophages are targeted54. This differential modulation by C5a was further explored in a 2013 study which showed that in response to lipopolysaccharide, C5a enhanced the secretion of proinflammatory cytokines by monocytes, but induced an anti-inflammatory response in macrophages, with production of anti-inflammatory IL-10 (REF. 63). Additional insight has been provided by the observation that CR3 positively modulates lipopolysaccharide-induced responses in myeloid dendritic cells, but not macrophages, by promoting endosome-mediated endocytosis of the TLR required for subsequent signalling64. In support of a bi-directional crosstalk between TLRs and complement, activation of TLR4 in a mouse model of sepsis leads to increased production of FB in tissues such as heart, lung, kidney, liver, and spleen; the resulting increase in complement activation in turn correlates with increased TLR signalling65.
Tuning of inflammasome activity
Several lines of evidence suggest that complement activation modulates inflammasome function66. For example, cooperation between C3aR, TLR, and NLRP3 has been demonstrated in human monocytes, in which C3a seems to modulate the TLR4-mediated production of IL-1β with subsequent NLRP3 inflammasome activation and induction of TH17 responses by T cells67. Sublytic amounts of MAC deposited on cell surfaces, which could reflect improper regulation in disease states, increases cytosolic Ca2+ and results in NLRP3 activation68,69. In addition, C5a has been indirectly implicated in the modulation of NLRP3 activation and inflammation induced by cholesterol crystals in atherosclerosis70. Finally, an inhibitory effect of C1q on inflammasome activation in response to apoptotic cells has been demonstrated in lipopolysaccharide-stimulated human macrophages71.
Crosstalk with adaptive immune systems
Complement participates in multifaceted collaborations between the innate and adaptive immune systems. This ‘bridging’ ability seems to have been conserved throughout evolution; in fish, among the most primitive species with both innate and adaptive immune systems, C3b has a role in the uptake of antigens by APCs, increasing the efficacy of antigen presentation and proliferation of B and T cells72–74. As mentioned earlier, iC3b and C3dg bind to CR2 (CD21), which is part of the B cell co-receptor complex (FIG. 1). The resulting co-ligation of CR2 and the B-cell receptor by iC3b/C3dg-coated antigens largely augments B-cell responses, especially at the outset of an immune response when limited amounts of antigen are available75,76. Moreover, opsonization of particles by iC3b and C3dg mediates the shuttling of antigens between B cells and follicular dendritic cells in the lymph nodes and is important for memory B-cell induction and maintenance in the germinal centres5,75,77. Importantly, robust antibody production against pathogens improves the innate immune response by facilitating C1q-mediated activation of complement.
Extensive collaboration also occurs between complement components and antibodies in providing effector functions. For example, complement receptors and Fc receptors (FcRs) coordinate to regulate phagocytosis and modulate various immune responses78,79. A 2012 study described an intricate crosstalk mechanism, whereby immune complexes carrying galactosylated IgG1, FcγRIIB, and the C-type lectin receptor Dectin-1 suppressed C5aR-derived inflammation in neutrophils, offering new mechanistic insights into the regulation of inflammation by immune complexes80.
In summary, complement determines the type and magnitude of immune responses in different tissues by communicating with other defence pathways and immune cells. Many of these events are likely to be context-specific, and the physiological relevance of each pathway remains to be defined. Ideally, such crosstalk should facilitate the rapid elimination of microbial intruders, damaged cargo, and injurious agents, and contribute to repair and the maintenance of homeostasis. However, as described below, these protective mechanisms can have deleterious effects when inappropriately activated.
Complex involvement in clinical disorders
Given that complement is a well-connected surveillance system (FIG. 2) it is not surprising that even small disturbances in its network, or an encounter with an ambiguous trigger such as a biomaterial, can provoke an unwanted response with clinical consequences. Despite the broad spectrum of clinical conditions that have been linked to complement dysregulation, there are only a few basic mechanisms that typically define the disease involvement of complement (FIG. 3). This section describes common principles of complement-based pathologies as a basis for the discussion of specific kidney-related disorders that have garnered particular attention over the past few years.
Figure 3. Pathological activation of complement.

Various events contribute to the development and progression of complement-related diseases. Excessive generation of triggers, such as the massive influx of pathogen-associated molecular patterns during sepsis or release of damage-associated molecular patterns (DAMPs) during trauma or tissue injury, leads to binding of recognition molecules and induces a complement response. These pronounced complement responses can lead to bystander activation of healthy host tissues in proximity to the initial trigger (for example, a pathogen). Moreover, the recognition of inappropriate targets, such as biomaterials (for example, implants or haemodialysis filters) or accumulating debris resulting from ageing or oxidative stress can induce misguided complement activation that contributes to inflammatory complications. Complement deficiencies can contribute to the generation of autoantibodies owing to impaired removal of immune complexes and other mechanisms, and the resulting antibodies can activate complement by binding to neoantigens on damaged cells or to self-antigens on healthy cells, with subsequent sensing by C1 complexes. Autoantibodies can also influence the activity of the complement system by interfering with the surface-recognition capacity of regulators of complement activation (RCA) proteins such as factor H (FH), or by stabilizing convertase complexes, thereby exacerbating complement activation. Gain-of-function mutations in complement components or loss-of-function mutations and deficiencies in complement regulators largely define the systemic activation profile of complement (marked by thick and thin arrows to indicate the level of activity), and can lead to attack of susceptible organs such as the kidney. In addition to circulating systemic complement, local secretion of complement components by tissue cells and infiltrating or tissue-resident immune cells contributes to activation events. The generation of complement effectors leads to the attraction and activation of immune cells, with release of proinflammatory mediators such as cytokines, reactive oxygen species (ROS) or reactive nitrogen species. The resulting cell damage further stimulates complement activation and fuels a vicious cycle of complement activation, inflammation, and tissue damage. Finally, strong crosstalk between complement and the coagulation system contributes to thrombotic events.
Overwhelming and inappropriate activation
Although much of the interest in complement-associated diseases has been driven by the identification of mutations in complement components by GWAS, in many clinical conditions complement is fully functional. However, as seen in conditions related to systemic inflammatory response syndrome (SIRS), too much of a good thing can ‘turn bad’. SIRS occurs when defence systems are suddenly presented with an overwhelming amount of PAMPs or DAMPs. As an early warning system and immune mediator, the induction of an intense complement response fuels hyperinflammatory events, which can be more devastating than the initial trigger. This fateful reaction has been described in sepsis but also after severe trauma, burns and in other SIRS-associated conditions. Numerous studies have identified complement as a key contributor to hyperinflammation in sepsis, with C5a in particular acting as an adverse effector through functions ranging from the release of cytokines and reactive oxygen species to consumptive coagulopathy, which contribute directly to lethality81,82. Genetic ablation or pharmacological blockade of C5a signalling or C3 activation improves outcomes in animal models of sepsis81,83,84. Moreover, in vitro and in vivo studies have provided evidence of synergistic effects of blocking complement and TLRs simultaneously85,86, underscoring the crosstalk that occurs between these systems in pathological events. In patients with severe trauma, the presentation of DAMPs after tissue injury can trigger complement activation, which induces SIRS and contributes to complications such as multi-organ dysfunction and/or death87–89. Given their simultaneous activation at the site of injury, crosstalk between complement, coagulation pathways and TLRs might be particularly relevant in this setting89. Interestingly, findings from animal studies suggest that complement might also influence the repair process after trauma by inducing osteoclastogenesis and contributing to wound healing89,90. A 2015 study of spinal cord injury in mice showed that early inhibition of C5aR1 during the acute phase of injury improved clinical outcomes, whereas extended blockade during the chronic phase of injury did not, indicating dual roles for complement in inflammation and repair91.
The presence of autoantibodies in the circulation and on self-surfaces can also trigger inflammation and tissue damage through inappropriate complement activation. For example, immune complexes present in systemic lupus erythematosus (SLE) cause consumption of complement components in the circulation and deposition of complement activation products in the kidney (lupus nephritis) and other organs92. Similar mechanisms are involved in antiphospholipid antibody syndrome, myasthenia gravis, and many other autoimmune diseases3,92. In patients with ANCA-associated vasculitis, neutrophil priming by C5a or other stimuli leads to the expression of proteins that are recognized by autoantibodies. This recognition further activates complement, immune cell recruitment, and neutrophils, causing a vicious cycle that induces inflammation and tissue damage to blood vessels, with the kidney being among the most affected organs93. Finally, the interaction of circulating natural antibodies with neoepitopes exposed during tissue damage can trigger antibody-mediated complement activation and contribute to conditions such as IRI94. Conversely, autoantibodies against complement components can directly modulate the complement response. Some examples, such as anti-FH antibodies that impair the surface-recognition properties of FH in atypical haemolytic uraemic syndrome (aHUS), or C3/C4 nephritic factors that stabilize assembled convertases and contribute to C3 glomerulopathies, are discussed in further detail below59,95.
Ineffective removal of debris
Another mechanism by which complement is inadvertently activated is through the failed removal of debris (FIG. 3), which is thought to contribute to the progression of age-related diseases. Ageing, lifestyle factors, and oxidative stress can lead to the formation of deposits and/or plaques that are sensed by PRRs, and the involvement of complement in this process is complex. During early stages of disease onset, the complement system has been attributed important roles in the clearance of debris and prevention of disease progression. Once debris begins to accumulate and cannot be efficiently cleared, however, perpetual activation of the complement cascade induces inflammation and tissue damage that drives degenerative processes. In models of Alzheimer disease, C1q and C3 exert neuroprotective effects via opsonic clearance of amyloid-β aggregates, whereas C1q-mediated complement activation by accumulating amyloid-β fibrils increasingly generate C5-derived effectors that maintain a focal inflammatory state, thus exacerbating disease progression96,97. Similar complement-related mechanisms have been described in other neurological and neurodegenerative diseases, as well as in atherosclerosis96–100. Another prominent example of a disease of ageing that is mediated by complement is age-related macular degeneration (AMD), a chronic degenerative eye disease, in which complement activation contributes to the geographic atrophy and choroidal neovascularization that causes loss of vision101. Although the aetiology of AMD is complex, the presence of complement components in lipoprotein deposits (known as drusen) often observed in the early stages of AMD suggests that insufficient clearance and accumulation of debris leading to activation of complement is an important factor2,101.
Misguided activation by foreign materials
Finally, complement can be inappropriately activated by the biomaterials and foreign cells that are increasingly used in modern medicine (FIG. 3). Polymers and metal surfaces present in haemodialysis filters, extracorporeal circuits, oxygenators, implants, and drug delivery devices can be recognized as foreign intruders by complement, which induces an inflammatory response that can affect the functionality of the device and patients’ quality-of-life2,102. The mechanisms of complement activation by biomaterials are diverse and material-dependent but often involve the adsorption of plasma proteins such as albumin or immunoglobulins, which bind and activate recognition molecules or complement components such as C3 (REF. 102). Cell and organ transplants, in contrast, typically express antigenic epitopes that trigger complement activation via antibody-dependent routes60. Foreign-body reactions in the context of kidney transplantation and haemodialysis are discussed in further detail below.
Missing and dysfunctional complement components
Whereas the above-described disease mechanisms are mainly attributed to excessive complement activation in the presence of an intact complement system, missing or dysfunctional components can induce or exacerbate an entirely new set of problems. Primary deficiencies have been reported for many key components of the complement system, but the frequencies and clinical impact of these deficiencies are diverse103. Deficiency of early complement components is commonly associated with autoimmune and immune complex-mediated diseases, with more than 90% of patients with deficiencies in C1 proteins developing SLE-like disorders92. Impaired scavenging and clearance of apoptotic cells and immune complexes are thought to be major causal factors. Moreover, since complement participates in the elimination of self-reactive lymphocytes, deficiencies in complement components might favour the production of autoantibodies by affecting B-cell tolerance92. Interestingly, and despite a prominent role for C3 opsonization in ‘waste disposal’ (that is, clearance of debris and apoptotic cells), C3 deficiencies are not strongly associated with autoimmune disease. These paradoxical roles of C1q and C3 have been explained by the observation that, whereas C1q primarily facilitates phagocytic uptake, C3 opsonins also mediate intracellular trafficking of apoptotic cargoes and enhance endocytic processing of antigens that are required for generating potentially adverse T-cell responses9.
Interestingly, especially in view of the intricate effector roles of C5a and MAC, primary deficiencies in terminal components (C5 to C9) seem to have limited clinical consequences, most prominently an increased risk of meningococcal infection. Similarly, C3 deficiency is mostly associated with increased susceptibility to certain pyogenic infections, such as those caused by meningococci, pneumococci and haemophilus influenzae. Finally, deficiencies in complement regulators such as FH or CD46, though rare, are typically associated with kidney diseases103. Paroxysmal nocturnal haemoglobinuria (PNH) is an ultra-rare but severe haematological disease in which somatic mutations in the genes responsible for glycosylphosphatidylinositol anchor synthesis lead to clonal populations of blood cells that lack several membrane proteins, including CD55 and CD59. As a consequence, PNH erythrocytes show increased susceptibility to complement-mediated haemolysis, for example, resulting from bystander activation during infection, with consequential anaemia and high thrombotic risk104.
More common than deficiencies are genetic variations in complement components that define the functionality of the complement system and often represent a tipping point if and when clinical issues manifest. In general, gain-of-function mutations in activator components (for example, in C3) or loss-of-function mutations in regulators (for example, in FH) predispose individuals to disease susceptibility11. The advent of GWAS in particular has led to a surge in newly identified mutations and polymorphisms that correlate with disease. In February 2016, the FH aHUS mutation database105 listed 1,146 genetic alterations in C3, FH, FI, and CD46 alone, some 400 of which were disease-related (mostly aHUS but also AMD and other conditions). Despite this wealth of information, the functional consequences of distinct genetic variations are only beginning to be understood. For example, a strong correlation between the common Tyr402His variation in FH and risk of developing AMD was described in 2005 (REF. 106), yet the exact pathological process is not yet clear. Current data indicate that the Try402 and His402 variants of FH mainly differ in their binding to self-pattern and oxidative stress markers such as malondialdehyde107.
Several of these genetic alterations might have only a small effect on the function of the affected component but might have considerable effects on the complement system as a whole by exacerbating an imbalance in complement activation, particularly in chronic conditions such as AMD. Furthermore, an increasing prevalence and diversity of complement mutations and polymorphisms increase the chance that an individual will carry a distinct set of variants, or ‘complotype’, which defines the reactivity of the complement system and an individual’s susceptibility to complement-related disease11. These concepts were underscored by studies showing that the combination of polymorphisms in C3, FB, and FH with weak individual consequences could result in a profound shift in complement activity108. Perhaps most intriguingly, mutations and/or polymorphisms, dysfunctions, and deficiencies in complement proteins are expected to lead to a systemic imbalance that can affect all cells and tissues, yet clinical manifestations are primarily detected in particular organs, such as the eye and, even more prominently, the kidneys.
Complement-mediated kidney disease
The reason for the kidney’s unique susceptibility to complement-induced damage is not fully understood but is probably influenced by its anatomical organization and functional specialization. For example, size and charge-sensitive filtration exposes glomeruli to circulating immune effectors and favours the deposition of immune complexes, and the high resorptive and secretory activity of tubular epithelial cells renders these cells susceptible to oxidative stress109. Moreover, the kidney seems to have an activated complement profile even under baseline conditions, which probably serves to remove immune complexes and provide protection from microorganisms of the urinary tract58. Glomerular filtration generally increases the local protein concentration in plasma, and it is well-appreciated that several complement components are locally produced by parenchymal cells of the glomeruli, tubules, and medulla110, with both these functions probably contributing to increased complement turnover.
The expression of complement regulators varies considerably between cell types, suggesting distinct levels of regulation within different sections of the kidney. For example, renal tubules express very low levels of complement regulators, and the apical membrane of proximal tubular cells is almost completely devoid of membrane-bound complement inhibitors111. The glomerular endothelium expresses lower levels of CD55 and CD59 than do endothelial cells from other tissues, making the glomerular endothelium more dependent on fluid-phase regulators, such as FH, to suppress complement activity, as evidenced in patients with aHUS. This dependence on FH for complement regulation is even more pronounced in the glomerular basement membrane (GBM), which is exposed to blood through the fenestrated endothelium and completely lacks membrane-bound complement regulators. Loss of endothelial cells and/or podocytes during acute kidney injury further exposes the ill-protected GBM. The glycocalyx, which coats the endothelial cells and the GBM is rich in glycosaminoglycans such as heparan sulfates, which recruit FH; the rapid loss of the glycocalyx, as observed in IRI, can increase the vulnerability of the glomerular filtration barrier. In this context, striking connections have been described between complement diseases in the kidney and eyes, as some patients with C3 glomerulopathies develop drusen in their macula. Basement membranes, such as the GBM and the subretinal Bruch’s membrane, constitute focal areas of complement activation and deposition that are largely affected by genetic variations in FH. Interestingly, however, the expression profile of heparan sulfate is distinct in the two tissues and may provide a ‘site-specific guidance’ for protection by FH and its related molecules that defines disease susceptibility. Indeed, FH seems to bind heparan sulfates in the eye mainly via its CCP7 domain, which contains the common AMD-related FH Tyr402His variation; by contrast, the C-terminal domain (CCP19–20) seem to be the major recognition site of FH for kidney heparan sulfate, explaining the frequency of aHUS-related variations in that region112. Furthermore, deregulation by factor H-related (FHR) proteins (BOX 1) is increasingly recognized as a disease-contributing factor, and genetic alterations in FHR proteins are associated with kidney diseases such as C3 glomerulopathies, aHUS, and IgA nephropathy113.
Other factors can also contribute to increased complement activation in the kidney. For example, the synthesis of ammonia by tubular epithelial cells can induce hydrolysis of C3 with subsequent formation of C3 convertases. Properdin seems to have a prominent role in kidney disease progression, but whether it acts as an initiator or modulator of complement activity is not yet clear28. Furthermore, complement activation induces the expression of P-selectin on endothelial cells, which in turn binds C3b and stabilizes the C3-convertase on the cell surface114. Since both the complement and coagulation systems are sensitive to acidic microenvironments, the comparatively low pH of the fluid within the lumen of the renal tubule can exacerbate activation of and crosstalk between the systems115. Finally, proinflammatory cytokines increase the local production of complement components by kidney cells, thereby fueling the complement response.
aHUS and other thrombotic microangiopathies
The strong connection between complement, coaglation pathways and endothelial cells and the consequences of imbalances therein are particularly obvious in thrombotic microangiopathies (TMAs). Although diseases with distinct aetiologies such as aHUS and thrombotic thrombocytopenic purpura (TTP) fall under the TMA spectrum, common features define their pathology, including microangiopathic haemolytic anaemia, thrombocytopenia, vascular damage with thrombosis, and organ injury114,116,117. Essential insights into TMA disease pathogenesis have been acquired from studies of aHUS, a rare kidney disorder associated with imbalanced complement activation, which results in damage to the kidney endothelium and thrombus formation. Notably, aHUS is strongly associated with polymorphisms and mutations of activators (C3 and FB) and regulators (CD46, FH, and FI) of the ‘alternative pathway’ (REF. 118) (BOX 1). More than 300 aHUS-associated genetic alterations are listed in the FH aHUS mutation database to date, with mutational hotspots primarily in the C-terminus of FH and the C3d-domain of C3 (REF. 105). Variations in C3 and FB enhance convertase-mediated generation of C3b, whereas alterations in genes that encode regulators of complement disturb the control of convertase activity and C3b deposition on renal endothelium which, as discussed previously, strongly relies on fluid-phase regulators. Importantly, the devastating effects of imbalanced complement activation on the renal endothelium are primarily mediated by excessive opsonization and formation of C5b–9 on the cell surface, rather than uncontrolled activation of the fluid phase119. Alongside activation of endothelial cells and platelets by C5b–9, the anaphylatoxins C3a and C5a induce a local proinflammatory environment that promotes the formation of microthrombi in the renal micro-vasculature114. The release of haeme during haemolytic periods might act as secondary ‘hit’ by further inducing complement activation, since haeme has been shown in vitro to enhance C3 activation, decrease CD55 and CD46 expression on endothelial cells, and increase the expression of P-selectin as a ligand for C3b120.
As a disease essentially conditioned by the excessive generation of C5-derived effectors, aHUS is successfully treated by the anti-C5 antibody eculizumab121. A longitudinal study in which patients with aHUS were treated for up to 1 year demonstrated that eculizumab normalized systemic levels of C5a and C5b–9 and markers of renal injury and endothelial damage; however, eculizumab decreased but did not normalize levels of the complement marker Ba (an FB fragment released during convertase formation) or soluble VCAM-1, a marker of endothelial cell activation122. Although this finding reflects ongoing complement activation despite blockage of C5, the implications for disease progression are not yet clear; therapeutic intervention at upstream levels, for example, by inhibiting MASP-2 are currently being evaluated123.
Although the involvement of complement in aHUS pathogenesis is well-established, studies over the past few years suggest that complement has contributory roles in other TMA diseases114,124. Typical HUS is induced by Shiga toxin-producing Escherichia coli (STEC), and increased complement activation has been linked to Shiga toxin-induced expression of the proposed C3b ligand P-selectin on endothelial cells125. Beneficial effects of eculizumab in STEC-induced HUS were reported in connection with the STEC outbreak in Germany in 2011, but the effect of this treatment on HUS remains controversial114,124,126. TTP is caused by decreased activity of ADAMTS13, a metalloproteinase that cleaves vWF. The influence of complement on TTP is not well defined, although the interaction between FH and vWF oligomers, endothelial damage, and microvascular thrombus formation might lead to secondary complement activation33,114,124. Thus far, complement-targeted therapies have only been explored in one case study of TTP with favourable outcome114,127. Of note, however, the clinical distinction between aHUS and TTP remains challenging in some cases, and individual patients may have clinical signs of both TTP and aHUS128,129; more research is needed to elucidate the effect of complement on TMA beyond aHUS.
C3 glomerulopathy
The term C3 glomerulopathy, introduced in 2010 as consensus nomenclature130, refers to a group of renal pathologies characterized by detection of C3 opsonins in glomeruli, with minimal or no deposition of immunoglobulins. These forms of renal disease are typically caused by abnormal control of the alternative pathway of complement activation, in contrast to forms of immunoglobulin-associated membranoproliferative glomerulonephritis, which can be caused by autoimmune reactions or infection and are characterized by strong IgG or IgM staining alongside C3 and C1q. C3 glomerulopathies can be subdivided into dense deposit disease (DDD) and C3 glomerulonephritis on the basis of histological observations95,130,131. C3 glomerulopathies develop as a consequence of imbalanced complement activation and lead to proteinuria and haematuria, with progression to renal failure within 10 years of diagnosis in approximately 50% of patients. The high mortality is a consequence of the lack of effective therapy and poor prognosis after kidney transplantation, with disease recurrence being observed in over 50% of all cases132.
In contrast to aHUS, in which endothelial cell damage and local deposition of complement fragments are considered driving factors, uncontrolled activation and consumption of complement in the fluid phase leading to bystander opsonization of ill-protected surfaces (such as the GBM) is considered the predominant mechanism in C3 glomerulopathy. The reasons for this hyperactivation of complement are diverse and can include autoimmune mechanisms, particularly the presence of C3/C4 nephritic factors that stabilize C3 convertases, and genetic alterations in C3, FH, FI and/or FHR proteins. Whereas C3 nephritic factor is associated with chronic activation of the amplification loop by stabilizing C3 convertases, a heterozygous C3 mutation associated with DDD was found to impair the FH-mediated degradation of C3b by FI133. Similarly, genetic variations in FH and FI observed in patients with C3 glomerulopathy result in the loss of regulatory functions. In contrast to aHUS-related mutations and polymorphisms in FH, which typically affect the surface-recognizing C-terminus, those associated with C3 glomerulopathy generally occupy a focal area in the regulatory domains. Genetic alterations in the region encoding FHR proteins, including point mutations, polymorphisms, deletions, and rearrangements, have been identified in patients with C3 glomerulopathy. The pathologic mechanisms by which alterations in FHR proteins affect complement activation remain elusive, but likely involve deregulation and other modulatory effects on the complement system95 (BOX 2). The increased complement turnover generates C5a that is recognized as a promoter of necrotic and inflammatory lesions in the glomeruli, which progress to renal fibrosis and impairment of kidney function121.
The vast diversity of pathologic mechanisms that underlie C3 glomerulopathies renders diagnosis and classification challenging, and has implications for the development of treatment strategies. Indeed, whereas anti-C5 therapy has developed into a gold standard for the treatment of aHUS, only select patients with C3 glomerulopathies will benefit from the same treatment126,134. Given the central involvement of excessive amplification loop activity in most patients, strategies that target C3 and/or the convertases are currently under investigation.
Transplantation and IRI
Many kidney disorders will eventually progress to end-stage renal disease (ESRD) requiring haemodialysis and/or kidney transplantion23. Although transplantation is the desirable treatment option for patients with ESRD, organ shortages and compatibility issues limit the number of transplantations performed worldwide. Complement has rapidly moved into the spotlight of transplantation medicine, with involvement in most complications that can arise during the procedure60. In deceased organ donors, complement activation occurs shortly after brain death or cardiac arrest. Clinical studies have shown a direct correlation between the presence of complement activation fragments in the plasma of deceased donors and poorer post-transplantation outcome, suggesting that inhibition of complement and coagulation pathways in the donor could result in decreased tissue damage and better transplant outcomes135.
In addition, the inevitable period of cold ischaemia during organ collection and storage induces hypoxia, inflammation, and tissue damage that contribute to rejection episodes and graft loss. During reperfusion, ischemia-induced DAMPs such as histones, heat-shock proteins, and biglycan activate TLRs and provide neoepitopes for natural antibodies, which subsequently activate complement56. The increased expression of complement proteins, the loss of glycocalyx and complement regulators, and the damage and detachment of endothelial cells as a consequence of acute kidney injury can exacerbate complement activation on tubular epithelial cells, contributing to the production of proinflammatory cytokines60. Interestingly, whereas mice that lack FB are protected from kidney IRI, Fb/Tlr2 double-knockout mice are not, suggesting counter-regulatory effects between complement and TLR2 during IRI136. While complement-related tissue damage during IRI is mainly dependent on C5-derived effectors with recruitment and activation of neutrophils by C5a and cell activation and damage by C5b–9, the initial triggers of complement activation in kidney IRI remain elusive60. In addition to antibody-mediated activation, components of the lectin pathway are increasingly identified as inducing factors. For example, a 2016 study showed that collectin-11 can recognize an abnormal L-fucose pattern on postischaemic renal tubule cells and activate complement via MASP-2137. Independent of the mechanisms, acute kidney injury caused by complement and other inadvertently triggered defence systems during donor death and IRI affects the quality of the graft and largely contributes to the rejection risk in transplantation.
The prominent role of complement in cell-mediated and antibody-mediated transplant rejection has long been recognized, with increased levels of C5a and soluble C5b–9 commonly detected in the plasma and urine of transplant recipients during rejection episodes60,138. Moreover, C4d-containing opsonins are among the most reliable biomarkers of rejection in kidney biopsy samples139. Several mechanisms link complement to kidney transplant injury, and local complement production is of particular importance in this context. Tubular epithelial cells are the main target but also a major source of complement in the kidney undergoing rejection, yet graft-infiltrating cells also contribute to local complement production60. In crossover transplantation studies in mice, allograft rejection was entirely dependent on complement production by the donor tissue; transplantation of kidneys from C3-knockout mice into wild-type mice was successful without any requirement for immunosuppression140.
The role of complement in cellular rejection seems to be complex and some aspects remain controversial. The interaction between APCs and alloreactive T cells leads to the secretion of complement proteins and local generation of C3a and C5a; signalling through C3a and C5a receptors on APCs, and potentially T cells, amplifies the TH1-cell response and graft rejection. Activation of C3aR and C5aR1 on APCs and CD4+ T cells sends proliferation, co-stimulatory, and survival signals for T cells; blockage of C3a and C5a-induced responses results in the endogenous production of TGF-β by APCs and T cells141. CD4+ T cells with a regulatory phenotype suppressed immune responses in a mouse model of graft-versus-host disease, providing proof-of-principle of the benefits of complement inhibition during transplantation141,142.
As improvements in immunosuppression have decreased the clinical impact of cellular rejection, attention has increasingly turned to antibody-mediated rejection (ABMR), which remains associated with high rates of graft loss. Complement integrally participates in ABMR by causing direct tissue damage, regulating the production of donor-specific antibodies (DSAs) in B cells, potentiating the injury caused by antibodies, and fostering thrombo-inflammatory complications. In fact, the combination of serum DSAs and positive C4d staining in the allograft is used as a criterion to define ABMR in renal allografts. The binding of antibodies to the allograft strongly activates complement, resulting in the generation of C5a and C5b–9, which activate neutrophils and endothelial cells, among others. Although a rare occurrence as a result of improved crossmatching, complement-mediated damage is particularly pronounced in hyperacute rejection due to the presence of pre-formed antibodies. Of note, however, complement also contributes to the induction of DSAs as opsonization with iC3b and C3dg decreases the threshold of B-cell activation, as described above, and complement activation is required for the alloantigen-induced priming of B cells143. Crosstalk between complement, coagulation, and FcR-mediated processes further potentiates the pro-coagulant and inflammatory responses, leading to endothelial damage, thrombosis, and graft loss144.
An area that has gained interest is the potential link between complement inhibition and graft accommodation through the sustained protection of endothelial cells from DSAs and complement. In monkeys that had been pre-sensitized with skin grafts, depletion of complement induced long-term survival of a subsequently transplanted kidney145. In this model, graft accommodation was associated with upregulation in the expression levels of complement regulatory proteins and anti-apoptotic genes. Many aspects of accommodation as an ‘acquired resistance’ to complement-mediated injury of endothelial cells remain elusive and might involve modulation of surface-regulation and cellular responses. Given the central involvement of complement in most aspects of transplant-related complications, and the potential prospect of allowing transplantation across ABO, HLA or species barriers, therapeutic complement inhibition is currently considered a promising strategy60,146.
Haemodialysis-related complications
Patients awaiting kidney transplantation and other patients with impaired kidney function must undergo dialysis. While essential, this procedure exacerbates clinical complications such as muscle wasting, endothelial and immune cell dysfunction, malnutrition, and anaemia; it also increases the risk of cardiovascular disease, which represents a major cause of mortality23,147.
During the contact of blood with the haemodialysis filters and extracorporeal circuits, the exposed biomaterials are recognized as foreign surfaces and activate complement. Although this effect was most obvious for older cellulose-based haemodialysis filters, modern synthetic filters with improved biocompatibility still induce substantial complement activation through all initiation pathways3,147. The generation of C5a during haemodialysis-induced complement activation is associated with neutrophil activation and an increase in TF expression as a primary initiator of coagulation. Indeed, patients on haemodialysis have an elevated risk of thrombosis and exhibit increased expression of oxidative stress markers and proinflammatory cytokines — factors that are all strongly correlated with mortality in this population147,148. The impact of haemodialysis-induced complement activation on overall inflammation, or even mortality, is difficult to assess given the complexity of the underlying diseases and treatments. Nevertheless, repetitive, chronic–acute inflammatory and complement-mediated triggers are likely to contribute to disease progression and/or affect quality of life. Further research into the biocompatibility of haemodialysis filters and potential treatment options might open new avenues for decreasing the burden of haemodialysis.
Complement-targeted therapy
Modulation of the complement system has long been recognized as a potential therapeutic strategy in inflammatory diseases but faced initial challenges (BOX 3); however, the newly discovered disease associations of complement have reinvigorated interest in the development of complement-targeted therapeutics. Although the currently available arsenal of complement inhibitors is limited, their clinical use has already changed the treatment landscape of selected disorders and showed great potential for many other indications3,123,149.
Box 3. The promises and challenges of therapeutic complement inhibition.
The concept of inhibiting complement for therapeutic purposes has been considered for decades but only resulted in broad availability of clinically approved drugs in the past 10 years. Indeed, complement-targeted drug discovery has long faced unique challenges, some of which were technical, such as the strong involvement of protein–protein interactions that are difficult to target, or the high plasma concentrations and turnover of certain components149,123,155. The introduction of biologics such as antibodies or peptides has overcome many of these hurdles. General concerns about the safety of inhibiting a defence system like complement have traditionally posed the biggest roadblocks. Such concerns long remained hypothetical and based on limited data from individuals with complement deficiencies, many of whom show increased susceptibility to pyogenic infections and/or immune complex-related diseases. The clinical impact of complement deficiencies is variable though, and depends on the type of deficiency. Importantly, pharmacologic control of complement targets cannot be directly compared with corresponding deficiencies in complement components, as deficiencies are irreversible and probably affect baseline immune responses. Conversely, therapies can be interrupted during adverse events, recovery of complement activity can be accelerated by plasma therapy, and common infectious risks can be managed by vaccination and/or antibiotics. Moreover, the antimicrobial relevance of complement seems to wane in adults with increasing strength of adaptive immunity. We suggest that the pathological consequences of exaggerated complement activation might outweigh the potential loss of physiological function in many disorders. Of course, this theory does not necessarily render therapeutic complement inhibition safe, and each target and drug will require careful evaluation. However, the considerations discussed above and the positive experiences with complement inhibitors currently on the market, in particular with eculizumab, have spurred new confidence in this approach. The ongoing clinical trials with compounds targeting almost all levels of the cascade will undoubtedly benefit the field and replace hypothetical considerations about safety with clinically validated data. The far greater challenge in the coming years will be to identify the right indications, therapeutic strategies and patient cohorts for complement-targeted therapeutics in order to facilitate clinical translation.
Alongside preparations of the broad serine protease inhibitor C1-INH, the only complement-specific drug on the market is eculizumab. This therapeutic antibody binds to and prevents activation of C5, thereby blocking the generation of C5a and MAC121. Originally introduced to prevent intravascular haemolysis in PNH, improving both anaemia and thrombotic complications and changing the natural history of this disease104, eculizumab was subsequently approved for the treatment of aHUS121. In clinical trials, eculizumab was well-tolerated and improved or normalized platelet counts and glomerular filtration rates for most patients with aHUS; in one trial, 19 of 20 patients achieved TMA-free status in the second year of treatment150. Although eculizumab is now established as a standard therapy for aHUS in many countries, the high treatment cost can be prohibitive and has spurred discussions about the required duration of anti-C5 therapy in patients with aHUS121,151. Despite the successful application of eculizumab in diseases that are driven by the damaging effects of C5-derived effectors, anti-C5 therapy does have limitations. Importantly, blockage of C5 activation does not prevent upstream activation of complement by recognition complexes, opsonization with C4 and C3-derived fragments, or the resulting amplification of the response. These considerations are most obvious in the case of C3-driven disorders such as C3 glomerulopathy, in which only a subset of patients seem to benefit from C5-targeted therapy134. Upstream complement inhibition could also prove beneficial in conditions with complex complement activation patterns, such as transplant rejection146,152. For instance, eculizumab did not reach its primary end point in a recent multi-center phase II trial with the drug for preventing ABMR in living-donor kidney transplant recipients requiring desensitization despite promising initial results153,154; other clinical trials with eculizumab in transplantation and kidney graft IRI are ongoing123,146,152.
Over the past few years, several new therapeutic strategies involving interventions at various points in the complement cascade have been developed, several of which are being evaluated in clinical trials3,123,149. For example, regulator-based therapeutics that target C3 and C5 convertases are being investigated in diseases driven by dysregulated complement and excessive amplification155. Short-term use of a soluble form of CR1 (CDX-1135 by Celldex Therapeutics, the development of which has now been discontinued) was well-tolerated, normalized levels of C3 and decreased levels of sC5b-9 in an 8-year-old patient with rapidly progressing DDD, providing important proof-of-concept of the benefits of C3-targeted intervention in this disease156. Another example is mirococept (APT070), which contains a recombinant fragment of CR1 linked to a membrane-tethering moiety, and is being evaluated for its ability to provide localized tissue protection during kidney transplantation. When perfused after organ collection, mirococept lines the endothelium of the donor organ to restrict complement mediated damage after transplantation123,157. Convertase-mediated amplification can also be impaired by inhibiting FD, which represents a bottle-neck in convertase formation. Genentech is currently enrolling patients with AMD for a phase III trial of the anti-FD antibody, lampalizumab158,159. Small-molecule strategies for inhibiting FD are being developed by Achillion Pharmaceuticals and Novartis123.
An alternative approach to inhibiting convertases is protecting native C3 from convertase-mediated activation, through the action of the peptidic inhibitor compstatin and its derivatives155. By binding to C3 and C3b, compstatin analogues prevent C3 opsonization by all initiation routes, thereby impairing amplification, downstream C5 activation, and effector generation. Among other indications155, the lead analogue Cp40 has shown efficacy in an in vitro model of C3 glomerulopathy160. Although systemic inhibition of C3 had been considered restrictive because of its high plasma concentration and turnover (BOX 3), a 2014 study demonstrated that target-saturating concentrations of Cp40 can be maintained in monkeys by repetitive subcutaneous injection161. Cp40 has also been used in a monkey model of haemodialysis-induced inflammation, in which a single injection of the drug abrogated filter-induced complement activation during the 4 h haemodialysis session and increased levels of the anti-inflammatory cytokine IL-10 (REF. 162). Add-on therapy with a broad complement inhibitor might prove useful in treating haemodialysis-related complications because of the potential involvement of several initiation pathways; the short-term use and synthetic nature of Cp40 might also address the cost sensitivity for this indication. A Cp40-based drug (AMY-101; Amyndas Pharmaceuticals) is scheduled for evaluation in phase I trials, with uses for PNH, C3 glomerulopathy, periodontitis, and transplantation, whereas inhibitors based on first-generation compstatin analogues (APL-1, APL-2; Apellis Pharmaceuticals) are in clinical trials for chronic obstructive pulmonary disease, AMD, and PNH123,163–165.
In instances where a single known initiation route or effector molecule determines disease onset, a more focused intervention could be considered. For example, a C1s-specific antibody (TNT009, TrueNorth Therapeutics) is currently being evaluated for auto-immune haemolytic anemia166, and Omeros is conducting phase II trials of an anti-MASP2 antibody (OMS721) for TMAs, including aHUS123,167. Purified or recombinant C1-INH preparations that target both C1s and MASP-2 as well as other proteases are already on the market for the treatment of hereditary angioedema24 and some have been evaluated in transplantation168. In diseases that are primarily caused by C5a activity, such as ANCA-associated vasculitis with its vicious cycle of complement and neutrophil activation, selective intervention at the C5a–C5aR1 axis might be sufficient to confer therapeutic benefit. Indeed, ongoing phase II clinical trials with the orally active C5aR1 antagonist CCX168 (Chemocentryx) are promising169, and Chemocentryx has now initiated phase II studies of CCX168 in aHUS and IgA nephropathy170,171. The anti-C5a antibody IFX-1 (InflaRx) is in clinical evaluation for acute inflammatory conditions, including a phase II trial in organ dysfunction during sepsis123,172.
The above-mentioned approaches only cover a fraction of the complement-targeted strategies that are currently in development and reflect a new confidence in the therapeutic modulation of the complement cascade123,149,155. As detailed above, the harmful effects of complement rapidly become dominant in many clinical disorders, and the protective functions against microbial threats become less important with the development of adaptive immunity. Still, the potentially increased risk of infection or other adverse effects needs to be considered and carefully evaluated during the clinical development of complement-targeted strategies (BOX 3). Overall, experience thus far with long-term treatment of patients with eculizumab and C1-INH has been positive173,174, and the ongoing clinical trials with various complement inhibitors acting at distinct stages of the cascade will shed more light on the limitations and opportunities of complement-targeted therapies.
Conclusions
The past few decades have provided insights into the complexity of the complement system; we have now arrived at a new perception of this evolutionarily refined pathway that reaches far beyond its role in host defence to tissue development, waste disposal, and immune modulation. The dynamic interplay between numerous components within the cascade and extensive crosstalk with other pathways defines this versatility in immune surveillance and homeostasis; however, it also makes complement vulnerable to inappropriate activation, with clinical consequences. Complement might not always be the driving force of disease progression, yet some degree of pathological involvement of complement is very likely whenever the body is confronted with damaged or altered cells, accumulating debris, or foreign materials. These situations are typically exacerbated by imbalances in complement resulting from genetic variation, deficiencies, or autoimmune reactions, often occurring in combination. The resulting inflammation, immune cell activation, and tissue damage can cause further complement activation and fuel a vicious cycle that spurs disease progression. Particularly in age-related and degenerative diseases, complement can assume a dual role of removing disease triggers at early stages but fueling inflammation once a ‘tipping point’ has been reached. Research over the past few decades also provides new insights into why some organs, especially the kidney, seem to be particularly susceptible to complement-mediated tissue damage, even though complement dysregulation occurs systemically. Indeed, increasing knowledge of the involvement of complement in kidney conditions has already changed the classification of disease spectra such as C3 glomerulopathies, furthering our understanding of pathological mechanisms and suggesting new avenues for treatment. Despite tremendous progress in the field, additional genetic, molecular, preclinical, and clinical studies are necessary to refine that insight and determine which indications will benefit most from complement-targeted therapy. Although the selection of clinically available complement inhibitors is currently limited, the success of eculizumab in PNH and aHUS illustrates clearly the potential of therapeutic interventions that target the complement system. Fortunately, a plethora of potent drug candidates covering a diverse set of targets within the cascade are already undergoing or awaiting clinical evaluation, likely making the next decades an even more transformative era for complement research in kidney and other diseases.
Acknowledgments
The authors thank Deborah McClellan for editing the manuscript before submission. J.D.L. also thanks Ralph and Sallie Weaver for the generous endowment of his professorship. Given the broad scope of this Review, we often refer to specialized review articles rather than primary literature, and we have only been able to include selected examples of the breadth of transformative work in the field; we therefore thank all our colleagues who are not specifically cited for their great contributions and their understanding. This work was supported by grants from the US NIH (AI068730, AI030040, DE021685) and the National Science Foundation (grant No. 1423304) and by funding from the European Community’s Seventh Framework Programme, under grant agreement number 602699 (DIREKT).
Glossary
- Convertase
A protein complex transiently formed by an opsonin (C3b, C4b) and a protease fragment (Bb, C2b), which activates complement components C3 or C5 and promotes opsonization, amplification and/or generation of effector molecules
- Anaphylatoxin
A widely used term for complement effector fragments C3a and C5a, which are released upon activation of C3 and C5, respectively, and exert numerous immunomodulatory, chemotactic and/or cell-activating functions
- Opsonization
The process of depositing complement proteins on a particle surface to facilitate its removal. In the complement system, C3b, C4b (and their degradation fragments) and C1q facilitate phagocytosis via their receptors on immune cells
- Amplification
A major function of the ‘alternative pathway’ of complement, whereby convertase-mediated deposition of C3b on a targeted surface enables the formation of additional C3 convertases and deposition of more C3b. In absence of regulators, these events lead to rapid opsonization of the attacked cell or particle
- Consumption
A result of continuous and poorly controlled activation of complement components in the circulation, leading to a depletion of the native plasma protein and affecting the responsiveness of the cascade
- Fluid phase
Refers to complement activity in the circulation rather than on the targeted surface. Fluid-phase activation maintains alertness of the complement system (tick-over) but needs to be tightly controlled by soluble complement regulators
Footnotes
Author contributions
All authors contributed to researching data for the article, discussion of the article’s content, writing, and review/editing of the manuscript before submission.
Competing interests statement
J.D.L and D.R. are inventors of patents or patent applications that describe the use of complement inhibitors for therapeutic purposes. J.D.L. is the founder of Amyndas Pharmaceuticals, which is developing complement inhibitors. E.S.R. declares no competing interests.
References
- 1.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ricklin D, Lambris JD. Complement in immune and inflammatory disorders: pathophysiological mechanisms. J Immunol. 2013;190:3831–3838. doi: 10.4049/jimmunol.1203487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reis ES, et al. Applying complement therapeutics to rare diseases. Clin Immunol. 2015;161:225–240. doi: 10.1016/j.clim.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I — molecular mechanisms of activation and regulation. Front Immunol. 2015;6:262. doi: 10.3389/fimmu.2015.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement system part II: role in immunity. Front Immunol. 2015;6:257. doi: 10.3389/fimmu.2015.00257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mastellos DC, Deangelis RA, Lambris JD. Complement-triggered pathways orchestrate regenerative responses throughout phylogenesis. Semin Immunol. 2013;25:29–38. doi: 10.1016/j.smim.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci. 2012;35:369–389. doi: 10.1146/annurev-neuro-061010-113810. [DOI] [PubMed] [Google Scholar]
- 8.Kohl J. The role of complement in danger sensing and transmission. Immunol Res. 2006;34:157–176. doi: 10.1385/IR:34:2:157. [DOI] [PubMed] [Google Scholar]
- 9.Scott D, Botto M. The paradoxical roles of C1q and C3 in autoimmunity. Immunobiology. 2015 doi: 10.1016/j.imbio.2015.05.001. http://dx.doi.org/10.1016/j.imbio.2015.05.001. [DOI] [PubMed]
- 10.Kolev M, Le Friec G, Kemper C. Complement — tapping into new sites and effector systems. Nat Rev Immunol. 2014;14:811–820. doi: 10.1038/nri3761. [DOI] [PubMed] [Google Scholar]
- 11.Harris CL, Heurich M, Rodriguez de Cordoba S, Morgan BP. The complotype: dictating risk for inflammation and infection. Trends Immunol. 2012;33:513–521. doi: 10.1016/j.it.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diebolder CA, et al. Complement is activated by IgG hexamers assembled at the cell surface. Science. 2014;343:1260–1263. doi: 10.1126/science.1248943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kouser L, et al. Emerging and novel functions of complement protein C1q. Front Immunol. 2015;6:317. doi: 10.3389/fimmu.2015.00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harboe M, et al. The down-stream effects of mannan-induced lectin complement pathway activation depend quantitatively on alternative pathway amplification. Mol Immunol. 2009;47:373–380. doi: 10.1016/j.molimm.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 15.Harboe M, Ulvund G, Vien L, Fung M, Mollnes TE. The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation. Clin Exp Immunol. 2004;138:439–446. doi: 10.1111/j.1365-2249.2004.02627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nilsson B, Nilsson Ekdahl K. The tick-over theory revisited: is C3 a contact-activated protein? Immunobiology. 2012;217:1106–1110. doi: 10.1016/j.imbio.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 17.Morgan BP. The membrane attack complex as an inflammatory trigger. Immunobiology. 2015 doi: 10.1016/j.imbio.2015.04.006. http://dx.doi.org/10.1016/j.imbio.2015.04.006. [DOI] [PubMed]
- 18.Taylor RP, Lindorfer MA. The role of complement in mAb-based therapies of cancer. Methods. 2014;65:18–27. doi: 10.1016/j.ymeth.2013.07.027. [DOI] [PubMed] [Google Scholar]
- 19.Klos A, Wende E, Wareham KJ, Monk PN International Union of Basic and Clinical Pharmacology. LXXXVII Complement peptide C5a, C4a, and C3a receptors. Pharmacol Rev. 2013;65:500–543. doi: 10.1124/pr.111.005223. erratum 66, 466, 2014. [DOI] [PubMed] [Google Scholar]
- 20.Coulthard LG, Woodruff TM. Is the complement activation product C3a a proinflammatory molecule? Re-evaluating the evidence and the myth. J Immunol. 2015;194:3542–3548. doi: 10.4049/jimmunol.1403068. [DOI] [PubMed] [Google Scholar]
- 21.Nordahl EA, et al. Activation of the complement system generates antibacterial peptides. Proc Natl Acad Sci USA. 2004;101:16879–16884. doi: 10.1073/pnas.0406678101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghebrehiwet B, Hosszu KK, Valentino A, Ji Y, Peerschke EI. Monocyte expressed macromolecular C1 and C1q receptors as molecular sensors of danger: implications in SLE. Front Immunol. 2014;5:278. doi: 10.3389/fimmu.2014.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Helmy KY, et al. CRIg: a macrophage complement receptor required for phagocytosis of circulating pathogens. Cell. 2006;124:915–927. doi: 10.1016/j.cell.2005.12.039. [DOI] [PubMed] [Google Scholar]
- 24.Zeerleder S. C1-inhibitor: more than a serine protease inhibitor. Semin Thromb Hemost. 2011;37:362–374. doi: 10.1055/s-0031-1276585. [DOI] [PubMed] [Google Scholar]
- 25.Matsushita M, Endo Y, Fujita T. Structural and functional overview of the lectin complement pathway: its molecular basis and physiological implication. Arch Immunol Ther Exp (Warsz) 2013;61:273–283. doi: 10.1007/s00005-013-0229-y. [DOI] [PubMed] [Google Scholar]
- 26.Clark SJ, Bishop PN. Role of factor H and related proteins in regulating complement activation in the macula, and relevance to age-related macular degeneration. J Clin Med. 2015;4:18–31. doi: 10.3390/jcm4010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reis ES, et al. C5a receptor-dependent cell activation by physiological concentrations of desarginated C5a: insights from a novel label-free cellular assay. J Immunol. 2012;189:4797–4805. doi: 10.4049/jimmunol.1200834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lesher AM, Nilsson B, Song WC. Properdin in complement activation and tissue injury. Mol Immunol. 2013;56:191–198. doi: 10.1016/j.molimm.2013.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kemper C, Atkinson JP, Hourcade DE. Properdin: emerging roles of a pattern-recognition molecule. Annu Rev Immunol. 2010;28:131–155. doi: 10.1146/annurev-immunol-030409-101250. [DOI] [PubMed] [Google Scholar]
- 30.Marsh JE, Zhou W, Sacks SH. Local tissue complement synthesis — fine tuning a blunt instrument. Arch Immunol Ther Exp (Warsz) 2001;49:S41–S46. [PubMed] [Google Scholar]
- 31.Conway EM. Reincarnation of ancient links between coagulation and complement. J Thromb Haemost. 2015;13:S121–S132. doi: 10.1111/jth.12950. [DOI] [PubMed] [Google Scholar]
- 32.Ritis K, et al. A novel C5a receptor-tissue factor crosstalk in neutrophils links innate immunity to coagulation pathways. J Immunol. 2006;177:4794–4802. doi: 10.4049/jimmunol.177.7.4794. [DOI] [PubMed] [Google Scholar]
- 33.Rayes J, et al. The interaction between factor H and VWF increases factor H cofactor activity and regulates VWF prothrombotic status. Blood. 2014;123:121–125. doi: 10.1182/blood-2013-04-495853. [DOI] [PubMed] [Google Scholar]
- 34.Speth C, et al. Complement and platelets: mutual interference in the immune network. Mol Immunol. 2015;67:108–118. doi: 10.1016/j.molimm.2015.03.244. [DOI] [PubMed] [Google Scholar]
- 35.Hamad OA, et al. Contact activation of C3 enables tethering between activated platelets and polymorphonuclear leukocytes via CD11b/CD18. Thromb Haemost. 2015;114:1207–1217. doi: 10.1160/TH15-02-0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ali H. Regulation of human mast cell and basophil function by anaphylatoxins C3a and C5a. Immunol Lett. 2010;128:36–45. doi: 10.1016/j.imlet.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bohlson SS, O’Conner SD, Hulsebus HJ, Ho MM, Fraser DA. Complement, c1q, and c1q-related molecules regulate macrophage polarization. Front Immunol. 2014;5:402. doi: 10.3389/fimmu.2014.00402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Camous L, et al. Complement alternative pathway acts as a positive feedback amplification of neutrophil activation. Blood. 2011;117:1340–1349. doi: 10.1182/blood-2010-05-283564. [DOI] [PubMed] [Google Scholar]
- 39.Wang H, Wang C, Zhao MH, Chen M. Neutrophil extracellular traps can activate alternative complement pathways. Clin Exp Immunol. 2015;181:518–527. doi: 10.1111/cei.12654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu MC, et al. The receptor for complement component C3a mediates protection from intestinal ischemia-reperfusion injuries by inhibiting neutrophil mobilization. Proc Natl Acad Sci USA. 2013;110:9439–9444. doi: 10.1073/pnas.1218815110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hsieh CC, et al. The role of complement component 3 (C3) in differentiation of myeloid-derived suppressor cells. Blood. 2013;121:1760–1768. doi: 10.1182/blood-2012-06-440214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Markiewski MM, et al. Modulation of the antitumor immune response by complement. Nat Immunol. 2008;9:1225–1235. doi: 10.1038/ni.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Son M, Santiago-Schwarz F, Al-Abed Y, Diamond B. C1q limits dendritic cell differentiation and activation by engaging LAIR-1. Proc Natl Acad Sci USA. 2012;109:E3160–E3167. doi: 10.1073/pnas.1212753109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li K, et al. Functional modulation of human monocytes derived DCs by anaphylatoxins C3a and C5a. Immunobiology. 2012;217:65–73. doi: 10.1016/j.imbio.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hawlisch H, et al. C5a negatively regulates toll-like receptor 4-induced immune responses. Immunity. 2005;22:415–426. doi: 10.1016/j.immuni.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 46.Kemper C, Kohl J. Novel roles for complement receptors in T cell regulation and beyond. Mol Immunol. 2013;56:181–190. doi: 10.1016/j.molimm.2013.05.223. [DOI] [PubMed] [Google Scholar]
- 47.Dunkelberger J, Zhou L, Miwa T, Song WC. C5aR expression in a novel GFP reporter gene knockin mouse: implications for the mechanism of action of C5aR signaling in T cell immunity. J Immunol. 2012;188:4032–4042. doi: 10.4049/jimmunol.1103141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karsten CM, et al. Monitoring and cell-specific deletion of C5aR1 using a novel floxed GFP–C5aR1 reporter knock-in mouse. J Immunol. 2015;194:1841–1855. doi: 10.4049/jimmunol.1401401. [DOI] [PubMed] [Google Scholar]
- 49.Cardone J, et al. Complement regulator CD46 temporally regulates cytokine production by conventional and unconventional T cells. Nat Immunol. 2010;11:862–871. doi: 10.1038/ni.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Le Friec G, et al. The CD46–Jagged1 interaction is critical for human TH1 immunity. Nat Immunol. 2012;13:1213–1221. doi: 10.1038/ni.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ghannam A, Fauquert JL, Thomas C, Kemper C, Drouet C. Human complement C3 deficiency: Th1 induction requires T cell-derived complement C3a and CD46 activation. Mol Immunol. 2014;58:98–107. doi: 10.1016/j.molimm.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 52.Liszewski MK, et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity. 2013;39:1143–1157. doi: 10.1016/j.immuni.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Iwasaki A, Medzhitov R. Control of adaptive immunity by the innate immune system. Nat Immunol. 2015;16:343–353. doi: 10.1038/ni.3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hajishengallis G, Lambris JD. Crosstalk pathways between Toll-like receptors and the complement system. Trends Immunol. 2010;31:154–163. doi: 10.1016/j.it.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Morgan BP, Gasque P. Extrahepatic complement biosynthesis: where, when and why? Clin Exp Immunol. 1997;107:1–7. doi: 10.1046/j.1365-2249.1997.d01-890.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leemans JC, Kors L, Anders HJ, Florquin S. Pattern recognition receptors and the inflammasome in kidney disease. Nat Rev Nephrol. 2014;10:398–414. doi: 10.1038/nrneph.2014.91. [DOI] [PubMed] [Google Scholar]
- 57.Cheung KP, Kasimsetty SG, McKay DB. Innate immunity in donor procurement. Curr Opin Organ Transplant. 2013;18:154–160. doi: 10.1097/MOT.0b013e32835e2b0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McCullough JW, Renner B, Thurman JM. The role of the complement system in acute kidney injury. Semin Nephrol. 2013;33:543–556. doi: 10.1016/j.semnephrol.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Noris M, Remuzzi G. Glomerular diseases dependent on complement activation, including atypical hemolytic uremic syndrome, membranoproliferative glomerulonephritis, and C3 glomerulopathy: core curriculum 2015. Am J Kidney Dis. 2015;66:359–375. doi: 10.1053/j.ajkd.2015.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sacks SH, Zhou W. The role of complement in the early immune response to transplantation. Nat Rev Immunol. 2012;12:431–442. doi: 10.1038/nri3225. [DOI] [PubMed] [Google Scholar]
- 61.Song WC. Crosstalk between complement and toll-like receptors. Toxicol Pathol. 2012;40:174–182. doi: 10.1177/0192623311428478. [DOI] [PubMed] [Google Scholar]
- 62.Bosmann M, et al. Complement activation product C5a is a selective suppressor of TLR4-induced, but not TLR3-induced, production of IL-27(p28) from macrophages. J Immunol. 2012;188:5086–5093. doi: 10.4049/jimmunol.1102914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Seow V, et al. Inflammatory responses induced by lipopolysaccharide are amplified in primary human monocytes but suppressed in macrophages by complement protein C5a. J Immunol. 2013;191:4308–4316. doi: 10.4049/jimmunol.1301355. [DOI] [PubMed] [Google Scholar]
- 64.Ling GS, et al. Integrin CD11b positively regulates TLR4-induced signalling pathways in dendritic cells but not in macrophages. Nat Commun. 2014;5:3039. doi: 10.1038/ncomms4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zou L, et al. Complement factor B is the downstream effector of TLRs and plays an important role in a mouse model of severe sepsis. J Immunol. 2013;191:5625–5635. doi: 10.4049/jimmunol.1301903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Triantafilou M, Hughes TR, Morgan BP, Triantafilou K. Complementing the inflammasome. Immunology. 2016;147:152–164. doi: 10.1111/imm.12556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Asgari E, et al. C3a modulates IL-1β secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood. 2013;122:3473–3481. doi: 10.1182/blood-2013-05-502229. [DOI] [PubMed] [Google Scholar]
- 68.Laudisi F, et al. Cutting edge: the NLRP3 inflammasome links complement-mediated inflammation and IL-1β release. J Immunol. 2013;191:1006–1010. doi: 10.4049/jimmunol.1300489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Triantafilou K, Hughes TR, Triantafilou M, Morgan BP. The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J Cell Sci. 2013;126:2903–2913. doi: 10.1242/jcs.124388. [DOI] [PubMed] [Google Scholar]
- 70.Samstad EO, et al. Cholesterol crystals induce complement-dependent inflammasome activation and cytokine release. J Immunol. 2014;192:2837–2845. doi: 10.4049/jimmunol.1302484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Benoit ME, Clarke EV, Morgado P, Fraser DA, Tenner AJ. Complement protein C1q directs macrophage polarization and limits inflammasome activity during the uptake of apoptotic cells. J Immunol. 2012;188:5682–5693. doi: 10.4049/jimmunol.1103760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pinto MR, Melillo D, Giacomelli S, Sfyroera G, Lambris JD. Ancient origin of the complement system: emerging invertebrate models. Adv Exp Med Biol. 2007;598:372–388. doi: 10.1007/978-0-387-71767-8_26. [DOI] [PubMed] [Google Scholar]
- 73.Flajnik MF. Re-evaluation of the immunological Big Bang. Curr Biol. 2014;24:R1060–R1065. doi: 10.1016/j.cub.2014.09.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sunyer JO, et al. Evolution of complement as an effector system in innate and adaptive immunity. Immunol Res. 2003;27:549–564. doi: 10.1385/IR:27:2-3:549. [DOI] [PubMed] [Google Scholar]
- 75.Carroll MC, Isenman DE. Regulation of humoral immunity by complement. Immunity. 2012;37:199–207. doi: 10.1016/j.immuni.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dempsey PW, Allison ME, Akkaraju S, Goodnow CC, Fearon DT. C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science. 1996;271:348–350. doi: 10.1126/science.271.5247.348. [DOI] [PubMed] [Google Scholar]
- 77.Humphrey JH, Grennan D, Sundaram V. The origin of follicular dendritic cells in the mouse and the mechanism of trapping of immune complexes on them. Eur J Immunol. 1984;14:859–864. doi: 10.1002/eji.1830140916. [DOI] [PubMed] [Google Scholar]
- 78.Karsten CM, Kohl J. The immunoglobulin, IgG Fc receptor and complement triangle in autoimmune diseases. Immunobiology. 2012;217:1067–1079. doi: 10.1016/j.imbio.2012.07.015. [DOI] [PubMed] [Google Scholar]
- 79.Huang ZY, et al. Interaction of two phagocytic host defense systems: Fcγ receptors and complement receptor 3. J Biol Chem. 2011;286:160–168. doi: 10.1074/jbc.M110.163030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Karsten CM, et al. Anti-inflammatory activity of IgG1 mediated by Fc galactosylation and association of FcγRIIB and dectin-1. Nat Med. 2012;18:1401–1406. doi: 10.1038/nm.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bosmann M, Ward PA. The inflammatory response in sepsis. Trends Immunol. 2013;34:129–136. doi: 10.1016/j.it.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ward PA. The dark side of C5a in sepsis. Nat Rev Immunol. 2004;4:133–142. doi: 10.1038/nri1269. [DOI] [PubMed] [Google Scholar]
- 83.Silasi-Mansat R, et al. Complement inhibition decreases early fibrogenic events in the lung of septic baboons. J Cell Mol Med. 2015;19:2549–2563. doi: 10.1111/jcmm.12667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Silasi-Mansat R, et al. Complement inhibition decreases the procoagulant response and confers organ protection in a baboon model of Escherichia coli sepsis. Blood. 2010;116:1002–1010. doi: 10.1182/blood-2010-02-269746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Barratt-Due A, et al. Combined inhibition of complement C5 and CD14 markedly attenuates inflammation, thrombogenicity, and hemodynamic changes in porcine sepsis. J Immunol. 2013;191:819–827. doi: 10.4049/jimmunol.1201909. [DOI] [PubMed] [Google Scholar]
- 86.Egge KH, et al. The anti-inflammatory effect of combined complement and CD14 inhibition is preserved during escalating bacterial load. Clin Exp Immunol. 2015;181:457–467. doi: 10.1111/cei.12645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Burk AM, et al. Early complementopathy after multiple injuries in humans. Shock. 2012;37:348–354. doi: 10.1097/SHK.0b013e3182471795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ganter MT, et al. Role of the alternative pathway in the early complement activation following major trauma. Shock. 2007;28:29–34. doi: 10.1097/shk.0b013e3180342439. [DOI] [PubMed] [Google Scholar]
- 89.Huber-Lang M, Kovtun A, Ignatius A. The role of complement in trauma and fracture healing. Semin Immunol. 2013;25:73–78. doi: 10.1016/j.smim.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 90.Rafail S, et al. Complement deficiency promotes cutaneous wound healing in mice. J Immunol. 2015;194:1285–1291. doi: 10.4049/jimmunol.1402354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brennan FH, et al. The complement receptor C5aR controls acute inflammation and astrogliosis following spinal cord injury. J Neurosci. 2015;35:6517–6531. doi: 10.1523/JNEUROSCI.5218-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chen M, Daha MR, Kallenberg CG. The complement system in systemic autoimmune disease. J Autoimmun. 2010;34:J276–286. doi: 10.1016/j.jaut.2009.11.014. [DOI] [PubMed] [Google Scholar]
- 93.Chen M, Kallenberg CG. ANCA-associated vasculitides — advances in pathogenesis and treatment. Nat Rev Rheumatol. 2010;6:653–664. doi: 10.1038/nrrheum.2010.158. [DOI] [PubMed] [Google Scholar]
- 94.Banz Y, Rieben R. Role of complement and perspectives for intervention in ischemia-reperfusion damage. Ann Med. 2012;44:205–217. doi: 10.3109/07853890.2010.535556. [DOI] [PubMed] [Google Scholar]
- 95.Zipfel PF, et al. The role of complement in C3 glomerulopathy. Mol Immunol. 2015;67:21–30. doi: 10.1016/j.molimm.2015.03.012. [DOI] [PubMed] [Google Scholar]
- 96.Orsini F, De Blasio D, Zangari R, Zanier ER, De Simoni MG. Versatility of the complement system in neuroinflammation, neurodegeneration and brain homeostasis. Front Cell Neurosci. 2014;8:380. doi: 10.3389/fncel.2014.00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Veerhuis R, Nielsen HM, Tenner AJ. Complement in the brain. Mol Immunol. 2011;48:1592–1603. doi: 10.1016/j.molimm.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hovland A, et al. The complement system and toll-like receptors as integrated players in the pathophysiology of atherosclerosis. Atherosclerosis. 2015;241:480–494. doi: 10.1016/j.atherosclerosis.2015.05.038. [DOI] [PubMed] [Google Scholar]
- 99.Morgan BP. The role of complement in neurological and neuropsychiatric diseases. Expert Rev Clin Immunol. 2015;11:1109–1119. doi: 10.1586/1744666X.2015.1074039. [DOI] [PubMed] [Google Scholar]
- 100.Patzelt J, Verschoor A, Langer HF. Platelets and the complement cascade in atherosclerosis. Front Physiol. 2015;6:49. doi: 10.3389/fphys.2015.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.McHarg S, Clark SJ, Day AJ, Bishop PN. Age-related macular degeneration and the role of the complement system. Mol Immunol. 2015;67:43–50. doi: 10.1016/j.molimm.2015.02.032. [DOI] [PubMed] [Google Scholar]
- 102.Ekdahl KN, et al. Innate immunity activation on biomaterial surfaces: a mechanistic model and coping strategies. Adv Drug Deliv Rev. 2011;63:1042–1050. doi: 10.1016/j.addr.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Grumach AS, Kirschfink M. Are complement deficiencies really rare? Overview on prevalence, clinical importance and modern diagnostic approach. Mol Immunol. 2014;61:110–117. doi: 10.1016/j.molimm.2014.06.030. [DOI] [PubMed] [Google Scholar]
- 104.Risitano AM. Paroxysmal nocturnal hemoglobinuria and other complement-mediated hematological disorders. Immunobiology. 2012;217:1080–1087. doi: 10.1016/j.imbio.2012.07.014. [DOI] [PubMed] [Google Scholar]
- 105.Rodriguez E, Rallapalli PM, Osborne AJ, Perkins SJ. New functional and structural insights from updated mutational databases for complement factor H, Factor I, membrane cofactor protein and C3. Biosci Rep. 2014;34:5. doi: 10.1042/BSR20140117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hageman GS, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA. 2005;102:7227–7232. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Weismann D, et al. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature. 2011;478:76–81. doi: 10.1038/nature10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Heurich M, et al. Common polymorphisms in C3, factor B, and factor H collaborate to determine systemic complement activity and disease risk. Proc Natl Acad Sci USA. 2011;108:8761–8766. doi: 10.1073/pnas.1019338108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kurts C, Panzer U, Anders HJ, Rees AJ. The immune system and kidney disease: basic concepts and clinical implications. Nat Rev Immunol. 2013;13:738–753. doi: 10.1038/nri3523. [DOI] [PubMed] [Google Scholar]
- 110.Song D, Zhou W, Sheerin SH, Sacks SH. Compartmental localization of complement component transcripts in the normal human kidney. Nephron. 1998;78:15–22. doi: 10.1159/000044876. [DOI] [PubMed] [Google Scholar]
- 111.Noris M, Remuzzi G. Overview of complement activation and regulation. Semin Nephrol. 2013;33:479–492. doi: 10.1016/j.semnephrol.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Clark SJ, et al. Tissue-specific host recognition by complement factor H is mediated by differential activities of its glycosaminoglycan-binding regions. J Immunol. 2013;190:2049–2057. doi: 10.4049/jimmunol.1201751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jozsi M, Tortajada A, Uzonyi B, Goicoechea de Jorge E, Rodriguez de Cordoba S. Factor H-related proteins determine complement-activating surfaces. Trends Immunol. 2015;36:374–384. doi: 10.1016/j.it.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 114.Noris M, Mescia F, Remuzzi G. STEC-HUS, atypical HUS and TTP are all diseases of complement activation. Nat Rev Nephrol. 2012;8:622–633. doi: 10.1038/nrneph.2012.195. [DOI] [PubMed] [Google Scholar]
- 115.Kenawy HI, Boral I, Bevington A. Complement-coagulation cross-talk: a potential mediator of the physiological activation of complement by low pH. Front Immunol. 2015;6:215. doi: 10.3389/fimmu.2015.00215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014;371:654–666. doi: 10.1056/NEJMra1312353. [DOI] [PubMed] [Google Scholar]
- 117.Riedl M, et al. Spectrum of complement-mediated thrombotic microangiopathies: pathogenetic insights identifying novel treatment approaches. Semin Thromb Hemost. 2014;40:444–464. doi: 10.1055/s-0034-1376153. [DOI] [PubMed] [Google Scholar]
- 118.Mele C, Remuzzi G, Noris M. Hemolytic uremic syndrome. Semin Immunopathol. 2014;36:399–420. doi: 10.1007/s00281-014-0416-x. [DOI] [PubMed] [Google Scholar]
- 119.Noris M, et al. Dynamics of complement activation in aHUS and how to monitor eculizumab therapy. Blood. 2014;124:1715–1726. doi: 10.1182/blood-2014-02-558296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Frimat M, et al. Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome. Blood. 2013;122:282–292. doi: 10.1182/blood-2013-03-489245. [DOI] [PubMed] [Google Scholar]
- 121.Zuber J, et al. Use of eculizumab for atypical haemolytic uraemic syndrome and C3 glomerulopathies. Nat Rev Nephrol. 2012;8:643–657. doi: 10.1038/nrneph.2012.214. [DOI] [PubMed] [Google Scholar]
- 122.Cofiell R, et al. Eculizumab reduces complement activation, inflammation, endothelial damage, thrombosis, and renal injury markers in aHUS. Blood. 2015;125:3253–3262. doi: 10.1182/blood-2014-09-600411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Morgan BP, Harris CL. Complement, a target for therapy in inflammatory and degenerative diseases. Nat Rev Drug Discov. 2015;14:857–877. doi: 10.1038/nrd4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Meri S. Complement activation in diseases presenting with thrombotic microangiopathy. Eur J Intern Med. 2013;24:496–502. doi: 10.1016/j.ejim.2013.05.009. [DOI] [PubMed] [Google Scholar]
- 125.Morigi M, et al. Alternative pathway activation of complement by Shiga toxin promotes exuberant C3a formation that triggers microvascular thrombosis. J Immunol. 2011;187:172–180. doi: 10.4049/jimmunol.1100491. [DOI] [PubMed] [Google Scholar]
- 126.Nester CM, Brophy PD. Eculizumab in the treatment of atypical haemolytic uraemic syndrome and other complement-mediated renal diseases. Curr Opin Pediatr. 2013;25:225–231. doi: 10.1097/MOP.0b013e32835df4a3. [DOI] [PubMed] [Google Scholar]
- 127.Pecoraro C, et al. Treatment of congenital thrombotic thrombocytopenic purpura with eculizumab. Am J Kidney Dis. 2015;66:1067–1070. doi: 10.1053/j.ajkd.2015.06.032. [DOI] [PubMed] [Google Scholar]
- 128.Phillips EH, et al. The role of ADAMTS-13 activity and complement mutational analysis in differentiating acute thrombotic microangiopathies. J Thromb Haemost. 2016;14:175–185. doi: 10.1111/jth.13189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tsai E, Chapin J, Laurence JC, Tsai HM. Use of eculizumab in the treatment of a case of refractory, ADAMTS13-deficient thrombotic thrombocytopenic purpura: additional data and clinical follow-up. Br J Haematol. 2013;162:558–559. doi: 10.1111/bjh.12387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pickering MC, et al. C3 glomerulopathy: consensus report. Kidney Int. 2013;84:1079–1089. doi: 10.1038/ki.2013.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cook HT, Pickering MC. Histopathology of MPGN and C3 glomerulopathies. Nat Rev Nephrol. 2015;11:14–22. doi: 10.1038/nrneph.2014.217. [DOI] [PubMed] [Google Scholar]
- 132.Servais A, et al. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int. 2012;82:454–464. doi: 10.1038/ki.2012.63. [DOI] [PubMed] [Google Scholar]
- 133.Martinez-Barricarte R, et al. Human C3 mutation reveals a mechanism of dense deposit disease pathogenesis and provides insights into complement activation and regulation. J Clin Invest. 2010;120:3702–3712. doi: 10.1172/JCI43343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Xiao X, Pickering MC, Smith RJ. C3 glomerulopathy: the genetic and clinical findings in dense deposit disease and C3 glomerulonephritis. Semin Thromb Hemost. 2014;40:465–471. doi: 10.1055/s-0034-1376334. [DOI] [PubMed] [Google Scholar]
- 135.Damman J, et al. Hypoxia and complement-and-coagulation pathways in the deceased organ donor as the major target for intervention to improve renal allograft outcome. Transplantation. 2015;99:1293–1300. doi: 10.1097/TP.0000000000000500. [DOI] [PubMed] [Google Scholar]
- 136.Amura CR, et al. Complement activation and toll-like receptor-2 signaling contribute to cytokine production after renal ischemia/reperfusion. Mol Immunol. 2012;52:249–257. doi: 10.1016/j.molimm.2012.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Farrar CA, et al. Collectin-11 detects stress-induced L-fucose pattern to trigger renal epithelial injury. J Clin Invest. 2016;126:1911–19. doi: 10.1172/JCI83000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Muller TF, Kraus M, Neumann C, Lange H. Detection of renal allograft rejection by complement components C5A and TCC in plasma and urine. J Lab Clin Med. 1997;129:62–71. doi: 10.1016/s0022-2143(97)90162-1. [DOI] [PubMed] [Google Scholar]
- 139.Stites E, Le Quintrec M, Thurman JM. The complement system and antibody-mediated transplant rejection. J Immunol. 2015;195:5525–5531. doi: 10.4049/jimmunol.1501686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pratt JR, Basheer SA, Sacks SH. Local synthesis of complement component C3 regulates acute renal transplant rejection. Nat Med. 2002;8:582–587. doi: 10.1038/nm0602-582. [DOI] [PubMed] [Google Scholar]
- 141.Strainic MG, Shevach EM, An F, Lin F, Medof ME. Absence of signaling into CD4+ cells via C3aR and C5aR enables autoinductive TGF-β1 signaling and induction of Foxp3+ regulatory T cells. Nat Immunol. 2013;14:162–171. doi: 10.1038/ni.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.van der Touw W, et al. Cutting edge: receptors for C3a and C5a modulate stability of alloantigen-reactive induced regulatory T cells. J Immunol. 2013;190:5921–5925. doi: 10.4049/jimmunol.1300847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Marsh JE, et al. The allogeneic T and B cell response is strongly dependent on complement components C3 and C4. Transplantation. 2001;72:1310–1318. doi: 10.1097/00007890-200110150-00022. [DOI] [PubMed] [Google Scholar]
- 144.Thomas KA, Valenzuela NM, Reed EF. The perfect storm: HLA antibodies, complement, FcγRs, and endothelium in transplant rejection. Trends Mol Med. 2015;21:319–329. doi: 10.1016/j.molmed.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chen Song S, et al. Complement inhibition enables renal allograft accommodation and long-term engraftment in presensitized nonhuman primates. Am J Transplant. 2011;11:2057–2066. doi: 10.1111/j.1600-6143.2011.03646.x. [DOI] [PubMed] [Google Scholar]
- 146.Fremeaux-Bacchi V, Legendre CM. The emerging role of complement inhibitors in transplantation. Kidney Int. 2015;88:967–973. doi: 10.1038/ki.2015.253. [DOI] [PubMed] [Google Scholar]
- 147.DeAngelis RA, Reis ES, Ricklin D, Lambris JD. Targeted complement inhibition as a promising strategy for preventing inflammatory complications in hemodialysis. Immunobiology. 2012;217:1097–1105. doi: 10.1016/j.imbio.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kourtzelis I, et al. Complement anaphylatoxin C5a contributes to hemodialysis-associated thrombosis. Blood. 2010;116:631–639. doi: 10.1182/blood-2010-01-264051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ricklin D, Lambris JD. Complement in immune and inflammatory disorders: therapeutic interventions. J Immunol. 2013;190:3839–3847. doi: 10.4049/jimmunol.1203200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Licht C, et al. Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2-year extensions of phase 2 studies. Kidney Int. 2015;87:1061–1073. doi: 10.1038/ki.2014.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wong EK, Kavanagh D. Anticomplement C5 therapy with eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. Transl Res. 2015;165:306–320. doi: 10.1016/j.trsl.2014.10.010. [DOI] [PubMed] [Google Scholar]
- 152.Eskandary F, Wahrmann M, Muhlbacher J, Bohmig GA. Complement inhibition as potential new therapy for antibody-mediated rejection. Transpl Int. 2016;29:392–402. doi: 10.1111/tri.12706. [DOI] [PubMed] [Google Scholar]
- 153.Stegall MD, et al. Terminal complement inhibition decreases antibody-mediated rejection in sensitized renal transplant recipients. Am J Transplant. 2011;11:2405–2413. doi: 10.1111/j.1600-6143.2011.03757.x. [DOI] [PubMed] [Google Scholar]
- 154.Cornell LD, Schinstock CA, Gandhi MJ, Kremers WK, Stegall MD. Positive crossmatch kidney transplant recipients treated with eculizumab: outcomes beyond 1 year. Am J Transplant. 2015;15:1293–2302. doi: 10.1111/ajt.13168. [DOI] [PubMed] [Google Scholar]
- 155.Ricklin D, Lambris JD. Therapeutic control of complement activation at the level of the central component C3. Immunobiology. 2015 doi: 10.1016/j.imbio.2015.06.012. http://dx.doi.org/10.1016/j.imbio.2015.06.012. [DOI] [PMC free article] [PubMed]
- 156.Zhang Y, et al. Soluble CR1 therapy improves complement regulation in C3 glomerulopathy. J Am Soc Nephrol. 2013;24:1820–1829. doi: 10.1681/ASN.2013010045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Sacks S, et al. Targeting complement at the time of transplantation. Adv Exp Med Biol. 2013;735:247–255. doi: 10.1007/978-1-4614-4118-2_17. [DOI] [PubMed] [Google Scholar]
- 158.US National Library of Science. ClinicalTrials.gov. 2016 https://clinicaltrials.gov/show/NCT02247531.
- 159.US National Library of Science. ClinicalTrials.gov. 2016 https://clinicaltrials.gov/show/NCT02247479.
- 160.Zhang Y, et al. Compstatin analog Cp40 inhibits complement dysregulation in vitro in C3 glomerulopathy. Immunobiology. 2015;220:993–998. doi: 10.1016/j.imbio.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Risitano AM, et al. Peptide inhibitors of C3 activation as a novel strategy of complement inhibition for the treatment of paroxysmal nocturnal hemoglobinuria. Blood. 2014;123:2094–2101. doi: 10.1182/blood-2013-11-536573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Reis ES, et al. Therapeutic C3 inhibitor Cp40 abrogates complement activation induced by modern hemodialysis filters. Immunobiology. 2015;220:476–482. doi: 10.1016/j.imbio.2014.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.US National Library of Science. ClinicalTrials.gov. 2016 https://clinicaltrials.gov/show/NCT02264639.
- 164.US National Library of Science. ClinicalTrials.gov. 2016 https://clinicaltrials.gov/show/NCT02503332.
- 165.US National Library of Science. ClinicalTrials.gov. 2016 https://clinicaltrials.gov/show/NCT02588833.
- 166.US National Library of Science. ClinicalTrials.gov. 2015 https://clinicaltrials.gov/show/NCT02502903.
- 167.US National Library of Science. ClinicalTrials.gov. 2015 https://clinicaltrials.gov/show/NCT02222545.
- 168.Vo AA, et al. A phase I/II placebo-controlled trial of C1-inhibitor for prevention of antibody-mediated rejection in HLA sensitized patients. Transplantation. 2015;99:299–308. doi: 10.1097/TP.0000000000000592. [DOI] [PubMed] [Google Scholar]
- 169.ChemoCentryx. ChemoCentryx announces positive results in phase II ANCA-associated vasculitis CLEAR trial of orally administered complement 5a receptor inhibitor CCX168. 2016 http://ir.chemocentryx.com/releasedetail.cfm?releaseid=948998.
- 170.US National Library of Science. ClinicalTrials.gov. 2016 https://clinicaltrials.gov/show/NCT02384317.
- 171.US National Library of Science. ClinicalTrials.gov. 2015 https://clinicaltrials.gov/show/NCT02464891.
- 172.US National Library of Science. ClinicalTrials.gov. 2016 https://clinicaltrials.gov/show/NCT02246595.
- 173.Hillmen P, et al. Long-term safety and efficacy of sustained eculizumab treatment in patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2013;162:62–73. doi: 10.1111/bjh.12347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wu MA, Zanichelli A, Mansi M, Cicardi M. Current treatment options for hereditary angioedema due to C1 inhibitor deficiency. Expert Opin Pharmacother. 2016;17:27–40. doi: 10.1517/14656566.2016.1104300. [DOI] [PubMed] [Google Scholar]
- 175.Banda NK, et al. Mechanisms of mannose-binding lectin-associated serine proteases-1/3 activation of the alternative pathway of complement. Mol Immunol. 2011;49:281–289. doi: 10.1016/j.molimm.2011.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Dobo J, et al. Multiple roles of complement MASP-1 at the interface of innate immune response and coagulation. Mol Immunol. 2014;61:69–78. doi: 10.1016/j.molimm.2014.05.013. [DOI] [PubMed] [Google Scholar]
- 177.Arnold JN, Royle L, Dwek RA, Rudd PM, Sim RB. Human immunoglobulin glycosylation and the lectin pathway of complement activation. Adv Exp Med Biol. 2005;564:27–43. doi: 10.1007/0-387-25515-X_9. [DOI] [PubMed] [Google Scholar]
- 178.Amara U, et al. Molecular intercommunication between the complement and coagulation systems. J Immunol. 2010;185:5628–5636. doi: 10.4049/jimmunol.0903678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Inforzato A, et al. PTX3 as a paradigm for the interaction of pentraxins with the complement system. Semin Immunol. 2013;25:79–85. doi: 10.1016/j.smim.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 180.Csincsi AI, et al. Factor H-related protein 5 interacts with pentraxin 3 and the extracellular matrix and modulates complement activation. J Immunol. 2015;194:4963–4973. doi: 10.4049/jimmunol.1403121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Trouw LA, Blom AM, Gasque P. Role of complement and complement regulators in the removal of apoptotic cells. Mol Immunol. 2008;45:1199–1207. doi: 10.1016/j.molimm.2007.09.008. [DOI] [PubMed] [Google Scholar]