

Abstract
Purpose of review
The purpose of this review is to provide a succinct description of recent findings that advance our understanding of the fundamental renal process of ammonia metabolism and transport in conditions relevant to the clinician.
Recent findings
Recent studies advance our understanding of renal ammonia metabolism. Mechanisms through which chronic kidney disease and altered dietary protein intake alter ammonia excretion have been identified. Lithium, although it can acutely cause distal RTA, was shown with long-term use to increase urinary ammonia excretion, and this appeared to be mediated, at least in part, by increased Rhcg expression. Gene deletion studies showed that the ammonia recycling enzyme, glutamine synthetase, has a critical role in normal and acidosis-stimulated ammonia metabolism and that the proximal tubule basolateral bicarbonate transporter, NBCe1, is necessary for normal ammonia metabolism. Finally, our understanding of the molecular ammonia species, NH3 versus NH4+, transported by Rh glycoproteins continues to be advanced.
Summary
Fundamental studies have been recently published that advance our understanding of the regulation of ammonia metabolism in clinically important circumstances and our understanding of the mechanisms and regulation of proximal tubule ammonia generation and the mechanisms through which Rh glycoproteins contribute to ammonia secretion.
Keywords: Dietary protein, lithium, chronic kidney disease, Rh glycoprotein, NBCe1
Introduction
Normal acid–base balance is critical for normal health, and failure to achieve this leads to a wide variety of clinical disorders, including growth retardation, nausea and vomiting, electrolyte disturbances, cardiac arrhythmias, impaired cardiovascular catecholamine sensitivity, osteoporosis and osteomalacia, nephrolithiasis, skeletal muscle atrophy and accelerated progression of chronic kidney disease (CKD) [1;2]. Recent studies also show an important correlation between abnormal acid-base homeostasis and increased mortality [3].
The kidneys contribute to acid-base homeostasis by net acid excretion which is achieved by ammonia1 and titratable acid excretion and is decreased by bicarbonate excretion. Ammonia excretion is the greatest component of basal net acid excretion and changes in ammonia excretion are the predominant component of changes in net acid excretion in response to acid-base disturbances [1;4].
Overview of renal ammonia metabolism
Renal ammonia metabolism differs fundamentally from the excretion of most other urinary solutes. Most urinary solutes are delivered to the kidneys via the renal artery, and then are excreted in the urine through a combination of glomerular filtration and renal tubular transport. In contrast, less than 5% of urinary ammonia derives from arterial delivery. Instead, kidneys produce ammonia in a metabolic process that results in equimolar bicarbonate generation. Under basal conditions, ~50% of the ammonia that is produced is excreted in urine and 50% is added to the systemic circulation via renal veins. Changes in urinary ammonia excretion can result either from changes in net ammonia generation or from changes in renal epithelial cell ammonia transport that determines the proportion that is excreted in the urine versus that delivered to the systemic circulation.
CKD and ammonia metabolism
CKD is a common cause of abnormal acid-base homeostasis. In addition to its many other adverse effects, CKD is an important risk factor for development of metabolic acidosis [5], if not explicitly a cause of it. As Figure 1 shows, ~40% of individuals with advanced CKD develop metabolic acidosis. This metabolic acidosis primarily results from decreased ammonia excretion. Importantly, both animal and human studies show acidosis worsens the progression of CKD and that correcting the acidosis slows the progression [6–9]. Thus, a positive feedback cycle can develop, where CKD causes metabolic acidosis, which leads to worsening of the CKD, leading to worsened metabolic acidosis, etc.
Figure 1. Correlation of metabolic acidosis with severity of chronic kidney disease.
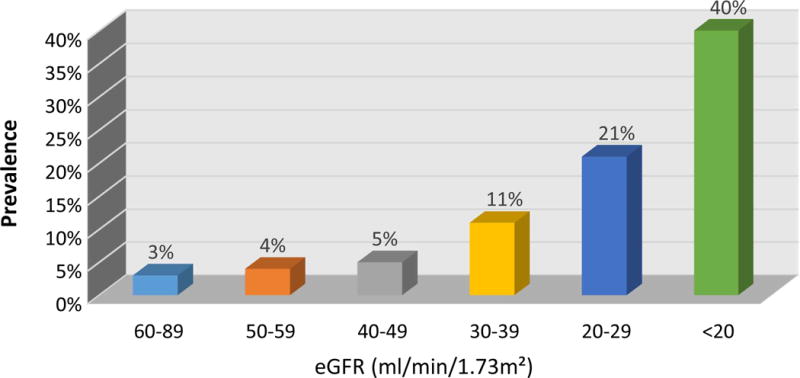
progressive decreases in renal function correlate with an increased risk of metabolic acidosis. Data from [5].
Altered ammonia metabolism likely contributes to the acidosis seen in patients with CKD. A recent study examined the effect of CKD on renal ammonia metabolism in a rat model and showed that the decreased ammonia excretion seen in advanced CKD was associated with impaired expression of multiple proteins involved in renal ammonia metabolism, including the glutamine transporter, SN1, the ammoniagenic enzymes, PDG and PEPCK, and the ammonia transporter, Rhcg [10]. Another, slightly older study using a less severe model of CKD, found that normal acid-base homeostasis in normal urinary ammonia excretion could be achieved through increased single nephron ammonia excretion [11], consistent with earlier studies [12], and that the increased single nephron ammonia excretion was associated with increased apical and basolateral polarization of Rhcg without changes in total expression [11].
Whether ammonia itself directly contributes to the progression of chronic kidney disease has been an intriguing question for many years. A classic study, one of the early studies to show that metabolic acidosis contributed to CKD progression, suggested that ammonia itself, possibly by activating complement, accelerated the progression of renal injury [6]. Although in vitro studies suggest that ammonia can activate complement directly, it is possible that reversible binding of interstitial ammonia by sulfatides [13] may limit unbound interstitial ammonia concentrations and thereby minimize this effect. Consistent with this conclusion is that a recent clinical study showed, after controlling for the degree of metabolic acidosis, that low urinary ammonia excretion, not high ammonia, correlated with an increased risk of worsening of renal function [14]. Thus, maintenance of adequate renal ammonia metabolism may be protective for patients with CKD.
Dietary protein and ammonia metabolism
Decreased dietary protein intake has multiple benefits for patients with chronic kidney disease. In addition, protein intake is a major determinant of endogenous acid production, and thus it is necessary that urinary ammonia excretion change in parallel with protein intake. A recent study has addressed the effect of dietary protein on ammonia metabolism in detail. Dietary protein restriction decreased urinary ammonia excretion by approximately 85% within two days [15]. Expression of PDG and PEPCK, key proteins involved in ammonia generation, decreased, whereas expression of the ammonia recycling protein, glutamine synthetase, increased [15]. Importantly, none of the individual changes could account quantitatively for the degree of decreased ammonia excretion; it was the coordinated effect that appeared to be necessary. Also, decreased glutamine synthetase expression could have an important role in nitrogen conservation in response to dietary protein restriction. Thus, dietary protein restriction induces coordinated expression consistent with decreasing urinary ammonia excretion.
Another, slightly older, study examined the converse condition, the response to high dietary protein intake. This study showed that it was metabolism to generate fixed acids, not the nitrogen content, that was responsible for the increased ammonia excretion. Secondly, it demonstrated increased expression of two key proteins involved in ammonia generation, glutamine transporter, SN1, and PDG [16]. Rhcg mRNA expression increased, and Rhcg gene deletion showed that Rhcg expression was necessary for the initial increase in ammonia excretion [16]. Rhcg’s role in the response to high protein diet contrasted with a lack of necessity for Rhcg in the response to dietary protein restriction [15].
These two studies, when taken together, indicate that changes in dietary protein induce coordinated changes in many aspects of renal ammonia metabolism. Whether the minor differences in the responses involved in adaptation to low versus high protein diet reflect fundamental differences or minor variations associated with slightly different models and mice cannot be differentiated at this time.
An unexpected finding in the studies of dietary protein restriction related to the ammonia transporter family member, Rhbg. Dietary protein restriction, even though it decreased ammonia excretion, increased Rhbg expression in intercalated cells in the inner stripe of the outer medullary collecting duct [15]. In general, Rhbg expression parallels changes in ammonia excretion, and thus the observation of increased Rhbg expression in a state of decreased ammonia excretion was not predicted. Increased Rhbg expression, however, did not appear necessary for the increase in ammonia excretion, as collecting duct-specific Rhbg deletion did not alter the response to dietary protein restriction [15]. This observation suggests Rhbg has an as yet unidentified biological role in addition to its contribution to transepithelial ammonia transport.
Lithium and ammonia excretion
Chronic lithium administration is another common clinical condition for which new information regarding ammonia metabolism is available. Acute lithium administration is well known to increase urine pH. However, urine pH does not necessarily correlate with net acid or ammonia excretion. A recent study [17], examining people treated chronically with lithium, showed increased basal rates of urinary ammonia excretion and an intact, if not increased, ability to increase ammonia excretion following acid-loading (Figure 2). Identical findings were observed in an animal model, and this increased ammonia excretion correlated with increased expression of the ammonia transporter, Rhcg [17]. Ammonia excretion requires parallel H+ and NH3 secretion in the collecting duct, and chronic lithium administration is known to increase both the number of acid-secreting intercalated cells and the expression of H+-ATPase [18;19]. Thus, not only does chronic lithium therapy not routinely induce distal RTA, it is actually associated with enhanced renal net acid excretion. These findings also emphasize the importance of not assuming that changes in urine pH predict changes in net acid excretion.
Figure 2. Effect of lithium therapy on urinary ammonia excretion in response to an acute acid load in humans.
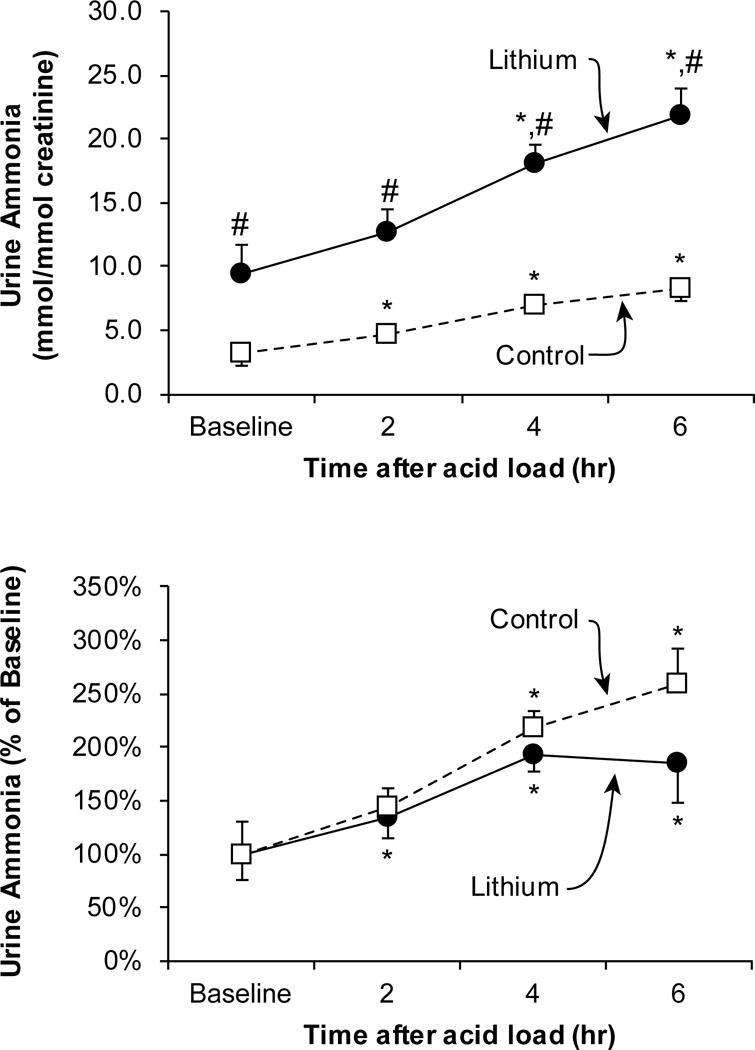
Top panel shows urinary ammonia excretion, expressed as mmol per mmol creatinine, at baseline, and following an acute acid load. Baseline urinary ammonia excretion was significantly higher in lithium-treated subjects than in control subjects. An acute acid load, induced by oral ammonium chloride loading, resulted in significant increases in urinary ammonia excretion in both groups at each time point. However, at each time point, urinary ammonia excretion was significantly greater in lithium-treated subjects than in control subjects. Bottom panel shows changes in urinary ammonia excretion relative to adjusted for baseline urinary ammonia excretion rates. An acute acid load resulted in significant increases in ammonia excretion relative to baseline excretion in both groups. *P < 0.05 versus baseline; #P < 0.05 versus control at same time point. Figure from [17], and used with permission.
Metabolic processes involved in “net” ammonia generation
The kidney is a net producer of ammonia, and this results from metabolism of amino acids, predominantly glutamine, that results in equimolar NH4+ and HCO3− generation. There also is a complementary mechanism through which kidneys “recycle” NH4+ and “regenerate” glutamine. This process involves the enzyme, glutamine synthetase, which catalyzes the reaction: NH4+ + glutamate + ATP → Glutamine + H+ + ADP + Pi. Several previous studies have shown that glutamine synthetase expression is negatively correlated with ammonia excretion, i.e., conditions that increase ammonia excretion decrease glutamine synthetase expression, and vice versa [15;20–24].
A recent study substantially advances our understanding of glutamine synthetase’s role in renal ammonia excretion. Because previous studies have shown that glutamine synthetase is expressed in cells involved both in ammonia production, i.e., proximal tubule cells, and cells involved in transepithelial ammonia transport, i.e., distal convoluted tubule and intercalated cells, and that glutamine synthetase expression can be differentially regulated in these different cell populations [20], the authors of this recent study used cell-specific glutamine synthetase deletion to determine the specific role of proximal tubule glutamine synthetase in renal ammonia metabolism. Proximal tubule-specific deletion increased basal ammonia excretion (Figure 3), consistent with decreased ammonia recycling leading to increased net ammonia generation and excretion [25]. There also was an impaired ability to respond to an acid load [25], indicating the ability to alter glutamine synthetase-mediated ammonia recycling is critical to increasing net ammoniagenesis. These findings demonstrate the presence of a “yin-yang” mechanism regulating “net” ammoniagenesis, one involving ammonia generation and another involving ammonia recycling. These two mechanisms likely act in concert in order to optimize renal ammonia generation.
Figure 3. Effect of proximal tubule specific glutamine synthetase deletion on urinary ammonia excretion.
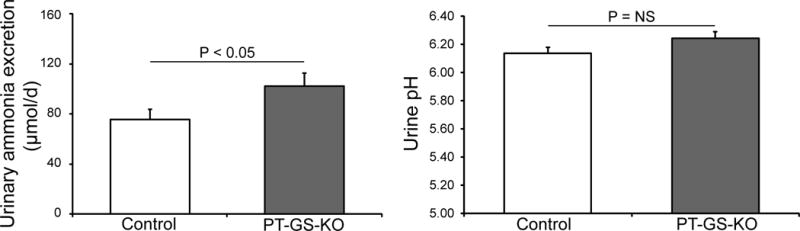
Left panel shows that proximal tubule-specific glutamine synthetase deletion increased basal urinary ammonia excretion significantly. Right panel shows that increased ammonia excretion occurred despite no change in urinary pH. Figure from [25] and reprinted with permission.
Regulation of ammonia generation
The recent finding that expression of the proximal tubule basolateral bicarbonate transporter, NBCe1, is necessary for normal renal ammonia metabolism is an important advance in understanding the mechanisms regulating ammonia generation. Proximal tubule NBCe1 mediates coupled co-transport of Na+ with three HCO3− equivalents, is the primary mechanism of proximal tubule basolateral bicarbonate exit and genetic abnormalities in it are the major genetic cause of proximal RTA [26]. NBCe1 deletion causes metabolic acidosis [27;28], which should increase ammonia excretion, yet recent studies show that NBCe1 deletion decreased ammonia excretion (Figure 4) [28]. This occurred despite intact urine acidification and intact expression of the collecting duct ammonia transport proteins, Rhbg and Rhcg [28]. Decreased ammonia excretion appeared to result from abnormal expression of multiple enzymes involved in ammonia metabolism. There was decreased expression of the major proximal tubule enzymes involved in ammonia generation, PDG, GDH and PEPCK, and increased expression of the ammonia recycling protein, glutamine synthetase [28;29], responses opposite that expected from metabolic acidosis. Thus, NBCe1 expression is necessary for normal proximal tubule ammonia metabolism.
Figure 4. Effect of NBCe1 deletion on urinary ammonia and pH.
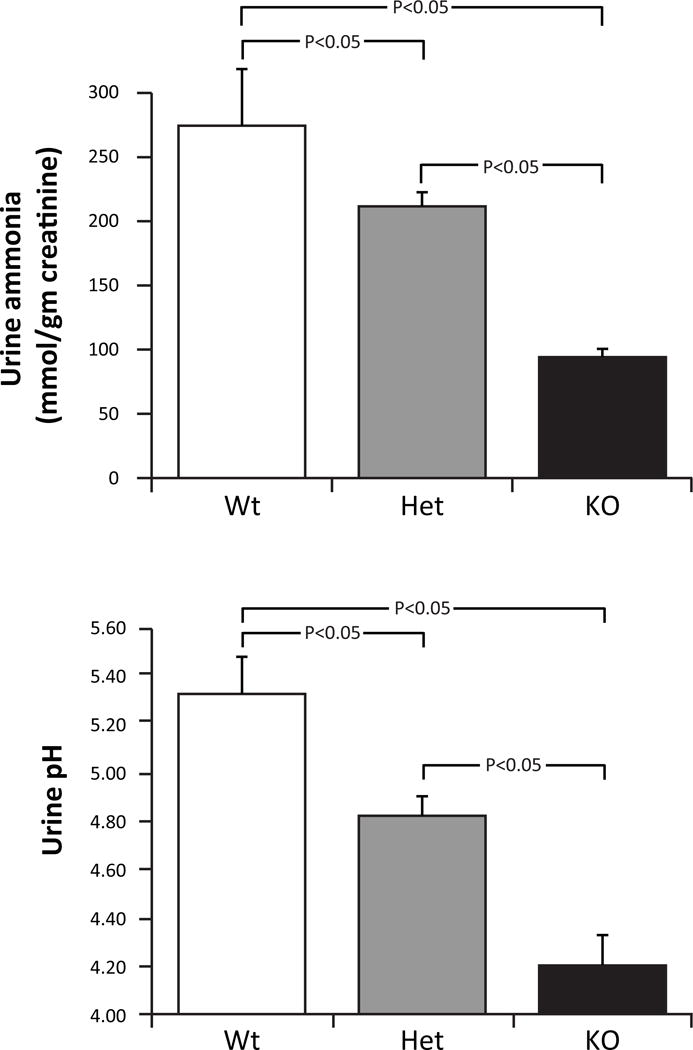
Top: effects on urine ammonia. Despite the metabolic acidosis associated with NBCe1 gene deletion, ammonia excretion decreased with heterozygous gene deletion and was suppressed significantly further with homozygous deletion. Bottom: effects on urine pH. Urine pH was significantly more acidic (lower) in mice with heterozygous NBCe1 deletion than in wild-type mice and was decreased significantly further by homozygous NBCe1 deletion. Figure originally published in [28] and reprinted with permission.
In addition to being necessary for normal ammonia metabolism, NBCe1 may also be important for organic anion transport. NBCe1 deletion caused abnormal regulation of expression of the primary proximal tubule citrate transporter, NaDC-1 (Figure 5) [29]. This did not alter net citrate excretion, possibly because of increased activity of a previously described acid-stimulated, calcium-sensitive citrate transport activity [30]. Excretion of another important organic anion, 2-oxoglutarate (also known as α-ketoglutarate), was also altered by NBCe1 deletion. Specifically, NBCe1 deletion dramatically increased 2-oxoglutarate excretion [29]. This change in 2-oxoglutarate excretion could not ascribed to the accompanying metabolic acidosis, as experimental metabolic acidosis in wild-type mice actually decreased 2-oxoglutarate excretion [29], as previously described by others [31;32]. Thus, NBCe1 is necessary for normal proximal tubule bicarbonate reabsorption, and also for two other major roles of the proximal tubule in acid-base homeostasis, namely ammonia generation and organic anion transport/metabolism.
Figure 5. Effect of NBCe1 deletion on NaDC1 expression.
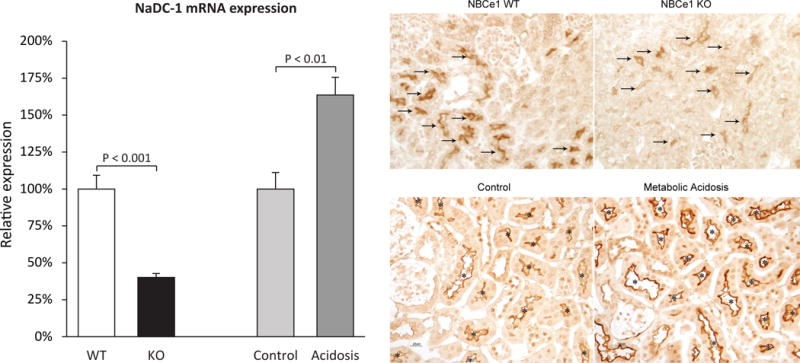
NaDC1 is an apical Na+-coupled citrate transporter present in the proximal tubule that is thought to the primary mechanism regulating urinary citrate excretion. NBCe1 deletion decreased NaDC1 mRNA (left panels) and immunolabel expression (right panels). This effect was unrelated to the associated metabolic acidosis, as experimental metabolic acidosis in the absence of NBCe1 gene deletion increased NaDC1 mRNA and immunolabel expression. Figures 2 (left panel) and 3 (right panel) from [29] were combined and are used with permission.
Advances in ammonia transport
Collecting duct ammonia secretion involves parallel NH3 and H+ transport, with little-to-no NH4+ permeability [33;34]. H+ secretion involves H+-ATPase and H+-K+-ATPase. The Rhesus (Rh) glycoproteins, Rhbg and Rhcg, in combination with basolateral Na+-K+-ATPase (in the IMCD), are involved directly in ammonia transport [4;35]. Figure 6 shows a current model of collecting duct ammonia secretion.
Figure 6. Model of collecting duct ammonia secretion.
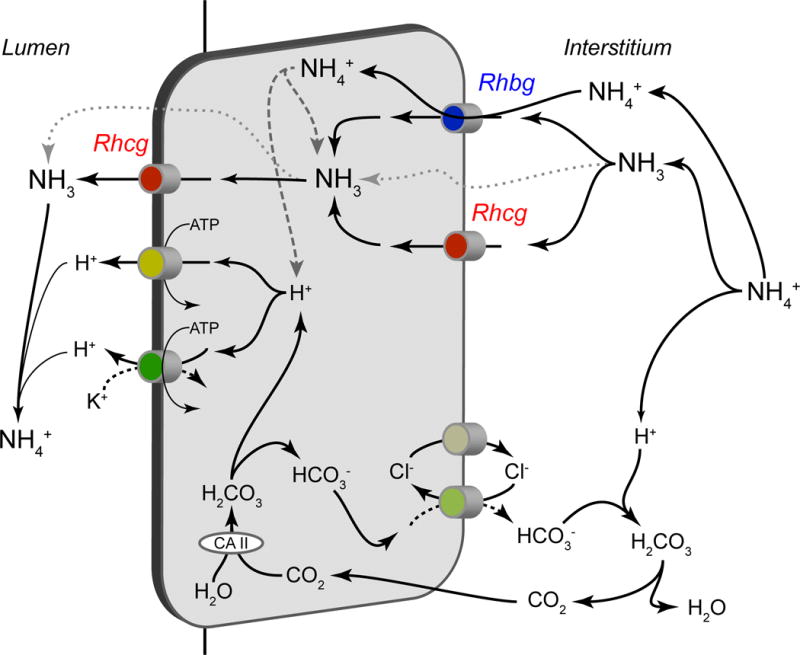
Ammonia uptake across the basolateral plasma membrane primarily involves transporter-mediated uptake by either Rhbg or Rhcg, with a component of diffusive NH3 absorption. Cytosolic NH3 is transported across the apical plasma membrane by a combination of Rhcg and diffusive transport. Not shown is that in the IMCD, but not the CCD, basolateral Na+-K+-ATPase also contributes to basolateral plasma membrane NH4+ uptake. Cytosolic H+ is generated by a carbonic anhydrase-II mediated mechanism, and is secreted across the apical plasma membrane via H+-ATPase and H+-K+-ATPase. Luminal H+ titrates luminal NH3, forming NH4+ and maintaining a low luminal NH3 concentration necessary for NH3 secretion. Figure from [35] with permission.
Molecular aspects of Rh glycoprotein ammonia transport
The molecular forms of ammonia, NH3 versus NH4+, that Rh glycoproteins transport continues to be actively investigated. One important finding from recent studies is the identification that results using the ammonia analogue, methylammonia, may not accurately predict results for ammonia [36]. A second important finding is that Rhbg, as does the erythroid-specific Rh glycoprotein, Rhag, primarily transports ammonia in the molecular form of NH4+, but that there may be a small component of NH3 transport [36]. Rhcg, in contrast, primarily appears to transport NH3, with no detectable NH4+ transport capability [36].
This difference in ammonia species transported by Rhbg and Rhcg is important in their role in collecting duct ammonia secretion. Rhbg is present in collecting duct basolateral plasma membrane, and its electrogenic transport of NH4+ results in a greater electrochemical gradient for uptake than would occur for NH3 transport. Rhcg’s transport of NH3 rather than NH4+ is critical for Rhcg to facilitate apical ammonia secretion. Specifically, the low luminal pH and the high pKà of the NH3 + H+ ↔ NH4+ buffer reaction keeps luminal [NH3] low, facilitating apical NH3 secretion. Electrogenic NH4+ secretion via Rhcg would be unlikely to occur because the high luminal [NH4+] in combination with intracellular electronegativity would cause the electrochemical gradient for NH4+ movement to be from lumen to cytoplasm, i.e., reabsorption, not secretion. Thus, the differential specificities of Rhbg and Rhcg for NH3 and NH4+ contributes to collecting duct ammonia secretion.
Conclusion
Ammonia metabolism is critical to acid-base homeostasis into the maintenance of normal health, both under basal conditions and in response to a variety of clinical circumstance. Recent studies have substantially advanced our understanding of ammonia metabolism in a variety of clinically important conditions. Additional studies have advanced understanding of the mechanism of ammonia metabolism by showing critical roles of glutamine synthetase and NBCe1, and by carefully determining the molecular ammonia species transported by Rh glycoproteins.
Key points.
In a model of chronic kidney disease, multiple components of renal ammonia metabolism, the primary component net acid excretion, were inhibited, thereby contributing to the development of metabolic acidosis.
Changes in dietary protein intake, both increases and decreases, induce a coordinated response of multiple components of renal ammonia metabolism that result in maintenance of nitrogen balance and acid-base homeostasis.
Chronic lithium intake results in a substantial stimulation of renal ammonia excretion and metabolism.
Cell-specific gene deletion shows that ammonia recycling via glutamine synthetase expression in the proximal tubule is necessary for basal and acidosis-stimulated renal ammonia metabolism.
The proximal tubule basolateral bicarbonate transporter, NBCe1, has a critical role in renal ammonia, and organic anion, metabolism.
The ammonia transporter family member, Rhbg, appears to transport both NH3 and NH4+, whereas the homolog, Rhcg only appears to transport NH3.
Acknowledgments
We thank Gina Cowsert for secretarial assistance.
Funding support and sponsorship
Funds from the NIH (R01–DK045788 and R01–DK107798) supported the generation of this review. This work, however, does not reflect official policy of either the NIH or the Department of Veterans Affairs.
Footnotes
Ammonia exists in two molecular forms, NH3 and NH4+, that are in equilibrium with each other. We use the term “ammonia” to refer to the combination of these two molecular forms. When referring to a specific molecular form, we state specifically either “NH3” or “NH4+.”
Conflicts of interest
The authors have no financial, consultant, institutional or other relationship that might lead to bias or a conflict of interest in regards to information presented in this review.
Literature Cited
- 1*.Weiner ID, Mitch WE, Sands JM. Urea and ammonia metabolism and the control of renal nitrogen excretion. Clin J Am Soc Nephrol. 2015;10:1444–1458. doi: 10.2215/CJN.10311013. Overview of renal urea and ammonia metabolism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mitch WE. Metabolic and clinical consequences of metabolic acidosis. J Nephrol. 2006;19:S70–S75. [PubMed] [Google Scholar]
- 3**.Raphael KL, Murphy RA, Shlipak MG, et al. Bicarbonate Concentration, Acid-Base Status, and Mortality in the Health, Aging, and Body Composition Study. Clin J Am Soc Nephrol. 2016;11:308–316. doi: 10.2215/CJN.06200615. Patients with abnormal serum bicarbonate and acid-base homeostasis have increased mortality. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weiner ID, Verlander JW. Renal Ammonia Metabolism and Transport. Comprehensive Physiology. 2013;3:201–220. doi: 10.1002/cphy.c120010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moranne O, Froissart M, Rossert J, et al. Timing of Onset of CKD-Related Metabolic Complications. J Am Soc Nephrol. 2009;20:164–171. doi: 10.1681/ASN.2008020159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nath KA, Hostetter MK, Hostetter TH. Pathophysiology of chronic tubulo-interstitial disease in rats. Interactions of dietary acid load, ammonia, and complement component C3. J Clin Invest. 1985;76:667–675. doi: 10.1172/JCI112020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goraya N, Simoni J, Jo CH, Wesson DE. A Comparison of Treating Metabolic Acidosis in CKD Stage 4 Hypertensive Kidney Disease with Fruits and Vegetables or Sodium Bicarbonate. Clin J Am Soc Nephrol. 2013;8:371–381. doi: 10.2215/CJN.02430312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kraut JA. Effect of Metabolic Acidosis on Progression of Chronic Kidney Disease. Am J Physiol Renal. 2011;300:F828–F829. doi: 10.1152/ajprenal.00074.2011. [DOI] [PubMed] [Google Scholar]
- 9.de Brito-Ashurst I, Varagunam M, Raftery MJ, Yaqoob MM. Bicarbonate Supplementation Slows Progression of CKD and Improves Nutritional Status. J Am Soc Nephrol. 2009;20:2075–2084. doi: 10.1681/ASN.2008111205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10**.Burki R, Mohebbi N, Bettoni C, et al. Impaired expression of key molecules of ammoniagenesis underlies renal acidosis in a rat model of chronic kidney disease. Nephrol Dial Transplant. 2014;30:770–781. doi: 10.1093/ndt/gfu384. Excellent study showing that CKD can impair renal ammonia excretion, not just because of decreased nephron number, but also because of impaired expression of multiple proteins involved in renal ammonia generation and transport. [DOI] [PubMed] [Google Scholar]
- 11.Kim HY, Baylis C, Verlander JW, et al. Effect of reduced renal mass on renal ammonia transporter family, Rh C glycoprotein and Rh B glycoprotein, expression. Am J Physiol Renal. 2007;293:F1238–F1247. doi: 10.1152/ajprenal.00151.2007. [DOI] [PubMed] [Google Scholar]
- 12.Buerkert J, Martin D, Trigg D, Simon E. Effect of reduced renal mass on ammonium handling and net acid formation by the superficial and juxtamedullary nephron of the rat. Evidence for impaired reentrapment rather than decreased production of ammonium in the acidosis of uremia. J Clin Invest. 1983;71:1661–1675. doi: 10.1172/JCI110921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stettner P, Bourgeois S, Marsching C, et al. Sulfatides are required for renal adaptation to chronic metabolic acidosis. Proc Natl Acad Sci U S A. 2013;110:9998–10003. doi: 10.1073/pnas.1217775110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14**.Vallet M, Metzger M, Haymann JP, et al. Urinary ammonia and long-term outcomes in chronic kidney disease. Kidney Int. 2015;88:137–145. doi: 10.1038/ki.2015.52. Patients with CKD, after adjustment for severity of CKD and acidosis, who have low urinary ammonia excretion are at increased risk of progressive nephropathy, suggesting that renal ammonia metabolism may be protective, not detrimental, in CKD. [DOI] [PubMed] [Google Scholar]
- 15*.Lee HW, Osis G, Handlogten ME, et al. Effect of dietary protein restriction on renal ammonia metabolism. American Journal of Physiology - Renal Physiology. 2015;308:F1463–F1473. doi: 10.1152/ajprenal.00077.2015. Decreased protein intake results in coordinated chagnes in multiple aspects of renal ammonia metabolism that decrease ammonia excretion and contribute to renal ammonia nitrogen conservation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bounoure L, Ruffoni D, Muller R, et al. The Role of the Renal Ammonia Transporter Rhcg in Metabolic Responses to Dietary Protein. J Am Soc Nephrol. 2014;25:2040–2052. doi: 10.1681/ASN.2013050466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17*.Weiner ID, Leader JP, Bedford JJ, Verlander JW, Ellis G, Vos F, de Jong S, Walker RJ. Effects of chronic lithium administration on renal acid excretion in humans and rats. Physiological Reports. 2014 Dec 1; doi: 10.14814/phy2.12242. Chronic lithium administration, both in people and in experimental animal model (rat), increase urinary ammonia excretion, and this increase is associated with, and likely partially mediated by, increased ammonia transporter family member, Rhcg, expression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Christensen BM, Marples D, Kim YH, et al. Changes in cellular composition of kidney collecting duct cells in rats with lithium-induced NDI. Am J Physiol Cell. 2004;286:C952–C964. doi: 10.1152/ajpcell.00266.2003. [DOI] [PubMed] [Google Scholar]
- 19.Kim YH, Kwon TH, Christensen BM, et al. Altered expression of renal acid-base transporters in rats with lithium-induced NDI. Am J Physiol Renal. 2003;285:F1244–F1257. doi: 10.1152/ajprenal.00176.2003. [DOI] [PubMed] [Google Scholar]
- 20.Verlander JW, Chu D, Lee HW, et al. Expression of glutamine synthetase in the mouse kidney: localization in multiple epithelial cell types and differential regulation by hypokalemia. Am J Physiol Renal. 2013;305:F701–F713. doi: 10.1152/ajprenal.00030.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee HW, Verlander JW, Handlogten ME, et al. Effect of collecting duct-specific deletion of both Rh B Glycoprotein (Rhbg) and Rh C Glycoprotein (Rhcg) on renal response to metabolic acidosis. Am J Physiol Renal. 2014;306:F389–F400. doi: 10.1152/ajprenal.00176.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee HW, Verlander JW, Bishop JM, et al. Collecting duct-specific Rh C Glycoprotein deletion alters basal and acidosis-stimulated renal ammonia excretion. Am J Physiol Renal. 2009;296:F1364–F1375. doi: 10.1152/ajprenal.90667.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xue Y, Liao SF, Son KW, et al. Metabolic acidosis in sheep alters expression of renal and skeletal muscle amino acid enzymes and transporters. J Anim Sci. 2010;88:707–717. doi: 10.2527/jas.2009-2101. [DOI] [PubMed] [Google Scholar]
- 24.Conjard A, Komaty O, Delage H, et al. Inhibition of glutamine synthetase in the mouse kidney: a novel mechanism of adaptation to metabolic acidosis. J Biol Chem. 2003;278:38159–38166. doi: 10.1074/jbc.M302885200. [DOI] [PubMed] [Google Scholar]
- 25*.Lee HW, Osis G, Handlogten ME, Lamers WH, Chaudhry FA, Verlander JW, Weiner ID. Proximal tubule-specific glutamine synthetase deletion alters basal and acidosis-stimulated ammonia metabolism. American Journal of Physiology - Renal Physiology. 2016 Mar 23; doi: 10.1152/ajprenal.00547.2015. Shows that proximal tubule glutamine synthetase expression is necessary for normal basal and acidosis-stimulated renal ammonia metabolism and excretion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kurtz I, Zhu Q. Structure, function, and regulation of the SLC4 NBCe1 transporter and its role in causing proximal renal tubular acidosis. Curr Opin Nephrol Hypertens. 2013;22:572–583. doi: 10.1097/MNH.0b013e328363ff43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gawenis LR, Bradford EM, Prasad V, et al. Colonic Anion Secretory Defects and Metabolic Acidosis in Mice Lacking the NBC1 Cotransporter. J Biol Chem. 2007;282:9042–9052. doi: 10.1074/jbc.M607041200. [DOI] [PubMed] [Google Scholar]
- 28*.Handlogten ME, Osis G, Lee HW, et al. NBCe1 expression is required for normal renal ammonia metabolism. Am J Physiol Renal. 2015;309:F658–F666. doi: 10.1152/ajprenal.00219.2015. NBCe1, the primary basolateral bicarbonate in the proximal tubule, which has a central role in filtered bicarbonate reabsorption, also has a critical role in ammonia metabolism. Deletion of NBCe1, despite causing a severe metabolic acidosis, due to the accompanying proximal RTA, substantially decreases urinary ammonia excretion. This appears to occur as a result of a generalized disturbance of mutliple components of proximal tubule ammonia generation and recycling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29*.Osis G, Handlogten ME, Lee H-W, et al. Effect of NBCe1 deletion on renal citrate and 2-oxoglutarate handling. Physiological Reports. 2016;4:e12778. doi: 10.14814/phy2.12778. NBCe1 deletion, in addition to causing abnormal proximal tubule ammonia metabolism, also disrupts normal NaDC1 expression and normal 2-oxoglutarate metabolism. Thus, NBCe1 is necessary for multiple proximal tubule roles in acid-base homeostasis, filtered bicarbonate reabsorption, ammonia metabolism and organic anion metabolism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hering-Smith KS, Gambala CT, Hamm LL. Citrate and succinate transport in proximal tubule cells. Am J Physiol Renal. 2000;278:F492–F498. doi: 10.1152/ajprenal.2000.278.3.F492. [DOI] [PubMed] [Google Scholar]
- 31.Tokonami N, Morla L, Centeno G, et al. α-Ketoglutarate regulates acid-base balance through an intrarenal paracrine mechanism. J Clin Invest. 2013;123:3166–3171. doi: 10.1172/JCI67562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheema-Dhadli S, Lin SH, Halperin ML. Mechanisms used to dispose of progressively increasing alkali load in rats. Am J Physiol Renal. 2002;282:F1049–F1055. doi: 10.1152/ajprenal.00006.2001. [DOI] [PubMed] [Google Scholar]
- 33.DuBose TD, Good DW, Hamm LL, Wall SM. Ammonium transport in the kidney: new physiological concepts and their clinical implications. J Am Soc Nephrol. 1991;1:1193–1203. doi: 10.1681/ASN.V1111193. [DOI] [PubMed] [Google Scholar]
- 34.Knepper MA. NH4+ transport in the kidney. Kidney Int. 1991;40:S95–S102. [PubMed] [Google Scholar]
- 35.Weiner ID, Verlander JW. Ammonia transport in the kidney by Rhesus glycoproteins. Am J Physiol Renal. 2014;306:F1107–F1120. doi: 10.1152/ajprenal.00013.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36**.Caner T, Abdulnour-Nakhoul S, Brown K, et al. Mechanisms of Ammonia and Ammonium Transport by Rhesus Associated Glycoproteins. American Journal of Physiology - Cell Physiology. 2015;309:C747–C758. doi: 10.1152/ajpcell.00085.2015. Elegant examination of molecular species of ammonia (NH3 and NH4+) that are transported by the three mammalian Rhesus glycoproteins, Rhag, Rhbg and Rhcg. [DOI] [PMC free article] [PubMed] [Google Scholar]