

Abstract
Background/Aim
Legalon SIL (SIL) is a chemically hydrophilized version of silibinin, an extract of milk thistle (Silybum marianum) seeds that has exhibited hepatoprotective and antiviral effectiveness against hepatitis C virus (HCV) in patients leading to viral clearance in combination with ribavirin. To elucidate the incompletely understood mode of action of SIL against HCV, mathematical modeling of HCV kinetics and human hepatocyte gene expression studies were performed in uPA-SCID-chimeric mice with humanized livers.
Methods
Chronically HCV-infected mice (n=15) were treated for 14 days with daily intravenous SIL at 469, 265 or 61.5 mg/kg. Serum HCV and human albumin (hAlb) were measured frequently and liver HCV RNA was analyzed at days 3 and 14. Microarray analysis of human hepatocyte gene expression was performed at days 0, 3, and 14 of treatment.
Results
While hAlb remained constant, a biphasic viral decline in serum was observed consisting of a rapid 1st phase followed by a 2nd slower phase (or plateau with the two lower SIL dosings). SIL effectiveness in blocking viral production was similar among dosing groups (median ε=77%). However, the rate of HCV-infected hepatocyte decline, δ, was dose-dependent. Intracellular HCV RNA levels correlated (r=0.66, P=.01) with serum HCV RNA. Pathway analysis revealed increased anti-inflammatory and anti-proliferative gene expression in human hepatocytes in SIL-treated mice.
Conclusions
The results suggest that SIL could lead to a continuous 2nd phase viral decline, i.e., potentially viral clearance, in the absence of adaptive immune response along with increased anti-inflammatory and anti-proliferative gene expression in human hepatocytes.
Keywords: viral kinetic modeling, uPA-SCID, chimeric mice with humanized livers, gene expression, anti-inflammatory
Introduction
Silymarin, an extract of milk thistle, and silymarin-derived compounds have long been considered hepatoprotective with antiviral, antioxidant, anti-inflammatory and immunomodulatory functions (1). A major component of silymarin is silibinin, which is an isomeric mixture of silybin A and silybin B. High doses of silibinin can be administered intravenously (i.v.) using an esterified modification silibinin-C-2′, 3-dihydrogen succinate, disodium salt (Legalon SIL). For over two decades, intravenous SIL has provided a well-tolerated and safe treatment for Amanita phalloides-induced acute liver failure (2).
Hepatitis C virus (HCV) infection affects over 150 million people worldwide causing approximately 350,000 deaths annually from cirrhosis and hepatocellular carcinoma (3, 4). While interferon (IFN)-based therapy was a mainstay of HCV treatment for decades, more effective IFN-free regimens with fewer side effects are the new treatment standard (5-8). Legalon SIL (SIL) has exhibited high antiviral efficacy against HCV in patients with both compensated and decompensated liver disease with minimal side effects (1, 9-16). Additionally, IFN-free therapy with SIL and ribavirin has resulted in sustained virologic response (i.e., viral clearance) (17).
Despite these potentially broad clinically-relevant hepatoprotective and antiviral effects, the mechanism of action (MOA) by which SIL exerts these effects is not completely understood. Morishima et al (18) demonstrated in vitro that silibinin inhibited T cell proliferation and pro-inflammatory cytokine secretion that could potentially control hepatic inflammation and subsequent fibrosis progression (19). Many antiviral MOA of SIL against HCV have been suggested (20, 21) including inhibition of replication (20, 22) and inhibition of viral entry (20, 23).
In this study HCV-infected uPA-SCID chimeric mice with humanized livers (24-26) were treated with SIL. Viral response kinetics in the serum and liver was monitored as was cellular gene expression. Mathematical modeling and gene profiling analysis were performed to shed light on the possible antiviral MOA of SIL and its effects on human hepatocytes.
Methods
Preparation of chimeric mice and infection with HCV
Human hepatocyte chimeric mice were prepared as described previously (27). Eighteen uPA+/+/SCID+/+ chimeric mice (PhoenixBio Co., Ltd., Higashihiroshima, Japan) were transplanted with cryopreserved human hepatocytes from the same donor. All mice used in the experiment had human hepatocyte repopulation rates greater than 90%. The repopulation index was determined by measurement of serum human albumin (hAlb) levels using the Human Albumin enzyme-linked immunosorbent assay (ELISA) Quantitation kit (Bethyl Laboratories Inc., Montgomery, TX) according to the instructions provided by the manufacturer. Mice were inoculated via the mouse tail vein with 100 μl of human serum containing 4×105 copies of HCV genotype 1b particles obtained from a patient with chronic hepatitis C who provided written informed consent to participate in the study. All mice developed measurable viremia 4 weeks after inoculation, and virus titers reached stable viremia (6-8 log10 copies/ml; mean 7.4 log10 copies/ml) 8 weeks after inoculation. Infection, extraction of serum samples, and sacrifice were performed under ether anesthesia, and all animals received humane care. The experimental protocol met the ethical guidelines of the 1975 Declaration of Helsinki and was performed in accordance with the guidelines of the local committee for animal experiments at Hiroshima University.
SIL treatment
Chimeric mice with established HCV infection were treated with daily intravenous SIL doses (solubilized in saline) of 469 mg/kg (n=5), 246 mg/kg (n=5) and 61.5 mg/kg (n=5). SIL doses were chosen to mimic the effective SIL 5-20 mg/kg doses of SIL used in patients (12). Blood was obtained at days 0, 0.5, 1, 2, 3, 5, 7, 10 and 14 in order to measure serum human albumin (hAlb) and HCV RNA levels (Roche COBAS AmpliPrep/COBAS TaqMan HCV test v2.0). Three mice were sacrificed at day 0 (d0) before treatment initiation, three mice in each group were sacrificed at day 3 (d3) of treatment, and two mice in each group were sacrificed at day 14 (d14) of treatment in order to monitor intracellular HCV RNA and hepatocyte gene expression.
Human albumin (hAlb) Assay
Mouse serum concentration of hAlb, which is correlated with the repopulation index (27) was measured based on latex agglutination immunonephelometry (LX Reagent ‘Eiken’ Alb II; Eiken Chemical, Tokyo, Japan) as described previously (28).
Analysis of intracellular HCV RNA
Human hepatocytes were finely dissected from chimeric mouse livers, and total cellular RNA was isolated using the RNeasy Mini Kit (Qiagen, Valencia, CA). One microgram of RNA was subjected to reverse transcription with random primers, and qPCR for HCV and human β-actin was performed. HCV primers were 5′-TTTATCCAAGAAAGGACCC-3′ and 5′-TTCACGCAGAAAGCGTCTAGC-3′. Quantitation of β-actin was performed using TaqMan Gene Expression Assay primer and probe sets (PE Applied Biosystems, Foster City, CA). HCV RNA levels are expressed as a ratio relative to β-actin levels.
Mathematical Modeling
HCV viral kinetics under SIL therapy was assumed to follow the standard biphasic model (29):
| Eq. 1 |
where T represents uninfected human hepatocytes (target cells), I, HCV-infected hepatocytes and V, free virus. Based on steady-state serum hAlb levels, we assumed that human hepatocyte numbers remained constant, represented by the pre-treatment level, T0=cδ/βp. Virus, V, infects human hepatocytes with rate constant β, generating infected cells, I, which produce new virus at rate p per infected hepatocyte. Infected hepatocytes are lost/cured at a rate δ per infected hepatocyte and virus is assumed to be cleared at rate c per virion. The effect of SIL on virus production/release is modeled by a factor (1-ε), where ε is defined as the effectiveness of the drug in blocking viral production/release. A pharmacological delay, t0, was estimated during which viral load remained at baseline level. After the delay, the viral load declined.
Parameter Estimation
Since the 2nd phase of serum HCV decline cannot be seen in mice that were treated for only 3 days, the biphasic model (Eq. 1) was fit only to serum HCV kinetic data obtained from the 6 mice which were treated for 14 days (2 in each dosing group). To avoid parameter identifiability issues, the HCV clearance rate constant from blood was set to c=6/day (as previously done in (9, 11)). In addition, a two-step fitting procedure was performed; first, the baseline viral load V0, and the pharmacokinetic delay t0 were estimated using Berkeley-Madonna software (v 8.3.18); then the values of V0 and t0 were fixed to estimate the values of the parameters ε and δ and their standard error using DEDiscover (https://www.dediscover.org/).
Statistical Analysis
A linear regression model was fit to hAlb levels using R software (v3.2.0). To estimate the transition time between the 1st and 2nd phases and their slopes, we used the segmented linear regression package called ‘segmented 0.5-0.0’ in R 3.2.0. Pearson correlation was calculated between serum HCV RNA and intrahepatic HCV RNA. The Mann-Whitney test was used to determine whether viral kinetics between groups was significantly different. A P-value≤0.05 was considered significant.
Microarray Analysis
The effect of SIL on gene expression in human hepatocytes was assessed by microarray analysis at d0 (n=3) in untreated mice, at d3 (n=3) in 469 mg/kg SIL-treated chimeric mice and at d14 (n=3) in two 469 mg/kg and one 265 mg/kg SIL-treated chimeric mice. Human hepatocytes were finely dissected, and total RNA was extracted as above for use in microarray analysis. As outlined in Fig. S1A, Amino Allyl aRNA was synthesized using the Amino Allyl MessageAmp II aRNA Amplification Kit (Ambion, Tokyo, Japan). CyeDye coupling and fragmentation were performed following the protocol supplied by the manufacturer (Toray Industries Inc., Tokyo, Japan). RNA samples were hybridized to Toray 3D-Gene Human 25kVer 2.10 chips (24460 probes) for 16 h at 37°C by rotary shaking (250 rpm). Microarrays were scanned using a 3D-Gene Scanner, and images were quantified using Toray Extraction software. Spot data was normalized by substitution with the mean intensity of the background signal, which was determined by the 95% confidence interval of the signal intensities over all blank spots. Duplicate spots with signal intensities greater than 2 standard deviations (SD) above background signal intensity were used for analysis. Gene expression levels were normalized across microarrays using quantile normalization (Fig. S1B). Probes for which one or more samples contained missing data were removed, resulting in a final set of 17084 probes. Differential expression was analyzed using the Limma package (30), which fits a linear model to each gene using moderated t statistics and empirical Bayes analysis. P-values were adjusted for multiple testing using the Benjamini–Hochberg false discovery rate (PFDR). Gene set enrichment in canonical pathways was analyzed using Ingenuity Pathway Analysis software (Ingenuity Systems, CA, USA; http://www.ingenuity.com). Notably, because there was no difference in gene expression observed in the SIL-treated mice analyzed on day 3 and day 14 as observed by hierarchical clustering (Fig. S3) data from the 3 control mice was compared to the 6 SIL treated mice as a single group. Microarray data generated in this study have been deposited in NCBI's Gene Expression Omnibus (GEO) and are accessible through GEO Series accession number GSE79103.
Results
HCV kinetic data
Baseline serum HCV RNA (median 7.5 [Q1-Q3:7.2-7.8] log10 IU/ml) and hAlb (median 7.1 [Q1-Q3:7.0-7.2] log10 mg/ml) were similar (P>0.4) among the three dosing groups (Table S1). Intravenous SIL infusion was well tolerated. In all mice hAlb levels remained constant during treatment, i.e., hAlb slopes were not different than zero, P≥0.10 (Fig. 1). The median viral decline from baseline to d1 or d2 was 0.4 log10 IU/ml with no difference (P>0.7) among the dosing groups (Table S1). A significantly (P=0.016) higher viral drop from baseline was observed in the 469 mg/kg dosing group at d3 (median 0.9 log10 IU/ml) compared to the 246 mg/kg (median 0.6 log10 IU/ml) and the 61.5 mg/kg (median 0 log10 IU/ml) groups. While the two lowest dosing groups showed a 2nd phase plateau in viral load from d2/d3 to the end of treatment, the 469 mg/kg dosing group exhibited continued viral decline with more than 1 log10 IU/ml decline at d10 and up to ∼1.5 log10 IU/ml decline by d14 (Fig. 1; Table S1). Consistent with this analysis, a segmented linear regression model indicates that serum HCV RNA declined in a biphasic manner, consisting of a rapid 1st phase (median 0.7 [Q1-Q3: 0.60-0.88] that lasted 1.3 days [Q1-Q3:0.7-1.7 day]. The two lower dosing groups exhibited a 2nd phase plateau, while mice in the 469 mg/kg dosing group (n=2) had a continuous 2nd phase viral decline with slopes of 0.1 and 0.12 log10 IU/ml/day/w (Table S2). Finally, a comparison of intracellular HCV RNA levels with serum HCV RNA levels in individual mice showed a significant positive correlation (r=0.66, P=0.01; Fig. S2).
Figure 1. Serum HCV RNA and human albumin (hAlb) kinetics with model curves.
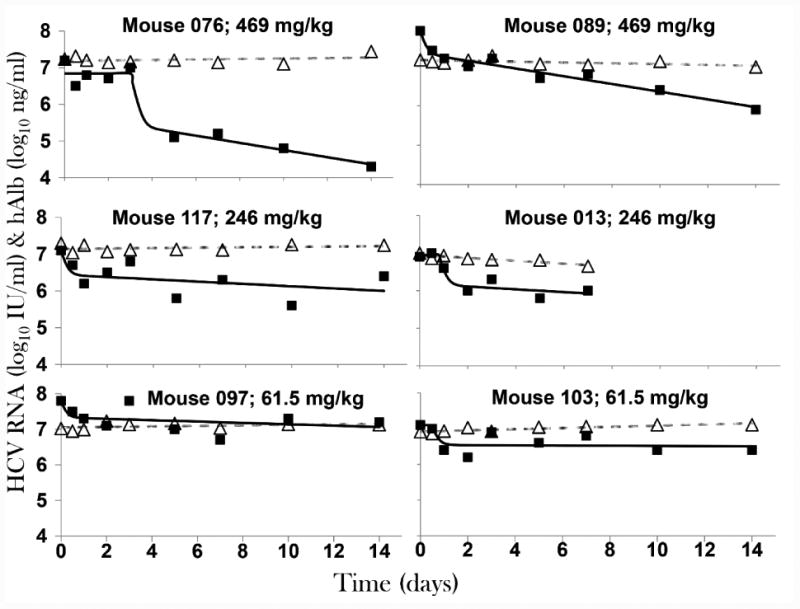
Chronically HCV infected uPA-SCID chimeric mice with humanized livers were treated intravenously with the indicated doses of SIL. Blood was drawn from mice daily. Serum HCV RNA level (solid black squares) and human albumin level (open grey triangles) are graphed as log10 IU/mL and ng/mL, respectively. HCV model curves (black lines) and hAlb linear regression (grey dashed lines) are shown. See Tables S1 and 1 for viral kinetic and estimated parameter values, respectively.
Mathematical modeling
Modeling was performed using the standard HCV biphasic model (29) (Equation 1). Results suggest a delay t0≥12 hr in 3 mice, one in each dosing group (Table 1). SIL effectiveness in blocking viral production was not different among the dosing groups (median ε=77% [Q1-Q3:72%-83%]. However, the rate of decrease in infected human hepatocytes, δ, was higher in mice (n=2) treated with 469 mg/kg SIL (0.23/day [standard error, se=0.06] and 0.31/day [se=0.04]) compared to mice (n=2) treated with 246 mg/kg SIL (0.09/day [se=0.10] and 0.10/day [se=0.00]) or mice (n=2) treated with 61.5 mg/kg SIL (0.07/day [se=0.07] and 0.01/day [se=0.08]).
Table 1. Modeling results.
V0, baseline viral load; t0, pharmacological delay; ε, SIL effectiveness in blocking viral production; δ, clearance of productively HCV-infected human hepatocytes; SE, standard error; Interquartile range:Q1-Q3; * Estimated t0 values <0.5 days were set to 0; **Fitting results based on data until day 7, i.e., death time. i.v., intravenous.
| Mouse ID | SIL Dosing [mg/kg, i.v.] |
V0 [Log10 IU/ml] |
t0 [days] |
ε (SE) [%] |
δ (SE) [1/day] |
|---|---|---|---|---|---|
| 076 | 469 | 6.8 | 3 | 96.3(1.3) | 0.23 (0.06) |
| 089 | 469 | 7.9 | 0.0* | 74.7 (3.9) | 0.31 (0.04) |
| 117 | 246 | 7.1 | 0.0* | 78.6 (10.3) | 0.09 (0.10) |
| 013** | 246 | 6.9 | 0.8 | 84.5 (3.8) | 0.10 (0.00) |
| 097 | 61.5 | 7.8 | 0.0* | 66.4 (0.0) | 0.07 (0.07) |
| 103 | 61.5 | 7.1 | 0.5 | 71.6 (10.4) | 0.01 (0.08) |
|
| |||||
| Median (Q1-Q3) | 7.0 (6.98-7.63) | 0.25 (0.0-0.5) | 76.7(72.4-83.0) | 0.10 (0.07-0.20) | |
Microarray analysis
Preliminary hierarchical clustering analysis initially revealed no significant differences in gene expression between mice treated with SIL for 3 days compared to 14 days (Fig. S3, moderated t-tests not shown) suggesting that changes in gene expression in response to SIL treatment that occurred by day 3 remained constant through day 14. When the SIL-treated mice (n=6) were compared as a group to the untreated mice (n=3), no genes were found to be significantly up- or down-regulated after correction for multiple testing using the false discovery rate. However, among the top 20 genes with the largest fold changes, most were down-regulated in SIL-treated mice (Table 2), suggesting an inhibitory trend. Also noteworthy, 30% of these top differentially expressed genes have been previously implicated in HCV infection in various ways (31-38). Although no genes were significantly differentially regulated, gene set enrichment analysis (http://www.broadinstitute.org/gsea/msigdb/annotate.jsp) of the top genes with the largest fold changes revealed significant over-representation of genes belonging to the “Genes involved in Chemokine receptor binding” signaling pathway (e.g., CXCL10, CCL20, and CXL6; P=5.3×10-4) (Table 2). Ingenuity Pathway Analysis (IPA) of linear models for microarray (limma) results suggested effects on immune-related pathways, including antigen presentation, granulocyte adhesion, and IL-17A signaling (Fig. 2). Consistent with previous findings (39, 40), IPA Upstream Regulator Analysis suggested that these results could be explained by inhibition of various inflammation-related cytokines and transcription regulators in SIL-treated mice, including TNFα (Fig. 3A), IFNγ, IL1A/B, IFNλ, and components of the NFκB signaling pathway (Fig. 3B, Table S3). Integrating predicted upstream regulators with projected downstream effects, the IPA Regulator Effects tool predicted that the gene expression profile of SIL-treated mice may result in inhibition of cellular immune responses and T lymphocyte chemotaxis in addition to suppression of myeloid and tumor cell movement and proliferation of connective tissue cells (Fig. 4).
Table 2. Differential gene expression.
No genes were significantly differentially expressed following correction for multiple testing. The top 20 genes with respect to fold change in SIL-treated versus control mice are shown.
| Gene ID | Gene | logFC | AveExpr | t | P | PFDR | B |
|---|---|---|---|---|---|---|---|
| 3484 | IGFBP1 | 2.25 | 8.52 | 5.40 | 0.0002 | 0.2181 | 0.58 |
| 6288 | SAA1 | -2.00 | 9.12 | -1.69 | 0.1187 | 0.7851 | -4.49 |
| 6289 | SAA2 | -1.88 | 8.76 | -1.59 | 0.1396 | 0.8080 | -4.62 |
| 3576 | IL8 | -1.75 | 6.86 | -4.13 | 0.0016 | 0.3859 | -1.00 |
| 4837 | NNMT | -1.74 | 7.20 | -3.27 | 0.0074 | 0.5378 | -2.23 |
| 9488 | PIGB | -1.73 | 7.23 | -4.64 | 0.0007 | 0.2725 | -0.33 |
| 100129193 | LOC100129193 | -1.71 | 6.97 | -1.37 | 0.1974 | 0.8352 | -4.88 |
| 3627 | CXCL10 | -1.53 | 10.97 | -3.19 | 0.0085 | 0.5378 | -2.34 |
| 57817 | HAMP | 1.52 | 8.19 | 1.31 | 0.2151 | 0.8493 | -4.94 |
| 56892 | C8orf4 | -1.45 | 6.13 | -4.15 | 0.0016 | 0.3859 | -0.97 |
| 1581 | CYP7A1 | 1.44 | 9.01 | 1.96 | 0.0755 | 0.7560 | -4.13 |
| 735 | C9 | -1.42 | 8.63 | -2.14 | 0.0558 | 0.7300 | -3.89 |
| 6696 | SPP1 | -1.38 | 8.45 | -3.00 | 0.0121 | 0.5600 | -2.63 |
| 963 | CD53 | -1.33 | 6.44 | -4.81 | 0.0005 | 0.2657 | -0.12 |
| 6277 | S100A6 | -1.30 | 7.84 | -5.54 | 0.0002 | 0.2181 | 0.74 |
| 100289597 | LOC100289597 | -1.29 | 5.84 | -5.07 | 0.0004 | 0.2181 | 0.20 |
| 100133941 | CD24L4 | -1.26 | 8.99 | -2.29 | 0.0426 | 0.7054 | -3.67 |
| 6364 | CCL20 | -1.25 | 8.06 | -2.47 | 0.0312 | 0.6762 | -3.41 |
| 1843 | DUSP1 | 1.22 | 8.12 | 1.91 | 0.0830 | 0.7650 | -4.21 |
| 6372 | CXCL6 | -1.22 | 6.22 | -2.18 | 0.0519 | 0.7212 | -3.83 |
logFC: log2 fold-change; AveExpr: The average log2 expression level; t: moderated t-statistics; P: uncorrected P-value for t-test; PFDR: P-value adjusted for multiple testing based on the false discovery rate; B: log-odds that the gene is differentially expressed.
Figure 2. Ingenuity Pathway Analysis (IPA) canonical pathway analysis.
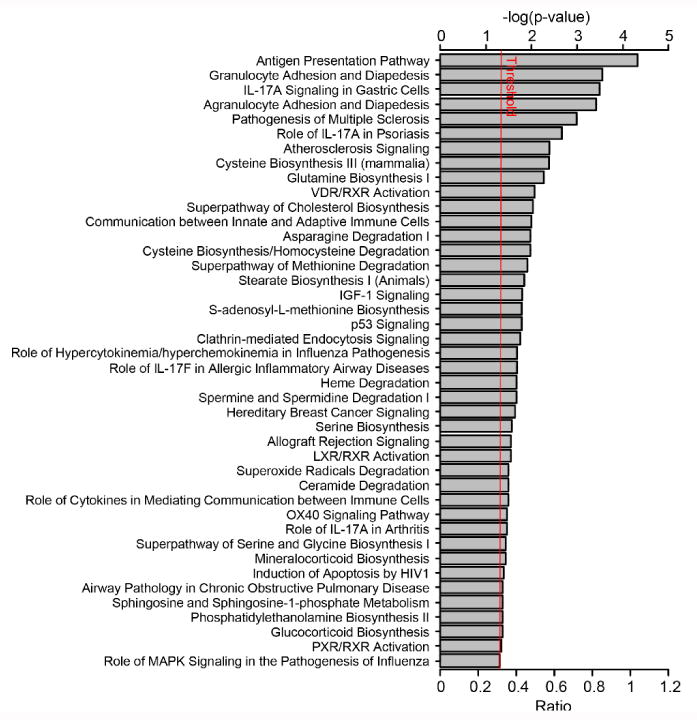
Linear Modeling of Microarray (limma) results were analyzed using IPA. P-values were calculated using the right-tailed Fisher exact test and reflect the likelihood that the association between SIL-related genes and genes in a given canonical pathway is due to random chance. Bar length represents the –log10 P-value; therefore, the longer the bar, the less likely the association is due to chance; i.e. the pattern of differentially expressed genes in SIL-treated versus control mice suggests involvement of the antigen presentation and granulocyte adhesion pathways. Bars crossing the red threshold line have P-values less than 0.05.
Figure 3. Predicted TNFα and NFκB –mediated regulatory effects downstream of SIL.
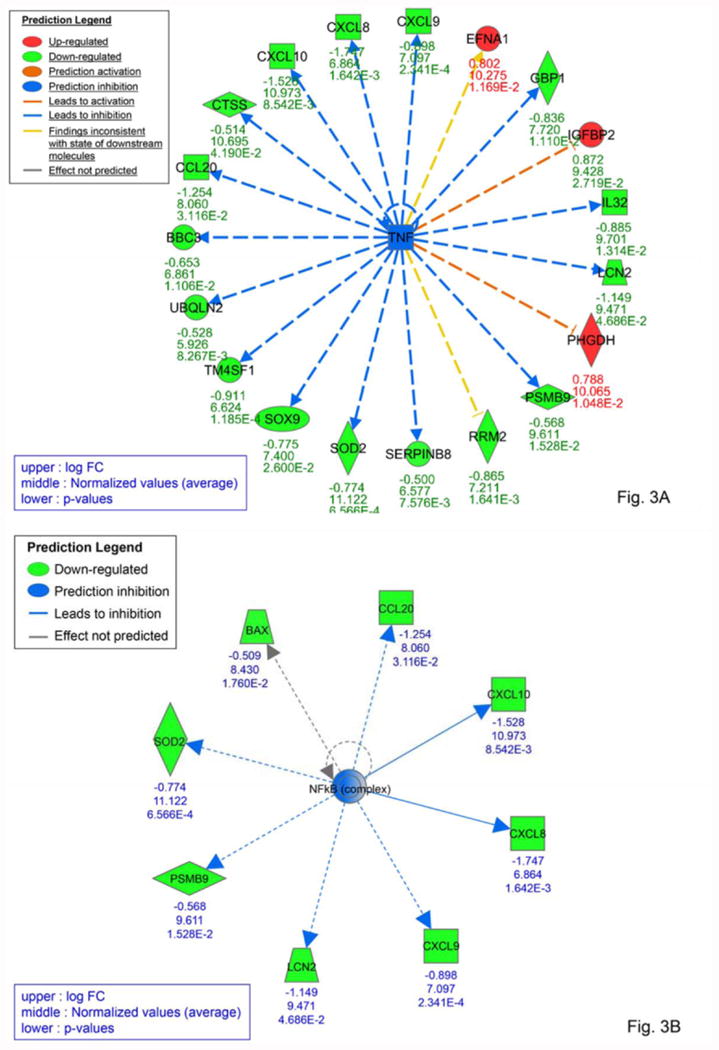
IPA software uses pathway information to identify potential upstream regulators that could explain the observed pattern of gene expression. The blue square in (A) and circle in (B) indicate that inhibition of the predicted upstream regulator TNFα (A) and NFκB (B) could directly or indirectly result in inhibition (blue arrows and green shapes) or activation (orange arrows and red shapes) of the target genes shown.
Figure 4. Predicted regulatory effects of gene expression profiles in response to SIL.
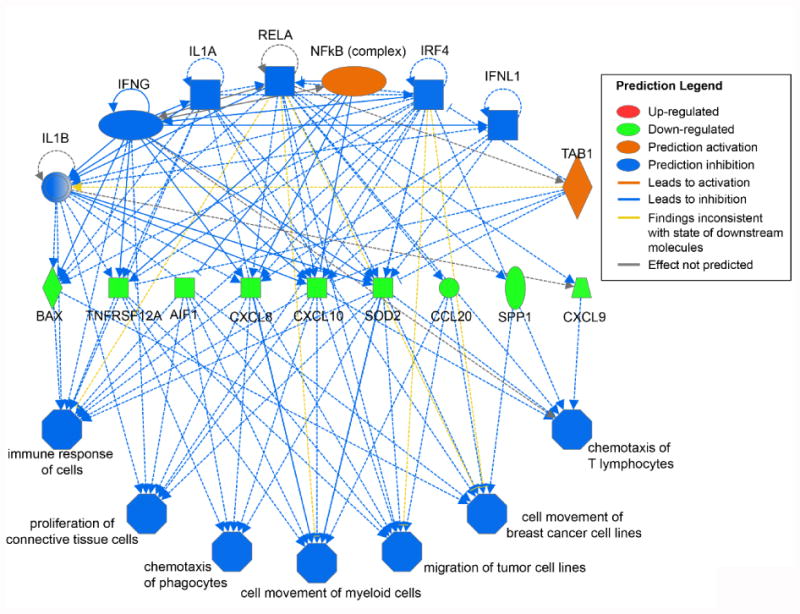
Differential gene expression patterns in SIL-treated versus control mice were used to infer perturbed upstream regulators and then predict the downstream effects on hepatocytes.
Discussion
Even with potent oral DAAs available for the treatment of HCV infection, intravenous SIL still may hold a niche as an efficacious IFN-free treatment option for chronically-infected patients who do not respond to or cannot be treated with DAA regimens including post-liver transplantation (10). Additionally, elucidating the enigmatic activities of SIL is potentially of interest for the treatment of other viruses such as HIV (41) and liver disorders, such as cirrhosis, cancer and liver damage by toxins (42, 43). The study of HCV treatment response in SIL-treated uPA-SCID-chimeric mice provides the opportunity to investigate the MOA of this drug in vivo in the absence of any secondary effects mediated by subsequent activation of the adaptive immune response.
Although the mouse numbers in this study are limited, the median estimate of SIL effectiveness in blocking viral production, ε=77%, has a high degree of confidence and indicates that the viral decline at d1 and d2 were similar among dosing groups. The estimates of HCV-infected human hepatocyte decline rates were higher in the two 469 mg/kg treated mice (median δ=0.27/day) than the mice in the other treatment groups (median δ=0.08/day). While the lower dosing groups' viral load reached a plateau, it is noteworthy that the first phase viral decline was similar among all dosing groups with the effect of increasing the dose being reflected by an increase in the second phase decline. The observed 2nd phase decline in mice treated with the highest SIL dosing might reflect a delayed effect of SIL on HCV RNA replication and/or on the stability of HCV replication complexes reminiscent of the proposed mechanism of action of high IFN dosing in vitro (44). That being said, the observed second phase loss rate (δ∼0.27/day) in the 469 mg/kg dosing group is much less than the loss rate (∼0.7/day) seen in SIL-treated patients (9, 11), which may reflect the importance of the adaptive immune effects of SIL in patients as these are missing in the uPA-SCID mouse model.
In the chimeric mouse model, serum hAlb is used as a surrogate measurement for human hepatocyte numbers. The observation that hAlb remained constant throughout therapy suggests that the 2nd phase viral decline is mediated by loss of intracellular HCV and not the death of infected cells. Another possibility, though less likely since there is no adaptive immune response, is that the hAlb levels remain constant due to the loss of infected cells being balanced by replication of non-infected human cells. In addition, due to the lack of SIL pharmacokinetic data in these chimeric mice, the effect of SIL pharmacokinetics and pharmacodynamics on the observed 2nd phase viral decline cannot be ruled out. Interestingly, while the intrahepatic HCV RNA data for this study is sparse, it is notable that the finding that HCV RNA decline in the serum and liver are correlated (r=0.66, P=0.01) is also consistent with the 2nd viral decline phase observed in serum being governed by an intrahepatic viral genome loss rather than death of HCV-infected hepatocytes.
Microarray analysis of SIL-treated versus non-treated control mice revealed no significant changes in gene expression after correction for multiple testing. The lack of significant changes may reflect the relatively small number of mice (3 controls and 6 SIL treated mice), or the subtle nature of the gene expression changes observed may reflect the fact that there is no adaptive immune response in these mice to amplify the more direct initial effects of the SIL treatment through secondary amplification effects of the innate immune response (e.g. NK cells). Regardless, the majority of the top most differentially expressed genes were down-regulated in SIL-treated mice, suggesting an inhibitory trend. Importantly, the results of pathway analysis suggest that SIL may exert anti-inflammatory effects through upstream inhibition of cytokine signaling as well as TNFα and NFκB-associated transcriptional regulation, which is consistent with previous studies demonstrating suppression of cell growth and metabolism in peripheral blood mononuclear cells (41) and inhibition of TNFα and NFκB-mediated pro-inflammatory gene expression in hepatic and other cell types (40, 45-47). While further experiments are needed to reveal the nature of the observed anti-inflammatory gene expression and whether the human hepatocytes were responding to the SIL directly and/or via signals from the mouse hepatic non-parenchymal cells, it is interesting that significant anti-HCV effects of SIL were observed in vivo in the absence of a secondary adaptive immune response and perhaps explains why SIL has been effective in HCV infected patients post-transplant (13, 14, 16).
Interestingly, quite a few of the genes that exhibited the largest change in expression in response to SIL (Table 2) have been previously implicated in HCV infection in various ways (31-38). Particularly notable, it has been reported that HCV infection up-regulates IL8 (33), NNMT (34), and SPP1 (i.e. osteopontin) (37) resulting in enhanced HCV replication and/or pathology, and all three of these genes were down-regulated in the mice treated with SIL. Also consistent with inhibition of HCV, the up regulation of HAMP (i.e. hepcidin) in SIL-treated mice would be expected to down regulate the HCV entry factor transferrin receptor 1 (35, 36, 48). While it is unclear if any of these gene expression changes were involved in the inhibition of HCV observed in the SIL treated mice, one could speculate that these perhaps more broadly hepatoprotective effects of SIL may also contribute to the antiviral effects observed.
In conclusion, the present study provides insights about HCV kinetics in blood and within human hepatocytes along with resulting gene expression changes in the human hepatocytes in mice treated with SIL. The viral kinetics indicate a biphasic viral kinetic pattern with continuous viral decline during the 2nd phase in the highest dosing group while gene expression analyses indicate that SIL treatment is associated with anti-inflammatory and anti-proliferative effects. The much slower 2nd phase viral decline observed in these SCID mice compared to in patients suggests that in addition to the reported direct antiviral effects of SIL (20, 22), perhaps in patients the adaptive immune response might also facilitate 2nd phase HCV clearance.
Supplementary Material
Acknowledgments
Financial support: Portions of this work were supported by PhoenixBio Co. Ltd., NIH grants P20-GM103452, R01-AI028433, R01-AI011095 and R01-AI078881 and done under the auspices of the U.S. Department of Energy under contract DE-AC52-06NA25396, USCB research grant award, and the UK Biotechnology and Biological Sciences Research Council - grant reference 1698:BB/L001330/1. SIL was provided by Rottapharm|Madaus Ltd.
Role of the Sponsor: None of the sponsors had any role in the design and conduct of the study; collection, management, analysis and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Footnotes
Conflict of Interest Disclosures: CT is an employee of PhoenixBio Co. Ltd. None of the authors has any financial interest or conflict of interest related to this research.
References
- 1.Polyak SJ, Ferenci P, Pawlotsky JM. Hepatoprotective and antiviral functions of silymarin components in hepatitis C virus infection. Hepatology. 2013;57(3):1262–71. doi: 10.1002/hep.26179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mengs U, Pohl RT, Mitchell T. Legalon(R) SIL: the antidote of choice in patients with acute hepatotoxicity from amatoxin poisoning. Curr Pharm Biotechnol. 2012;13(10):1964–70. doi: 10.2174/138920112802273353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.World Health Organization. Hepatitis C fact sheet no 164. 2013 http://who.int/mediacentre/factsheets/fs164/en/
- 4.Kwong JC, Ratnasingham S, Campitelli MA, Daneman N, Deeks SL, Manuel DG, et al. The impact of infection on population health: results of the Ontario burden of infectious diseases study. PloS One. 2012;7(9):e44103. doi: 10.1371/journal.pone.0044103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bourliere M, Bronowicki JP, de Ledinghen V, Hezode C, Zoulim F, Mathurin P, et al. Ledipasvir-sofosbuvir with or without ribavirin to treat patients with HCV genotype 1 infection and cirrhosis non-responsive to previous protease-inhibitor therapy: a randomised, double-blind, phase 2 trial (SIRIUS) Lancet Infect Dis. 2015;15(4):397–404. doi: 10.1016/S1473-3099(15)70050-2. [DOI] [PubMed] [Google Scholar]
- 6.Reddy KR, Bourliere M, Sulkowski M, Omata M, Zeuzem S, Feld JJ, et al. Ledipasvir and sofosbuvir in patients with genotype 1 hepatitis C virus infection and compensated cirrhosis: An integrated safety and efficacy analysis. Hepatology. 2015;62(1):79–86. doi: 10.1002/hep.27826. [DOI] [PubMed] [Google Scholar]
- 7.Sulkowski MS, Naggie S, Lalezari J, Fessel WJ, Mounzer K, Shuhart M, et al. Sofosbuvir and ribavirin for hepatitis C in patients with HIV coinfection. JAMA. 2014;312(4):353–61. doi: 10.1001/jama.2014.7734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mizokami M, Yokosuka O, Takehara T, Sakamoto N, Korenaga M, Mochizuki H, et al. Ledipasvir and sofosbuvir fixed-dose combination with and without ribavirin for 12 weeks in treatment-naive and previously treated Japanese patients with genotype 1 hepatitis C: an open-label, randomised, phase 3 trial. Lancet Infect Dis. 2015;15(6):645–53. doi: 10.1016/S1473-3099(15)70099-X. [DOI] [PubMed] [Google Scholar]
- 9.Canini L, DebRoy S, Mariño Z, Conway JM, Crespo G, Navasa M, et al. Severity of liver disease affects HCV kinetics in patients treated with intravenous silibinin monotherapy. Antivir Ther. 2015;20(2):149–55. doi: 10.3851/IMP2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marino Z, Crespo G, D'Amato M, Brambilla N, Giacovelli G, Rovati L, et al. Intravenous silibinin monotherapy shows significant antiviral activity in HCV-infected patients in the peri-transplantation period. Journal of hepatology. 2013;58(3):415–20. doi: 10.1016/j.jhep.2012.09.034. [DOI] [PubMed] [Google Scholar]
- 11.Guedj J, Dahari H, Pohl RT, Ferenci P, Perelson AS. Understanding silibinin's modes of action against HCV using viral kinetic modeling. J Hepatol. 2012;56(5):1019–24. doi: 10.1016/j.jhep.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferenci P, Scherzer TM, Kerschner H, Rutter K, Beinhardt S, Hofer H, et al. Silibinin is a potent antiviral agent in patients with chronic hepatitis C not responding to pegylated interferon/ribavirin therapy. Gastroenterology. 2008;135(5):1561–7. doi: 10.1053/j.gastro.2008.07.072. [DOI] [PubMed] [Google Scholar]
- 13.Socha L, Karpinska E, Jurczyk K, Laurans L, Bander D, Lachtara M, et al. Rescue therapy with intravenous silibinin in liver transplant recipients with recurrent HCV hepatitis - two case reports. Ann Transplant. 2014;19:161–4. doi: 10.12659/AOT.889779. [DOI] [PubMed] [Google Scholar]
- 14.Rendina M, D'Amato M, Castellaneta A, Castellaneta NM, Brambilla N, Giacovelli G, et al. Antiviral activity and safety profile of silibinin in HCV patients with advanced fibrosis after liver transplantation: a randomized clinical trial. Transpl Int. 2014;27(7):696–704. doi: 10.1111/tri.12324. [DOI] [PubMed] [Google Scholar]
- 15.Rutter K, Scherzer TM, Beinhardt S, Kerschner H, Stattermayer AF, Hofer H, et al. Intravenous silibinin as ‘rescue treatment’ for on-treatment non-responders to pegylated interferon/ribavirin combination therapy. Antivir Ther. 2011;16(8):1327–33. doi: 10.3851/IMP1942. [DOI] [PubMed] [Google Scholar]
- 16.Neumann UP, Biermer M, Eurich D, Neuhaus P, Berg T. Successful prevention of hepatitis C virus (HCV) liver graft reinfection by silibinin mono-therapy. J Hepatol. 2010;52(6):951–2. doi: 10.1016/j.jhep.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 17.Dahari H, Shteingart S, Gafanovich I, Cotler SJ, D'Amato M, Pohl RT, et al. Sustained virological response with intravenous silibinin: individualized IFN-free therapy via real-time modelling of HCV kinetics. Liver Int. 2015;35(2):289–94. doi: 10.1111/liv.12692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morishima C, Shuhart MC, Wang CC, Paschal DM, Apodaca MC, Liu Y, et al. Silymarin inhibits in vitro T-cell proliferation and cytokine production in hepatitis C virus infection. Gastroenterology. 2010;138(2):671–81. 81 e1–2. doi: 10.1053/j.gastro.2009.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghany MG, Kleiner DE, Alter H, Doo E, Khokar F, Promrat K, et al. Progression of fibrosis in chronic hepatitis C. Gastroenterology. 2003;124(1):97–104. doi: 10.1053/gast.2003.50018. [DOI] [PubMed] [Google Scholar]
- 20.Wagoner J, Negash A, Kane OJ, Martinez LE, Nahmias Y, Bourne N, et al. Multiple effects of silymarin on the hepatitis C virus lifecycle. Hepatology. 2010;51(6):1912–21. doi: 10.1002/hep.23587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Esser-Nobis K, Romero-Brey I, Ganten TM, Gouttenoire J, Harak C, Klein R, et al. Analysis of hepatitis C virus resistance to silibinin in vitro and in vivo points to a novel mechanism involving nonstructural protein 4B. Hepatology. 2013;57(3):953–63. doi: 10.1002/hep.26260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahmed–Belkacem A, Ahnou N, Barbotte L, Wychowski C, Pallier C, Brillet R, et al. Silibinin and Related Compounds Are Direct Inhibitors of Hepatitis C Virus RNA-Dependent RNA Polymerase. Gastroenterology. 2010;138(3):1112–22. doi: 10.1053/j.gastro.2009.11.053. [DOI] [PubMed] [Google Scholar]
- 23.Blaising J, Levy PL, Gondeau C, Phelip C, Varbanov M, Teissier E, et al. Silibinin inhibits hepatitis C virus entry into hepatocytes by hindering clathrin-dependent trafficking. Cell Microbiol. 2013;15(11):1866–82. doi: 10.1111/cmi.12155. [DOI] [PubMed] [Google Scholar]
- 24.Kamiya N, Iwao E, Hiraga N, Tsuge M, Imamura M, Takahashi S, et al. Practical evaluation of a mouse with chimeric human liver model for hepatitis C virus infection using an NS3-4A protease inhibitor. J Genl Virol. 2010;91(7):1668–77. doi: 10.1099/vir.0.019315-0. [DOI] [PubMed] [Google Scholar]
- 25.Ohara E, Hiraga N, Imamura M, Iwao E, Kamiya N, Yamada I, et al. Elimination of hepatitis C virus by short term NS3-4A and NS5B inhibitor combination therapy in human hepatocyte chimeric mice. J Hepatol. 2011;54(5):872–8. doi: 10.1016/j.jhep.2010.08.033. [DOI] [PubMed] [Google Scholar]
- 26.Shi N, Hiraga N, Imamura M, Hayes CN, Zhang Y, Kosaka K, et al. Combination therapies with NS5A, NS3 and NS5B inhibitors on different genotypes of hepatitis C virus in human hepatocyte chimeric mice. Gut. 2013;62(7):1055–61. doi: 10.1136/gutjnl-2012-302600. [DOI] [PubMed] [Google Scholar]
- 27.Tateno C, Yoshizane Y, Saito N, Kataoka M, Utoh R, Yamasaki C, et al. Near completely humanized liver in mice shows human-type metabolic responses to drugs. Am J Pathol. 2004;165(3):901–12. doi: 10.1016/S0002-9440(10)63352-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsuge M, Hiraga N, Takaishi H, Noguchi C, Oga H, Imamura M, et al. Infection of human hepatocyte chimeric mouse with genetically engineered hepatitis B virus. Hepatology. 2005;42(5):1046–54. doi: 10.1002/hep.20892. [DOI] [PubMed] [Google Scholar]
- 29.Neumann AU, Lam NP, Dahari H, Gretch DR, Wiley TE, Layden TJ, et al. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science. 1998;282(5386):103–7. doi: 10.1126/science.282.5386.103. [DOI] [PubMed] [Google Scholar]
- 30.Smyth GK. Limma: linear models for microarray data. In: Gentleman VC R, Dudoit S, Irizarry R, Huber W, editors. Bioinformatics and Computational Biology Solutions using R and Bioconductor. New York: Springer; 2005. pp. 397–420. [Google Scholar]
- 31.Cai Z, Cai L, Jiang J, Chang KS, van der Westhuyzen DR, Luo G. Human serum amyloid A protein inhibits hepatitis C virus entry into cells. J Virol. 2007;81(11):6128–33. doi: 10.1128/JVI.02627-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lavie M, Voisset C, Vu-Dac N, Zurawski V, Duverlie G, Wychowski C, et al. Serum amyloid A has antiviral activity against hepatitis C virus by inhibiting virus entry in a cell culture system. Hepatology. 2006;44(6):1626–34. doi: 10.1002/hep.21406. [DOI] [PubMed] [Google Scholar]
- 33.Balasubramanian A, Munshi N, Koziel MJ, Hu Z, Liang TJ, Groopman JE, et al. Structural proteins of Hepatitis C virus induce interleukin 8 production and apoptosis in human endothelial cells. J Gen Virol. 2005;86(Pt 12):3291–301. doi: 10.1099/vir.0.81056-0. [DOI] [PubMed] [Google Scholar]
- 34.Li K, Prow T, Lemon SM, Beard MR. Cellular response to conditional expression of hepatitis C virus core protein in Huh7 cultured human hepatoma cells. Hepatology. 2002;35(5):1237–46. doi: 10.1053/jhep.2002.32968. [DOI] [PubMed] [Google Scholar]
- 35.Du F, Qian C, Qian ZM, Wu XM, Xie H, Yung WH, et al. Hepcidin directly inhibits transferrin receptor 1 expression in astrocytes via a cyclic AMP-protein kinase A pathway. Glia. 2011;59(6):936–45. doi: 10.1002/glia.21166. [DOI] [PubMed] [Google Scholar]
- 36.Du F, Qian ZM, Gong Q, Zhu ZJ, Lu L, Ke Y. The iron regulatory hormone hepcidin inhibits expression of iron release as well as iron uptake proteins in J774 cells. J Nutr Biochem. 2012;23(12):1694–700. doi: 10.1016/j.jnutbio.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 37.Choi SS, Claridge LC, Jhaveri R, Swiderska-Syn M, Clark P, Suzuki A, et al. Osteopontin is up-regulated in chronic hepatitis C and is associated with cellular permissiveness for hepatitis C virus replication. Clin Sci (Lond) 2014;126(12):845–55. doi: 10.1042/CS20130473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Choi JE, Kwon JH, Kim JH, Hur W, Sung PS, Choi SW, et al. Suppression of dual specificity phosphatase I expression inhibits hepatitis C virus replication. PloS One. 2015;10(3):e0119172. doi: 10.1371/journal.pone.0119172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Agarwal R, Agarwal C, Ichikawa H, Singh RP, Aggarwal BB. Anticancer potential of silymarin: from bench to bed side. Anticancer Res. 2006;26(6B):4457–98. [PubMed] [Google Scholar]
- 40.Polyak SJ, Morishima C, Shuhart MC, Wang CC, Liu Y, Lee DY. Inhibition of T-cell inflammatory cytokines, hepatocyte NF-kappaB signaling, and HCV infection by standardized Silymarin. Gastroenterology. 2007;132(5):1925–36. doi: 10.1053/j.gastro.2007.02.038. [DOI] [PubMed] [Google Scholar]
- 41.McClure J, Margineantu DH, Sweet IR, Polyak SJ. Inhibition of HIV by Legalon-SIL is independent of its effect on cellular metabolism. Virology. 2014;449:96–103. doi: 10.1016/j.virol.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Agarwal C, Wadhwa R, Deep G, Biedermann D, Gazak R, Kren V, et al. Anti-cancer efficacy of silybin derivatives -- a structure-activity relationship. PloS One. 2013;8(3):e60074. doi: 10.1371/journal.pone.0060074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pradhan SC, Girish C. Hepatoprotective herbal drug, silymarin from experimental pharmacology to clinical medicine. Indian J Med Res. 2006;124(5):491–504. [PubMed] [Google Scholar]
- 44.Dahari H, Sainz B, Jr, Perelson AS, Uprichard SL. Modeling subgenomic hepatitis C virus RNA kinetics during treatment with alpha interferon. J Virol. 2009;83(13):6383–90. doi: 10.1128/JVI.02612-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yousefi M, Ghaffari SH, Soltani BM, Nafissi S, Momeny M, Zekri A, et al. Therapeutic efficacy of silibinin on human neuroblastoma cells: Akt and NF-kappaB expressions may play an important role in silibinin-induced response. Neurochem Res. 2012;37(9):2053–63. doi: 10.1007/s11064-012-0827-9. [DOI] [PubMed] [Google Scholar]
- 46.Salamone F, Galvano F, Cappello F, Mangiameli A, Barbagallo I, Li Volti G. Silibinin modulates lipid homeostasis and inhibits nuclear factor kappa B activation in experimental nonalcoholic steatohepatitis. Transl Res. 2012;159(6):477–86. doi: 10.1016/j.trsl.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 47.Lovelace ES, Wagoner J, MacDonald J, Bammler T, Bruckner J, Brownell J, et al. Silymarin Suppresses Cellular Inflammation By Inducing Reparative Stress Signaling. J Nat Prod. 2015;78(8):1990–2000. doi: 10.1021/acs.jnatprod.5b00288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martin DN, Uprichard SL. Identification of transferrin receptor 1 as a hepatitis C virus entry factor. Proc Natl Acad Sci U S A. 2013;110(26):10777–82. doi: 10.1073/pnas.1301764110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.