

Abstract
Purpose of Review
Renal potassium (K) secretion plays a key role in maintaining K homeostasis. The classic mechanism of renal K secretion is focused on the connecting tubule (CNT) and cortical collecting duct (CCD) in which K is uptaken by basolateral Na-K-ATPase and it is secreted into lumen by apical ROMK (Kir1.1) and Ca2+ –activated big conductance K channel (BK). Recently, genetic studies and animal models have indicated that inwardly rectifying K channel 4.1 (Kir4.1 or Kcnj10) in distal convoluted tubule (DCT) may play a role in the regulation of K secretion in the aldosterone-sensitive distal nephron (ASDN) by targeting NaCl cotransporter (NCC). This review summarizes recent progresses regarding the role of Kir4.1 in the regulation of NCC and K secretion.
Recent Findings
Kir4.1 is expressed in the basolateral membrane of the DCT and plays a predominant role in contributing to the basolateral K conductance and in participating in the generation of negative membrane potential. Kir4.1 is also the substrate of src-family tyrosine kinase (SFK) and the stimulation of SFK activates Kir4.1 activity in the DCT. The genetic deletion or functional inhibition of Kir4.1 depolarizes the membrane of the DCT, inhibits ste20-proline-alanine rich kinase (SPAK) and suppresses NCC activity. Moreover, the down-regulation of Kir4.1 increases ENaC expression in the collecting duct and urinary K excretion. Finally, the mice with low Kir4.1 activity in the DCT are hypomagnesemia and hypokalemia.
Summary
Recent progress in exploring the regulation and the function of Kir4.1 in the DCT strongly indicates that Kir4.1plays an important role in initiating the regulation of renal K secretion by targeting NCC and it may serves as a K sensor in the kidney.
Keywords: Kir.5.1, ROMK, ENaC, NCC, With-No-lysine kinase, Ste20-proline-alanine rich kinase
Introduction
Hyperkalemia, a potentially fatal disorder, occurs commonly in the setting of chronic kidney disease and heart failure. Its incidence appears to have increased because the most effective cardio- and reno-protective agents that block the renin/angiotensin/aldosterone system (RAAS), all impair renal K disposition. To prevent hyperkalemia induced by using RAAS inhibitors has led to the development of new agents to treat hyperkalemia [1]. Although these agents appear to be effective, they do not address the fundamental cause of the hyperkalemia, which is inappropriate kidney K retention. The aldosterone-sensitive distal nephron (ASDN) including the late distal convoluted tubule (DCT2), connecting tubule (CNT) and cortical collecting duct (CCD) is responsible for K secretion [2;3]. The classic mechanism for the K secretion is that K ions are secreted to the lumen of the CNT and CCD via ROMK (Kir1.1) and Ca2+-activated BK channels along a favorable electrochemical gradient created by Na absorption via ENaC [3–8]. Recent paradigm-shifting discoveries concerning the control of systemic K balance have identified a previously unrecognized system that maintains K homeostasis normally. Surprisingly, the key player in this system is the thiazide-sensitive NCC of the DCT [9–12]. The role of NCC in regulating renal K secretion and K homeostasis is also convincingly established by human genetic and clinical studies demonstrating that an abnormal NCC activity is responsible for causing hyperkalemia or hypokalemia. For instance, pseudohypoaldosteronism type II (PHAII) or familial hyperkalemic hypertension (FHHt), is caused by high activity of NCC [13–15] whereas hypokalemia in patients with Gitelman syndrome is due to the loss-function mutations of NCC [16]. Although numerous factors have been shown to regulate NCC activity [17–19], we previously demonstrated that the depletion of Kir.4.1 activity in the DCT inhibited the expression of NCC[20**]. Loss-of-function mutations of Kcnj10 cause EAST/SeSAME syndrome in humans (seizures, sensorineural deafness, ataxia, mental retardation and electrolyte imbalance) [21–24]. The renal phenotype of the disease is reminiscent to Gitelman syndrome including hypomagnesemia, hypokalemia and metabolic alkalosis, suggesting that the disruption of Kir4.1 mainly impairs transport in the DCT [25;26]. Thus, the aim of the current review is an attempt to provide an overview regarding the role of Kir4.1 in regulating renal Na and K transport.
Expression of Kir4.1 along the nephron segments
Fig. 1A is a nephron scheme illustrating the expression of Kir4.1 along the nephron in the mouse kidney and the corresponding native tubule is shown in Fig. 1B. Kir4.1 is expressed in the basolateral membrane of the cortical TAL (cTAL), DCT, CNT and CCD. Fig. 1C is a scheme showing the expression of NKCC2, NCC, ENaC and ROMK in the apical membrane of the cTAL, DCT and CNT/CCD, respectively. However, experiments performed in post-neonatal 9 day (p9) mice have showed that the expression of Kir4.1 in the cTAL is mainly limited in the late part of cTAL rather than in the whole length of the cTAL of the mouse kidney [27*]. Kir4.1 expression was also detected in human TAL, however, loss-of-function mutations of Kir4.1 in humans did not have phenotype of defective membrane transport in the TAL, suggesting that Kir4.1 function in the TAL may not be indispensable [21–23]. The possibility that Kir4.1 function in the cTAL could be compensated is also supported by patch-clamp experiments performed in p9 neonatal Kcnj10−/− mice demonstrating that the disruption of Kir4.1 stimulates the activity of Na+ and Cl−-activated 80- to 150-pS K+ channel (Kca4.1 or slo2.2) in the cTAL [27;28]. The upregulation of Na-activated 80–150 pS K channel in the TAL may be induced by high vasopressin level in p9 neonatal Kir4.1 knockout mice as evidenced by the finding that AQP2 expression was upregulated [29].
Fig. 1.
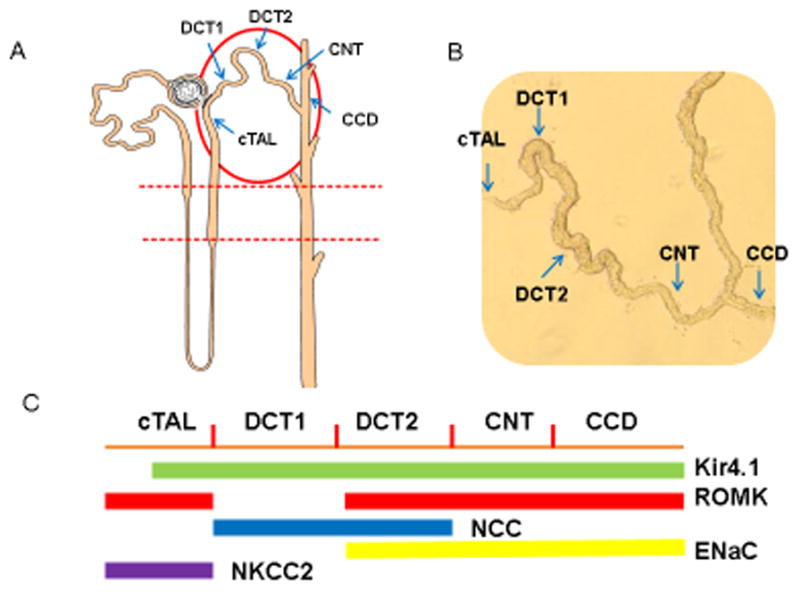
(A) A nephron scheme illustrating the expression of Kir4.1 in nephron segments circled by a red oval. (B) An image of isolated nephron segments which corresponds to the portion indicated by a red oval in Fig. 1A. The kir4.1 is expressed in the basolateral membrane from cortical thick ascending limb (cTAL) to the cortical collecting duct (CCD). (C) A scheme illustrating that NKCC2, NCC, ENaC and ROMK are expressed in the apical membrane of tubules from cTAL to CCD where Kir4.1 is expressed.
Kir.4.1 is also expressed in the basolateral membrane of the DCT along the whole length [22]. The DCT reabsorbs 5–9% of filtered Na load and it is functionally divided into the early part (DCT1) and the late portion (DCT2) [30–32]. While thiazide-sensitive NCC is expressed in the apical membrane of both DCT1 and DCT2, ROMK and ENaC activity are only detected in the apical membrane of DCT2 [33;34]. In the DCT1, Na and Cl enter the cells across the apical membrane through the NCC and Na is then pumped out of the cell through the basolateral Na-K-ATPase while Cl exits the cell along its electrochemical gradient by basolateral Cl channels (ClC-kb) or KCl cotransporter [35;35–37]. In the DCT2, Na enters the cell across the apical membrane not only through NCC but also by ENaC [38;39].
Kir4.1 interacts with Kir5.1 to form an inwardly-rectifying 40 pS K channel in the basolateral membrane of DCT1 and DCT2. Moreover, the 40 pS K channel is the only K channel type detected in the basolateral membrane of the DCT. Our previous experiments performed in p9 neonatal WT and Kcnj10−/− mice have demonstrated that Kir4.1 plays a dominant role in determining the basolateral K conductance in the DCT1 because the disruption of Kir4.1 almost completely eliminates the basolateral K conductance [20**]. The notion that Kir4.1 is a major type of K channel and it determines the membrane potential in the DCT is also supported by our recent unpublished observations made from kidney-specific Kir4.1 knockout mice (Wang’s observation). We have observed that Kir4.1/Kir5.1 heterotetramer is also the only K channel type detected in the basolateral membrane of DCT1 and DCT2 of adult mice.
In addition to cTAL and DCT, immunostaining and the patch-clamp experiments have detected Kir4.1 expression and activity in the basolateral membrane of the CNT and CCD [40*]. Like in the DCT, Kir4.1 interacts with Kir5.1 to form a 40–45 pS inwardly-rectifying K channels in the CNT and CCD [41;42]. However, experiments performed in p9 c57/bl6 mice have showed that the positive staining of Kir4.1 was limited in the top portion of the renal cortex but was almost absent in the low portion of the renal cortex. Moreover, K channels other than Kir4.1/5.1 heterotetramer were also detected in the basolateral membrane of the CNT and CCD of both adult and p9 neonatal mice [40*]. Therefore, unlike in the DCT, Kir4.1 participates only partially in generating the membrane potential in the CNT and CCD [40]. This notion is suggested by experiments performed in p9 neonatal Kir4.1 knockout mice in which the K reversal potential (an index of the cell membrane potential) in the CNT/CCD was only modestly depolarized in the knockout mice in comparison to those of WT littermates.
Regulation of Kir4.1
Kir4.1 is inhibited by protein kinase C (PKC) and stimulated by src-family protein tyrosine kinase (SFK) which phosphorylates Kir4.1 at tyrosine residues 8 and 9 of the N-terminus [43;44]. PKC has been shown to mediate the inhibitory effect of dopamine on the basolateral Kir4.1–Kir5.1 heterotetramer in the CCD [42]. On the other hand, the SFK-induced tyrosine phosphorylation of Kir4.1 stimulates the basolateral K conductance in the DCT [44]. Moreover, caveolin-1 plays a role in mediating the stimulatory effect of SFK on Kir4.1 [45**]. Caveolin-1 is highly expressed in the basolateral membrane of the DCT, CNT and CCD and the disruption of caveolin-1 inhibits the Kir4.1/5.1 activity in the mouse DCT. The basolateral Kir4.1–Kir5.1 channel activity was inhibited by acidic pH [41] and Kir5.1 may play a role in the regulation of pH sensitivity of Kir4.1 [46;47]. This notion is supported by the finding that the pH sensitivity of Kir4.1 is blunted in Kcnj16−/− mice [48]. Kir4.1 was immunoprecipitated from rat renal tissue extracts with Ca2+-sensing receptor (CaSR) and the expression of CaSR decreased the surface expression of Kir4.1 in HEK293 cells, suggesting the possibility that CaSR may regulate the activity of the basolateral 40 pS K channel [49;50].
Kir4.1 regulates NCC expression
Our recent experiments have demonstrated that Kir4.1 activity in the DCT determines the expression of NCC [20**;45**]. The role of NCC in the regulation of K excretion and K homeostasis has been well recognized by several independent studies [9–11;51**]. It has been demonstrated that an increase in dietary K intake inhibits the expression and activity of NCC and increases ENaC [9]. This should inhibit Na transport in the DCT thereby increasing Na delivery to the distal nephron segments such as DCT2 and CNT. Since high K intake stimulates ENaC activity [9], this should enhance K secretion in the CNT and CCD without the stimulation of overall Na absorption. On the other hand, a decrease in dietary K intake has been shown to increase the expression and activity of NCC thereby decreasing Na delivery to the distal nephron segments, leading to suppressing K secretion [9]. Thus, NCC activity plays a key role in controlling Na delivery to the DCT2, CNT and CCD and in the regulation of K secretion in the ASDN. It is well documented the NCC activity is regulated by with-no-lysine kinase (WNK), ste20-proline-alanine rich kinase (SPAK) and oxidative-sensitive responsive kinase (OSR) [18;52–55]. WNK phosphorylates and activates SPAK or OSR which in turn stimulates NCC activity by phosphorylation [54;56]. Gain-of-function mutations of WNK1 and WNK4 are responsible for increasing NCC activity in the DCT thereby causing familial hyperkalemic hypertension [57;58]. Thus, WNK-SPAK-mediated NCC phosphorylation plays a key role in the regulation of K excretion and K homeostasis. Recently, an elegant experiment by Terker et al has convincingly demonstrated that SPAK-induced NCC phosphorylation is closely controlled by plasma K level such as hyperkalemia inhibits while hypokalemia stimulates NCC phosphorylation [51**].
Although the role of plasma K level in the regulation of NCC activity is well established, it is not clear how NCC could sense the change in dietary K intake. Because Kir4.1 is the only type K channel in the basolateral membrane of the DCT, we hypothesize that Kir4.1 is actually a mediator between NCC and plasma K. This possibility is strongly suggested by the finding that the deletion or the inhibition of Kir4.1 decreases NCC expression [20**;45**]. Fig. 2 illustrates the mechanism by which the changes in Kir4.1 activity regulate NCC activity. The inhibition of the basolateral K conductance depolarizes the membrane and decreases Cl exit across the basolateral membrane of the DCT thereby increasing the intracellular Cl concentrations (Fig. 2A). Because WNK is a Cl-sensitive kinase such that a high intracellular Cl (Cli) concentration inhibits WNK activity [59*;60*], the inhibition of WNK leads to suppression of SPAK activity thereby inhibiting the NCC expression. On the other hand, the activation of Kir4.1 should decrease Cli and it activates WNK-SPAK/OSR pathway which in turn stimulates NCC activity by phosphorylation (Fig. 2B). It has been shown that SPAK-induced phosphorylation not only activates NCC but also increases NCC expression by inhibiting degradation [18;52;54;61]. The activity of Kir4.1 plays a role in the regulation of NCC activity has also been demonstrated by Terker et al and they have demonstrated that the change in the intracellular Cl concentration induced by membrane voltage is responsible for the inhibition of NCC activity in DCT cells expressing loss-function of Kcnj10 mutants [62**]. Thus, it is conceivable that the activity of the basolateral Kir4.1 activity in the DCT controls the NCC activity through a Cl-sensitive WNK pathway. Because the activity of NCC determines the Na transport in the DCT and controls the Na delivery to the distal nephron including DCT2, CNT and CCD, a change in Kir4.1 activity is expected to affect K secretion in the distal nephron segments. This also explains why hypokalemia is an essential feature of the tubulopathy in SeSAME syndrome [22].
Fig. 2.
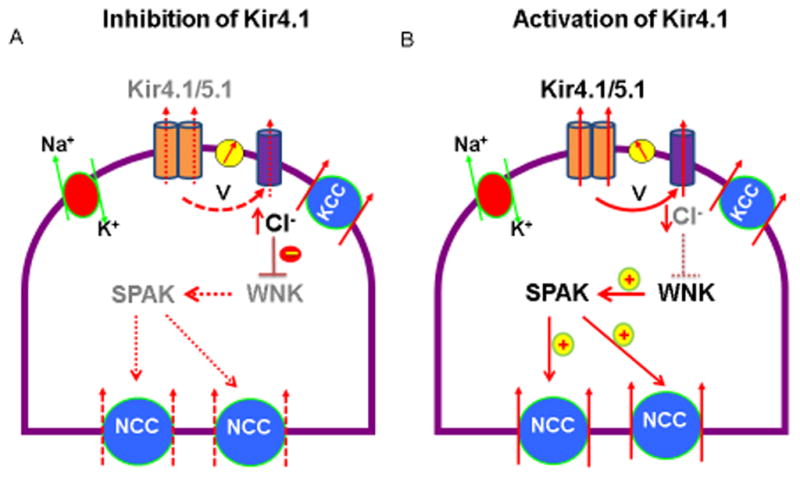
A scheme illustrating the mechanism by which the inhibition of Kir4.1 decreases (A) or the stimulation of Kir4.1 increases activity of NCC in the DCT (B). Dotted and solid lines represent a diminished and an enhanced function, respectively. Gray font means an inhibition or a decrease. Abbreviation: V, cell voltage; WNK, with-no-lysine kinase, SPAK, ste20-proline-alanine rich kinase.
Inhibition of Kir4.1 stimulates ENaC activity
Experiments performed in global Kir4.1 knockout mice have shown that the depletion of Kir4.1 stimulates the expression of ENaC-β, γ subunits and cleaved ENaC-α in the collecting duct [40*]. The upregulation of ENaC expression may explain the fact why patients with EAST/SeSAME syndrome have a modest phenotype of salt wasting despite of the inhibition of Na transport in the DCT [23]. We suspect that increased expression of ENaC-β, γ subunits and cleaved ENaC-α should be a compensation action as consequence of the down-regulation of NCC. It is possible that the inhibition of NCC is expected to cause a volume depletion which should increase the aldosterone and vasopressin level thereby stimulating ENaC expression. Moreover, we reasoned that a high vasopressin level may be mainly responsible for the stimulation of ENaC-β and γ expression in p9 neonatal Kcnj10−/− mice because vasopressin has been shown to stimulate the expression of ENaC-β subunit [63]. Furthermore, our previous studies showed that the disruption of Kir4.1 increased AQP2 expression, suggesting Kcnj10−/− mice have a high vasopressin level [29].
Inhibition of Kir4.1 impairs magnesium absorption
Hypomagnesemia is one phenotype of tubulopathy in SeSAME syndrome, suggesting the role of Kir4.1 in the regulation of magnesium absorption in the DCT [22]. Although the mechanism by which the inhibition of Kir4.1 causes hypomagnesemia is not completely understood, a decrease in the membrane potential in the DCT should partially contribute to inhibiting magnesium absorption in the DCT because transcellular magnesium transport process is electrogenic. Thus, a decrease in the basolateral K conductance in the DCT should diminish the driving force for the entry of magnesium ions across the apical membrane thereby inhibiting their absorption in the DCT. This notion is supported by the experiments performed in caveolin-1 knockout mice which have a low basolateral Kir4.1 activity and less negative membrane potential in the DCT in comparison to WT mice [45**]. Consequently, the caveolin-1 knockout mice are associated with increased magnesium excretion and have hypomagnesemia. Because intracellular magnesium ions have a strong inhibitory effect on ROMK outward current, a decrease in intracellular magnesium ion concentrations should increase ROMK outward currents and cause K wasting [64]. Thus, in addition to the regulation of NCC, Kir4.1 activity may affect renal K secretion through the modulation of magnesium absorption in the DCT.
Conclusion
The emerged evidence suggests that NCC plays a central role in the regulation of renal K secretion in response to hypokalemia or hyperkalemia and that Kir4.1 in the DCT mediates the effect of dietary K intake on NCC activity. Since Kir4.1 determines the basolateral K conductance and the membrane potential in the DCT, an alteration of Kir4.1 activity should affect WNK-SPAK activity through an intracellular Cl-sensitive mechanism thereby modulating NCC activity. Thus, Kir4.1 in the DCT may be a plasma K sensor and the regulation of Kir4.1 initiates the regulation of K secretion in the ASDN.
Key points.
Kir4.1 is a main contributor to the basolateral K conductance in the DCT.
The inhibition of Kir4.1 suppresses the expression of SPAK and NCC.
The down-regulation of Kir4.1 causes hypokalemia and hypomagnesimia.
Acknowledgments
The author would like to thank Drs. Jacques Teulon and Marc Paulais for their assistance with the study.
Financial support and sponsorship
The work is supported by NIH grant DK54983 and HL34100.
Footnotes
Conflicts of Interest
The author has no conflicts of interest
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
*of special interest
**of outstanding interest
- 1.Pitt B, Bakris GL. New Potassium Binders for the Treatment of Hyperkalemia: Current Data and Opportunities for the Future. Hypertension. 2015;66:731–738. doi: 10.1161/HYPERTENSIONAHA.115.04889. [DOI] [PubMed] [Google Scholar]
- 2.Hebert SC, Desir G, Giebisch G, Wang W. Molecular diversity and regulation of renal potassium channels. Physiol Rev. 2005;85:319–371. doi: 10.1152/physrev.00051.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Giebisch G, Hebert SC, Wang WH. New aspects of renal potassium transport. Pflugers Arch. 2003;446:289–297. doi: 10.1007/s00424-003-1029-8. [DOI] [PubMed] [Google Scholar]
- 4.Satlin LM. Developmental regulation of expression of renal potassium secretory channels. Curr Opin Nephrol Hypertens. 2004;13:445–450. doi: 10.1097/01.mnh.0000133979.17311.21. [DOI] [PubMed] [Google Scholar]
- 5.Pluznick JL, Sansom SC. BK channels in the kidney: role in K+ secretion and localization of molecular components. American Journal of Physiology - Renal Physiology. 2006;291:F517–F529. doi: 10.1152/ajprenal.00118.2006. [DOI] [PubMed] [Google Scholar]
- 6.Wang WH. View of K+ secretion through the apical K channel of cortical collecting duct. Kidney Internat. 1995;48:1024–1030. doi: 10.1038/ki.1995.385. [DOI] [PubMed] [Google Scholar]
- 7.Palmer LG, Frindt G. Aldosterone and potassium secretion by the cortical collecting duct. Kidney Int. 2000;57:1324–1328. doi: 10.1046/j.1523-1755.2000.00970.x. [DOI] [PubMed] [Google Scholar]
- 8.Ho K, Nichols CG, Lederer WJ, Lytton J, Vassilev PM, Kanazirska MV, Hebert SC. Cloning and expression of an inwardly rectifying ATP-regulated potassium channel. Nature. 1993;362:31–38. doi: 10.1038/362031a0. [DOI] [PubMed] [Google Scholar]
- 9.Frindt G, Palmer LG. Effects of dietary K on cell-surface expression of renal ion channels and transporters. American Journal of Physiology - Renal Physiology. 2010;299:F890–F897. doi: 10.1152/ajprenal.00323.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sorensen MV, Grossmann S, Roesinger M, Gresko N, Todkar AP, Barmettler G, Ziegler U, Odermatt A, Loffing-Cueni D, Loffing J. Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney Int. 2013;83:811–824. doi: 10.1038/ki.2013.14. [DOI] [PubMed] [Google Scholar]
- 11.van der Lubbe N, Moes AD, Rosenbaek LL, Schoep S, Meima ME, Danser AHJ, Fenton RA, Zietse R, Hoorn EJ. K+-induced natriuresis is preserved during Na+ depletion and accompanied by inhibition of the Na+-Cl cotransporter. American Journal of Physiology - Renal Physiology. 2013;305:F1177–F1188. doi: 10.1152/ajprenal.00201.2013. [DOI] [PubMed] [Google Scholar]
- 12.Sandberg MB, Maunsbach AB, McDonough AA. Redistribution of distal tubule Na+-Cl− cotransporter (NCC) in response to a high-salt diet. American Journal of Physiology - Renal Physiology. 2006;291:F503–F508. doi: 10.1152/ajprenal.00482.2005. [DOI] [PubMed] [Google Scholar]
- 13.Lalioti MD, Zhang J, Volkman HM, Kahle KT, Hoffmann KE, Toka HR, Nelson-Williams C, Ellison DH, Flavell R, Booth CJ, Lu Y, Geller DS, Lifton RP. Wnk4 controls blood pressure and potassium homeostasis via regulation of mass and activity of the distal convoluted tubule. Nat Genet. 2006;38:1124–1132. doi: 10.1038/ng1877. [DOI] [PubMed] [Google Scholar]
- 14.Take C, Ikeda K, Kurasawa T, Kurokawa K. Increased Chloride Reabsorption as an Inherited Renal Tubular Defect in Familial Type II Pseudohypoaldosteronism. N Engl J Med. 1991;324:472–476. doi: 10.1056/NEJM199102143240707. [DOI] [PubMed] [Google Scholar]
- 15.Shibata S, Zhang J, Puthumana J, Stone KL, Lifton RP. Kelch-like 3 and Cullin 3 regulate electrolyte homeostasis via ubiquitination and degradation of WNK4. Proceedings of the National Academy of Sciences. 2013;110:7838–7843. doi: 10.1073/pnas.1304592110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, Molina AM, Vaara I, Iwata F, Cushner HM, Koolen M, Gainza FJ, Gitleman HJ, Lifton RP. Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nature Genetics. 1996;12:24–30. doi: 10.1038/ng0196-24. [DOI] [PubMed] [Google Scholar]
- 17.Boyden LM, Choi M, Choate KA, Nelson-Williams CJ, Farhi A, Toka HR, Tikhonova IR, Bjornson R, Mane SM, Colussi G, Lebel M, Gordon RD, Semmekrot BA, Poujol A, Valimaki MJ, De Ferrari ME, Sanjad SA, Gutkin M, Karet FE, Tucci JR, Stockigt JR, Keppler-Noreuil KM, Porter CC, Anand SK, Whiteford ML, Davis ID, Dewar SB, Bettinelli A, Fadrowski JJ, Belsha CW, Hunley TE, Nelson RD, Trachtman H, Cole TRP, Pinsk M, Bockenhauer D, Shenoy M, Vaidyanathan P, Foreman JW, Rasoulpour M, Thameem F, Al-Shahrouri HZ, Radhakrishnan J, Gharavi AG, Goilav B, Lifton RP. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature. 2012;482:98–102. doi: 10.1038/nature10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCormick JA, Mutig K, Nelson JH, Saritas T, Hoorn EJ, Yang C-L, Rogers S, Curry J, Delpire E, Bachmann S, Ellison DH. A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metabolism. 2011;14:352–364. doi: 10.1016/j.cmet.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ronzaud C, Loffing-Cueni D, Hausel P, Debonneville A, Malsure SR, Fowler-Jaeger N, Boase NA, Perrier R, Maillard M, Yang B, Stokes JB, Koesters R, Kumar S, Hummler E, Loffing J, Staub O. Renal tubular NEDD4-2 deficiency causes NCC-mediated salt-dependent hypertension. J Clin Invest. 2013;123:657–665. doi: 10.1172/JCI61110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20**.Zhang C, Wang L, Zhang J, Su X-T, Lin DH, Scholl UI, Giebisch G, Lifton RP, Wang WH. KCNJ10 determines the expression of the apical Na-Cl cotransporter (NCC) in the early distal convoluted tubule (DCT1) Proc Natl Acad Sci USA. 2014;111:11864–11869. doi: 10.1073/pnas.1411705111. Depletion of Kir4.1 in the DCT inhibits SPAK expression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bandulik S, Schmidt K, Bockenhauer D, Zdebik AA, humberg E, Kleta R, Warth R, Reichold M. The salt-wasting phenotype of EAST syndrome, a disease with multifaceted symptoms linked to the KCNJ10 K channel. Pflugers Arch. 2011;461:423–435. doi: 10.1007/s00424-010-0915-0. [DOI] [PubMed] [Google Scholar]
- 22.Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, Tobin J, Lieberer E, Sterner C, Landoure G, Arora R, Sirimanna T, Thompson D, Cross JH, van’t Hoff W, Al Masri O, Tullus K, Yeung S, Anikster Y, Klootwijk E, Hubank M, Dillon MJ, Heitzmann D, Arcos-Burgos M, Knepper MA, Dobbie A, Gahl WA, Warth R, Sheridan E, Kleta R. Epilepsy, Ataxia, Sensorineural Deafness, Tubulopathy, and KCNJ10 Mutations. New England Journal of Medicine. 2009;360:1960–1970. doi: 10.1056/NEJMoa0810276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scholl UI, Choi M, Liu T, Ramaekers VT, Hausler MG, Grimmer J, Tobe SW, Farhi A, Nelson-Williams C, Lifton RP. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proceedings of the National Academy of Sciences. 2009;106:5842–5847. doi: 10.1073/pnas.0901749106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reichold M, Zdebik AA, Lieberer E, Rapedius M, Schmidt K, Bandulik S, Sterner C, Tegtmeier I, Penton D, Baukrowitz T, Hulton SA, Witzgall R, Ben Zeev B, Howie AJ, Kleta R, Bockenhauer D, Warth R. KCNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel function. Proceedings of the National Academy of Sciences. 2010;107:14490–14495. doi: 10.1073/pnas.1003072107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ellison DH. Divalent cation transport by the distal nephron: insights from Bartter’s and Gitelman’s syndromes. Am J Physiol Renal Physiol. 2000;279:F616–F625. doi: 10.1152/ajprenal.2000.279.4.F616. [DOI] [PubMed] [Google Scholar]
- 26.Simon DB. Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet. 1996;12:24–30. doi: 10.1038/ng0196-24. [DOI] [PubMed] [Google Scholar]
- 27*.Zhang C, Wang L, Su XT, Lin DH, Wang WH. KCNJ10 (Kir4.1) is expressed in the basolateral membrane of the cortical thick ascending limb. American Journal of Physiology - Renal Physiology. 2015;308:F1288–F1296. doi: 10.1152/ajprenal.00687.2014. Depletion of Kir4.1 in the cTAL stimulates Na and Cl activated potassium channels. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paulais M, Lachheb S, Teulon J. A Na+ and Cl-activated K+ Channel in the Thick Ascending Limb of Mouse Kidney. The Journal of General Physiology. 2006;127:205–215. doi: 10.1085/jgp.200509360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fan L, Wang X, Zhang D, Duan X, Zhao C, Zu M, Meng X, Zhang C, Su XT, Wang MX, Wang WH, Gu R. Vasopressin-induced stimulation of the Na+-activated K+ channels is responsible for maintaining the basolateral K+ conductance of the thick ascending limb (TAL) in EAST/SeSAME syndrome. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2015;1852:2554–2562. doi: 10.1016/j.bbadis.2015.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ellison DH, Velazquez H, Wright FS. Mechanisms of sodium, potassium and chloride transport by the renal distal tubule. Miner Electrolyte Metab. 1987;13:422–432. [PubMed] [Google Scholar]
- 31.Kim GH, Masilamani S, Turner R, Mitchell C, Wade JB, Knepper MA. The thiazide-sensitive Na-Cl cotransporter is an aldosterone-induced protein. Proc Natl Acad Sci U S A. 1998;95:14552–14557. doi: 10.1073/pnas.95.24.14552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Obermuller N, Bernstein P, Velazquez H, Reilly R, Moser D, Ellison DH, Bachmann S. Expression of the thiazide-sensitive Na-Cl cotransporter in rat and human kidney. Am J Physiol. 1995;269:F900–F910. doi: 10.1152/ajprenal.1995.269.6.F900. [DOI] [PubMed] [Google Scholar]
- 33.Bachmann S, Velazquez H, Obermuller N, Reilly RF, Moser D, Ellison DH. Expression of the thiazide-sensitive Na-Cl cotransporter by rabbit distal convoluted tubule cells. J Clin Invest. 1995;96:2510–2514. doi: 10.1172/JCI118311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wade JB, Fang L, Coleman RA, Liu J, Grimm PR, Wang T, Welling PA. Differential regulation of ROMK (Kir1.1) in distal nephron segments by dietary potassium. American Journal of Physiology - Renal Physiology. 2011;300:F1385–F1393. doi: 10.1152/ajprenal.00592.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vitzthum H, Castrop H, Meier-Meitinger M, Riegger GA, Kurtz A, Kramer BK, Wolf K. Nephron specific regulation of chloride channel CLC-K2 mRNA in the rat. Kidney Int. 2002;61:547–554. doi: 10.1046/j.1523-1755.2002.00165.x. [DOI] [PubMed] [Google Scholar]
- 36.Mount DB, Mercado A, Song L, Xu J, George AL, Jr, Delpire E, Gamba G. Cloning and characterization of KCC3 and KCC4, new members of the cation-chloride cotransporter gene family. J Biol Chem. 1999;274:16355–16362. doi: 10.1074/jbc.274.23.16355. [DOI] [PubMed] [Google Scholar]
- 37.Lourdel S, Paulais M, Marvao P, Nissant A, Teulon J. A Chloride Channel at the Basolateral Membrane of the Distal-convoluted Tubule: a Candidate CIC-K Channel. J Gen Physiol. 2003;121:287–300. doi: 10.1085/jgp.200208737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmitt R, Ellison DH, Farman N, Rossier BC, Reilly RF, Reeves WB, Oberbñumer I, Tapp R, Bachmann S. Developmental expression of sodium entry pathways in rat nephron. American Journal of Physiology - Renal Physiology. 1999;276:F367–F381. doi: 10.1152/ajprenal.1999.276.3.F367. [DOI] [PubMed] [Google Scholar]
- 39.Loffing J, Pietri L, Aregger F, Bloch-Faure M, Ziegler U, Meneton P, Rossier BC, Kaissling B. Differential subcellular localization of ENaC subunits in mouse kidney in response to high- and low-Na diets. American Journal of Physiology - Renal Physiology. 2000;279:F252–F258. doi: 10.1152/ajprenal.2000.279.2.F252. [DOI] [PubMed] [Google Scholar]
- 40*.Su XT, Zhang C, Wang L, Gu R, Lin DH, Wang WH. The disruption of KCNJ10 (Kir4.1) stimulates the expression of ENaC in the collecting duct. American Journal of Physiology - Renal Physiology. 2016 doi: 10.1152/ajprenal.00584.2015. Depletion of Kir4.1 increases the expression of ENaC in collecting duct. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lachheb S, Cluzeaud F, Bens M, Genete M, Hibino H, Lourdel S, Kurachi Y, Vandewalle A, Teulon J, Paulais M. Kir4.1/Kir5.1 channel forms the major K+ channel in the basolateral membrane of mouse renal collecting duct principal cells. AJP - Renal Physiology. 2008;294:F1398–F1407. doi: 10.1152/ajprenal.00288.2007. [DOI] [PubMed] [Google Scholar]
- 42.Zaika OL, Mamenko M, Palygin O, Boukelmoune N, Staruschenko A, Pochynyuk O. Direct inhibition of basolateral Kir4.1/5.1 and Kir4.1 channels in the cortical collecting duct by dopamine. American Journal of Physiology - Renal Physiology. 2013;305:F1277–F1287. doi: 10.1152/ajprenal.00363.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rojas A, Su J, Yang L, Lee M, Cui N, Zhang X, Fountain D, Jiang C. Modulation of the heteromeric Kir4.1–Kir5.1 channel by multiple neurotransmitters via Gαq-coupled receptors. Journal of Cellular Physiology. 2008;214:84–95. doi: 10.1002/jcp.21169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang C, Wang L, Thomas S, Wang K, Lin DH, Rinehart J, Wang WH. Src-family protein tyrosine kinase regulates the basolateral K channel in the distal convoluted tubule (DCT) by phosphorylation of KCNJ10. Journal of Biological Chemistry. 2013;288:26135–26146. doi: 10.1074/jbc.M113.478453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45**.Wang L, Zhang C, Su X, Lin DH, Wang W. Caveolin-1 Deficiency Inhibits the Basolateral K+ Channels in the Distal Convoluted Tubule and Impairs Renal K+ and Mg2+ Transport. Journal of the American Society of nephrology. 2015;26 doi: 10.1681/ASN.2014070658. The depletion of caveolin-1 inhibits Kir4.1 thereby increasing urinaru excretion of potassium and magnesium. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pessia M, Imbrici P, D’Adamo MC, Salvatore L, Tucker SJ. Differential pH sensitivity of Kir4.1 and Kir4.2 potassium channels and their modulation by heteropolymerisation with Kir5.1. J Physiol. 2001;532:359–367. doi: 10.1111/j.1469-7793.2001.0359f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tanemoto M, Kittaka N, Inanobe A, Kurachi Y. In vivo formation of a proton-sensitive K+ channel by heteromeric subunit assembly of Kir5.1 with Kir4.1. J Physiol. 2000;525(Pt 3):587–592. doi: 10.1111/j.1469-7793.2000.00587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paulais M, Bloch-Faure M, Picard N, Jacques T, Ramakrishnan SK, Keck M, Sohet F, Eladari D, Houillier P, Lourdel Sp, Teulon J, Tucker SJ. Renal phenotype in mice lacking the Kir5.1 (Kcnj16) K+ channel subunit contrasts with that observed in SeSAME/EAST syndrome. Proceedings of the National Academy of Sciences. 2011;108:10361–10366. doi: 10.1073/pnas.1101400108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cha SK, Huang C, Ding Y, Qi X, Huang CL, Miller RT. Calcium-sensing Receptor Decreases Cell Surface Expression of the Inwardly Rectifying K+ Channel Kir4.1. Journal of Biological Chemistry. 2011;286:1828–1835. doi: 10.1074/jbc.M110.160390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang C, Sindic A, Hill CE, Hujer KM, Chan KW, Sassen M, Wu Z, Kurachi Y, Nielsen S, Romero MF, Miller RT. Interaction of the Ca2+-sensing receptor with the inwardly rectifying potassium channels Kir4.1 and Kir4.2 results in inhibition of channel function. AJP - Renal Physiology. 2007;292:F1073–F1081. doi: 10.1152/ajprenal.00269.2006. [DOI] [PubMed] [Google Scholar]
- 51**.Terker AS, Zhang C, Erspamer KJ, Gamba G, Yang CL, Ellison DH. Unique chloride-sensing properties of WNK4 permit the distal nephron to modulate potassium homeostasis. Kidney Int. 2015 doi: 10.1038/ki.2015.289. Plasma K concentrations are closely correelated with NCC phosphorylation and WNK4 activity is regulated by plasma K at the physiological relevant ranges. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang SS, Lo YF, Wu CC, Lin SW, Yeh CJ, Chu P, Sytwu HK, Uchida S, Sasaki S, Lin SH. SPAK-Knockout Mice Manifest Gitelman Syndrome and Impaired Vasoconstriction. Journal of the American Society of nephrology. 2010;21:1868–1877. doi: 10.1681/ASN.2009121295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lalioti MD, Zhang J, Volkman HM, Kahle KT, Hoffmann KE, Toka HR, Nelson-Williams C, Ellison DH, Flavell R, Booth CJ, Lu Y, Geller DS, Lifton RP. Wnk4 controls blood pressure and potassium homeostasis via regulation of mass and activity of the distal convoluted tubule. Nat Genet. 2006;38:1124–1132. doi: 10.1038/ng1877. [DOI] [PubMed] [Google Scholar]
- 54.Piechotta K, Lu J, Delpire E. Cation Chloride Cotransporters Interact with the Stress-related Kinases Ste20-related Proline-Alanine-rich Kinase (SPAK) and Oxidative Stress Response 1 (OSR1) J Biol Chem. 2002;277:50812–50819. doi: 10.1074/jbc.M208108200. [DOI] [PubMed] [Google Scholar]
- 55.Liu Z, Xie J, Wu T, Truong T, Auchus RJ, Huang CL. Downregulation of NCC and NKCC2 cotransporters by kidney-specific WNK1 revealed by gene disruption and transgenic mouse models. Human Molecular Genetics. 2011;20:855–866. doi: 10.1093/hmg/ddq525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.San Cristobal P, Pacheco-Alvarez D, Richardson C, Ring AM, Vazquez N, Rafiqi FH, Chari D, Kahle KT, Leng Q, Bobadilla NA, Hebert SC, Alessi DR, Lifton RP, Gamba G. Angiotensin II signaling increases activity of the renal Na-Cl cotransporter through a WNK4-SPAK-dependent pathway. Proceedings of the National Academy of Sciences. 2009;106:4384–4389. doi: 10.1073/pnas.0813238106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bazua-Valenti S, Gamba G. Revisiting the NaCl cotransporter regulation by with-no-lysine kinases. American Journal of Physiology - Cell Physiology. 2015;308:C779–C791. doi: 10.1152/ajpcell.00065.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kahle KT, Ring AM, Lifton RP. Molecular Physiology of the WNK Kinases. Annual Review of Physiology. 2008;70:329–355. doi: 10.1146/annurev.physiol.70.113006.100651. [DOI] [PubMed] [Google Scholar]
- 59*.piala AT, Moon TM, Akella R, He HX, Cobb MH, Goldsmith EJ. Chloride sensing by WNK1 involves inhibition of autophosphosphorylation. Sciencesignaling. 2014;7:ra41. doi: 10.1126/scisignal.2005050. WNK1 activity is regulated by the intracellular Cl concentrations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60*.Bazua-Valenti S, Chavez-Canales M, Rojas-Vega L, Gonzalez-Rodriguez X, Vazquez N, Rodriguez-Gama A, Argaiz ER, Melo Z, Plata C, Ellison DH, Garcia-Valdes J, Hadchouel J, Gamba G. The Effect of WNK4 on the Na+Cl− Cotransporter Is Modulated by Intracellular Chloride. J Am Soc Nephrol. 2014;26:1781–1786. doi: 10.1681/ASN.2014050470. Intracellular Cl concentrations regulate the effect of WNK4 on NCC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rosenbaek LL, Kortenoeven MLA, Aroankins TS, Fenton RA. Phosphorylation Decreases Ubiquitylation of the Thiazide-sensitive Cotransporter NCC and Subsequent Clathrin-mediated Endocytosis. J Biol Chem. 2014;289:13347–13361. doi: 10.1074/jbc.M113.543710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62**.Terker A-S, Zhang C, McCormick J-A, Lazelle R-A, Zhang C, Meermeier N-P, Siler D-A, Park H-J, Fu Y, Cohen D-M, Weinstein A-M, Wang WH, Yang CL, Ellison D-H. Potassium Modulates Electrolyte Balance and Blood Pressure through Effects on Distal Cell Voltage and Chloride. Cell Metabolism. 2015;21:39–50. doi: 10.1016/j.cmet.2014.12.006. Membrane potential affected the intracellular Cl concentrations and the effect of WNK-SPAK on NCC phosphorylation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ecelbarger CA, Kim GH, Terris J, Masilamani S, Mitchell C, Reyes I, Verbalis JG, Knepper MA. Vasopressin-mediated regulation of epithelial sodium channel abundance in rat kidney. American Journal of Physiology - Renal Physiology. 2000;279:F46–F53. doi: 10.1152/ajprenal.2000.279.1.F46. [DOI] [PubMed] [Google Scholar]
- 64.Yang L, Frindt G, Palmer LG. Magnesium Modulates ROMK Channel-ÇôMediated Potassium Secretion. J Am Soc Nephrol. 2010;21:2109–2116. doi: 10.1681/ASN.2010060617. [DOI] [PMC free article] [PubMed] [Google Scholar]