

Abstract
Background and objective
Matrix metalloproteinases (MMPs) mediate blood–brain barrier dysfunction in inflammatory disease states. Our objective was to compare circulating MMPs in children with diabetic ketoacidosis (DKA) to children with type 1 diabetes mellitus without DKA.
Research design and methods
This was a prospective study performed at five tertiary-care pediatric hospitals. We measured plasma MMP-2, MMP-3, and MMP-9 early during DKA (time 1; within 2 h of beginning intravenous fluids) and during therapy (time 2; median 8 h; range: 4–16 h). The primary outcome was MMP levels in 34 children with DKA vs. 23 children with type 1 diabetes without DKA. Secondary outcomes included correlations between MMPs and measures of DKA severity.
Results
In children with DKA compared with diabetes controls, circulating MMP-2 levels were lower (mean 77 vs. 244 ng/mL, p < 0.001), MMP-3 levels were similar (mean 5 vs. 4 ng/mL, p = 0.57), and MMP-9 levels were higher (mean 67 vs. 25 ng/mL, p = 0.002) early in DKA treatment. MMP-2 levels were correlated with pH at time 1 (r = 0.45, p = 0.018) and time 2 (r = 0.47, p = 0.015) and with initial serum bicarbonate at time 2 (r = 0.5, p = 0.008). MMP-9 levels correlated with hemoglobin A1c in DKA and diabetes controls, but remained significantly elevated in DKA after controlling for hemoglobin A1c (β = −31.3, p = 0.04).
Conclusions
Circulating MMP-2 levels are lower and MMP-9 levels are higher in children during DKA compared with levels in children with diabetes without DKA. Alterations in MMP expression could mediate BBB dysfunction occurring during DKA.
Keywords: biomarkers, cerebral edema, cerebral injury, type 1 diabetes mellitus
Diabetic ketoacidosis (DKA) is a frequent complication of type 1 diabetes mellitus in children, and cerebral injury is the most common serious complication of DKA (1–4). The pathophysiology of DKA-related cerebral injury is not well understood. Cerebral injury in DKA is characterized by vasogenic edema (5), with an influx of fluid and possible influx of neurotoxic proteins into the central nervous system (CNS). The spectrum of cerebral injury ranges from clinically overt, life threatening events in <1% of DKA cases (3, 4, 6, 7), to more subtle and common cerebral injury, manifested as permanent deficits in memory function (2, 8, 9). Because DKA is characterized by profound systemic inflammation, it has been postulated that pro-inflammatory mediators may play a role in cerebral injury and vasogenic edema via disruption of the blood–brain barrier (BBB) (10, 11). Identification of these mediators may allow for development of novel targeted therapeutics to prevent cerebral injury in children with DKA.
Matrix metalloproteinases (MMPs) have been implicated in the pathophysiology of disease states that are characterized by inflammation, oxidative stress, and cerebral injury (12–16). Animal models of diabetes have suggested that increased MMP activity may be a mediator of increased BBB permeability (17). MMPs are endopeptidases that break peptide bonds in tight junction proteins and endothelial basement membranes. MMP-2, MMP-3, and MMP-9 in particular have been shown to actively degrade tight junction proteins at the BBB (18, 19). Tight junction complexes span intercellular clefts and are linked to the extracellular cytoskeleton to maintain structural integrity of the endothelial barrier (20). Degradation of tight junction proteins increase BBB permeability, allowing fluid and blood-borne pro-inflammatory or neurotoxic proteins to enter the CNS (21). In inflammatory diseases and following ischemic events, circulating MMP levels are increased both as a result of endogenous production in the CNS and of production by recruited neutrophils and mononuclear cells (22, 23). Patterns of MMP expression differ according to disease process. For example, in neuroinflammatory lesions produced by lipopolysaccharide injection in animal models, MMP-3 and MMP-9 expression are markedly increased (24). In contrast, in animal models of neuroinflammation resulting from ischemia, MMP-2 activity is increased in very early stages and appears to be responsible for early BBB disruption (25). This is characterized by a decrease in MMP-2 and an increase in MMP-9 activity that results in secondary injury to the BBB microvasculature.
The pattern of circulating MMP levels can yield insight into underlying pathophysiological processes resulting in cerebral injury. In this study, we measured circulating MMP levels in children with DKA at two separate times within the first 24 h of presentation, and compared these with levels of MMPs in a group of children with type 1 diabetes without DKA. We correlated MMP levels with laboratory markers of DKA severity such as pH and serum bicarbonate levels to determine the relationship between disease severity and MMP levels.
Methods
This study was conducted as an ancillary study to the Pediatric Emergency Care Applied Research Network (PECARN) FLUID trial (U01HD062417, PIs: Kuppermann/Glaser). The FLUID trial is a factorial-design, randomized-controlled trial comparing the effects of four fluid treatment protocols on both acute and long-term neurological and neurocognitive outcomes of DKA in children (26).
FLUID study participants with DKA from five PECARN study sites were eligible for enrollment in the current ancillary study. Both the FLUID parent study and the current ancillary study were approved by the institutional review boards at each site in accordance with the Declaration of Helsinki. Informed consent was obtained from the guardians of all participants prior to participation, and assent was obtained from study participants themselves when possible according to local guidelines. Children with DKA were eligible for inclusion if they were younger than 18 yr old, had serum glucose concentrations >300 mg/dL and either venous pH < 7.25 or serum bicarbonate concentrations <15 mmol/L. Children were excluded from participation in the following circumstances: (1) pre-existing neurological disease that impacted mental status determination or neurocognitive examination; (2) conditions that affected neurological function and assessment (e.g. alcohol or drug use, head trauma, meningitis); (3) transfer to a PECARN site after administration of more than 10 mL/kg IV fluid bolus; (4) known pregnancy; (5) two or more previous enrollments in the FLUID study; (6) treating physician believed a specific treatment regimen was warranted; (7) patient had been receiving IV fluids at weight-based maintenance rate or greater for more than 2 h, or (8) more than 4 h elapsed since initiation of DKA therapy (IV fluids or IV insulin).
We collected blood samples from children with DKA at two time points (1) time 1: within 2 h of emergency department presentation with DKA and (2) time 2: during DKA therapy. The sample collected during DKA therapy was targeted to occur 8 h after IV fluid therapy was initiated, but allowed for a range of 4–16 h so sampling could coincide with routine phlebotomy for clinical care. For each participant, we recorded the following laboratory variables from the first serum sample collected: pH, glucose, bicarbonate, sodium, blood urea nitrogen (BUN), partial pressure of carbon dioxide (pCO2), and peripheral white blood cell (WBC) count.
We recruited a comparison group of children with type 1 diabetes without recent DKA (diabetes controls) from the Pediatric Endocrinology clinic at Hasbro Children’s Hospital. Children were eligible as diabetes controls if they were younger than 18-yr-old, had established diabetes of over 1-yr duration, were undergoing phlebotomy for routine diabetes care and had no DKA episodes within the preceding 3 months (determined by review of medical records and confirmed with the treating endocrinologist). Phlebotomy occurred following the clinic visit.
Blood samples were cooled, centrifuged within 20 min of collection, and the plasma layer was separated and stored. At secondary sites, DKA samples were stored in a −20°C freezer before shipping. Samples were subsequently shipped to the primary site on dry ice, and stored at −80°C until analyzed. MMP assays were performed within 1 yr of plasma collection, and therefore no significant degradation of MMP would be expected in this timeframe (27). We used enzyme-linked immunosorbent assays (ELISA) to measure MMP-2, MMP-3, and MMP-9.
Statistical analysis
We performed all statistical analyses using Stata 12.0 (StataCorp. 2011, College Station, TX, USA). Protein concentrations were expressed as means ± standard deviations (SD). We performed comparisons for data using the Mann–Whitney U test. We analyzed paired protein levels (sample 1 and sample 2 in the DKA group) using the Wilcoxon signed-rank test for comparisons. We measured correlations between MMP levels at different time points and between MMP levels and laboratory measures (pH, glucose, bicarbonate, sodium, BUN, and pCO2) using Pearson’s correlation coefficient. We inspected non-linear relationships and potential outliers visually and by leave-one-out analyses. For all comparisons, we considered differences to be statistically significant when p-values were <0.05.
We conducted subanalyses to determine if differences in chronic glycemic control between children with DKA and diabetes controls affected MMP levels. The subanalyses included the following factors as covariates: (1) new onset vs. known diabetes; (2) differences in length of time since diagnosis; and (3) differences in most recent hemoglobin A1c measurements.
Results
We measured MMP-2 and MMP-3 levels in 34 children with DKA and 23 diabetes controls. Based on remaining sample availability, MMP-9 levels were measured in 19 children with DKA and 17 diabetes controls (see Fig. 1). Diabetes controls and children with DKA were similar in age (12.2 vs. 12.1 yr, p = 0.86) and gender (60% male vs. 56% male, p = 0.75). Relevant biochemical data for patients with DKA are presented in Table 1.
Fig. 1.
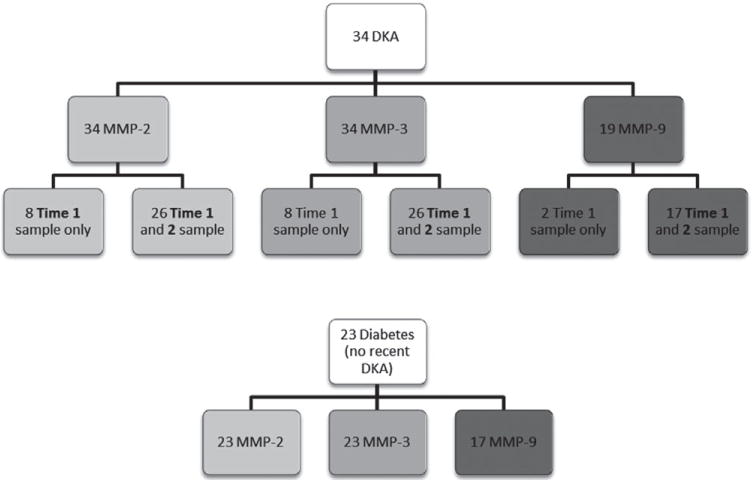
Shown is the number of assays for the three matrix metalloproteinases (MMPs) that were performed in the diabetic ketoacidosis (DKA) and diabetes control groups based on the volume of blood available. In the DKA group, the number of participants with only a time 1 sample (intravenous access discontinued prior to second sample) vs. both time 1 and time 2 samples is shown.
Table 1.
Biochemical data at initial presentation for children in DKA (n = 34)
| Serum pH | 7.14±0.11 |
| Serum glucose | 538±148 mg/dL |
| Serum bicarbonate | 10.0±3.9 mEq/L |
| Serum sodium | 134.1±4.1 mEq/L |
| Serum blood urea nitrogen | 17.8±6.8 mg/dL |
| Serum partial pressure of carbon dioxide | 25.7±7.5 mmHg |
| Peripheral white blood cell count* | 22.8±9.8 k/mm3 |
| Most recent hemoglobin A1C†(IFCC) | 10.6%±2.1% (92.36±18.30 mmoL/moL) |
DKA, diabetic ketoacidosis.
All data presented as means±standard deviation unless noted.
Available for 15 patients.
Available for 13 of 14 children with known diabetes.
Intravenous access was discontinued in 8 of the 34 children prior to collection of the second blood sample (2 of the 19 children in the subset for whom MMP-9 was measured). The severity of DKA in children with only one sample did not differ from that of children with two samples (pH at presentation 7.12 vs. 7.15, p = 0.43; serum glucose 537 vs. 539 mg/dL, p = 0.98; serum bicarbonate 9.3 vs. 10.2 mEq/L, p = 0.51).
Circulating MMP-2 levels were significantly lower in children with DKA compared with diabetes controls at time 1 (mean 77 vs. 244 ng/mL, p < 0.001, Fig. 2). In children with DKA, MMP-2 levels rose during DKA treatment (time 2; mean Δ = +31 ng/mL, SD ± 60 ng/mL, r = 0.47, p = 0.03), but remained significantly lower (mean 112 ng/mL) than values from diabetes controls (p < 0.001). Correlation between circulating MMP levels and serum biomarkers of DKA severity measured at first blood draw are presented in Table 2. Lower MMP-2 levels at both time points were significantly correlated with the severity of acidosis (lower pH) at presentation of DKA (time 1; r = 0.45, p = 0.008, time 2; r = 0.47, p = 0.015, see Fig. 3). MMP-2 levels were not correlated with the initial (presentation) serum bicarbonate at time 1, but at time 2 MMP-2 levels were correlated with the initial serum bicarbonate (r = 0.51, p = 0.008). Correlations between MMP-2 levels and serum glucose, sodium, BUN, or pCO2 did not reach the level of statistical significance.
Fig. 2.
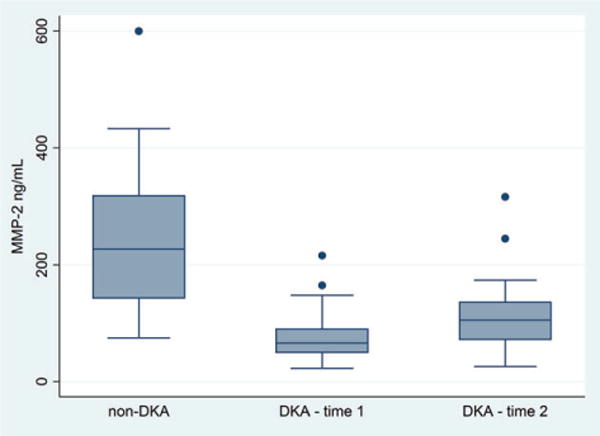
Plasma matrix metalloproteinase (MMP)-2 levels are presented for diabetes controls (n = 23), children with diabetic ketoacidosis (DKA) at ED presentation (time 1) (n = 34), and children with DKA during therapy (time 2) (n = 26). Box plots show the interquartile range (IQR) (shaded box), median (box center line), with fences extending to 1.5 times the IQR. Circulating MMP-2 levels were lower in children with DKA compared with diabetes controls at time 1 (mean 77 vs. 244 ng/mL, p < 0.001) and time 2 (mean 112 vs. 244 ng/mL, p < 0.001).
Table 2.
Correlation between circulating MMP levels and serum biomarkers of DKA severity
| pH | Glucose | Bicarbonate | Sodium | BUN | pCO2 | |
|---|---|---|---|---|---|---|
| MMP-2 time 1 | 0.45* (n= 34, p=0.008) | −0.16 (n=33, p=0.36) | 0.22 (n =34, p= 0.21) | 0.1 (n =34, p= 0.57) | 0.19 (n=34, p =0.27) | 0.3 (n = 30, p =0.11) |
| MMP-2 time 2 | 0.47* (n =26, p=0.015) | 0.11 (n= 25, p=0.61) | 0.51* (n =26, p= 0.008) | 0.08 (n =26, p= 0.70) | −0.13 (n=26, p =0.53) | −0.13 (n= 22, p =0.56) |
| MMP-3 time 1 | 0.01 (n= 34, p=0.96) | −0.16 (n=33, p=0.38) | −0.15 (n =34, p= 0.41) | −0.43* (n =34, p= 0.011) | 0.15 (n=34, p =0.41) | −0.24 (n= 30, p =0.21) |
| MMP-3 time 2 | 0.03 (n= 26, p=0.89) | −0.14 (n=25, p=0.49) | −0.06 (n =26, p= 0.77) | −0.52* (n =26, p= 0.007) | 0.06 (n=26, p =0.76) | −0.34 (n= 22, p =0.12) |
| MMP-9 time 1 | −0.05 (n= 19, p=0.84) | −0.2 (n =19, p=0.41) | −0.24 (n =19, p= 0.32) | 0.43 (n =19, p= 0.068) | −0.35 (n=19, p =0.14) | 0.23 (n=17, p =0.37) |
| MMP-9 time 2 | 0.07 (n=17, p=0.78) | −0.04 (n=17, p=0.87) | −0.03 (n =17, p=0.90) | 0.33 (n =17, p=0.19) | −0.22 (n=17, p=0.40) | 0.31 (n=15, p=0.27) |
BUN, blood urea nitrogen; DKA, diabetic ketoacidosis; MMP, matrix metalloproteinase; pCO2, partial pressure of carbon dioxide.
p < 0.05.
Fig. 3.
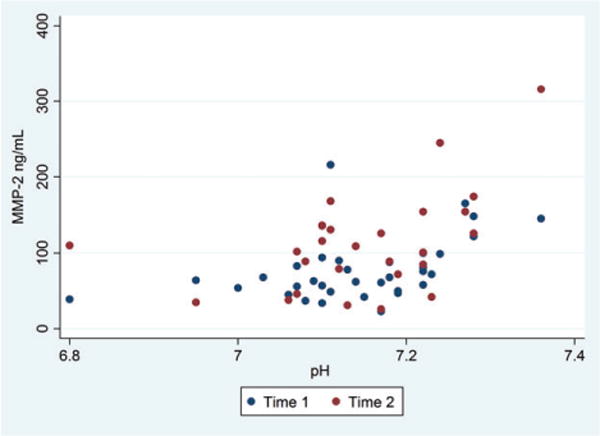
This scatterplot shows plasma matrix metalloproteinase (MMP)-2 concentrations and initial serum pH for children with diabetic ketoacidosis (DKA). Time 1 samples were obtained at presentation (n = 34), and time 2 samples were obtained during DKA therapy (n = 26). Lower MMP-2 levels at both time points were correlated with the severity of acidosis at presentation of DKA (time 1; r = 0.45, p = 0.008, time 2; r = 0.47, p = 0.015).
Circulating MMP-3 levels were not significantly different in children with DKA compared with diabetes controls at time 1 (mean 5 vs. 4 ng/mL, p = 0.81, Fig. 4), but were significantly higher at time 2 (mean 8 vs. 4 ng/mL, p = 0.013). In children with DKA, circulating MMP-3 levels increased slightly during therapy (mean Δ = +2.8 ng/mL, SD ±5.2 ng/mL, r = 0.93, p = 0.009). MMP-3 levels were negatively correlated with presenting serum sodium concentrations for children with DKA at time 1 (r = −0.43, p = 0.011) and time 2 (r = −0.52, p = 0.07). There were no other significant correlations between MMP-3 levels and serum pH, glucose, bicarbonate, BUN, or pCO2.
Fig. 4.
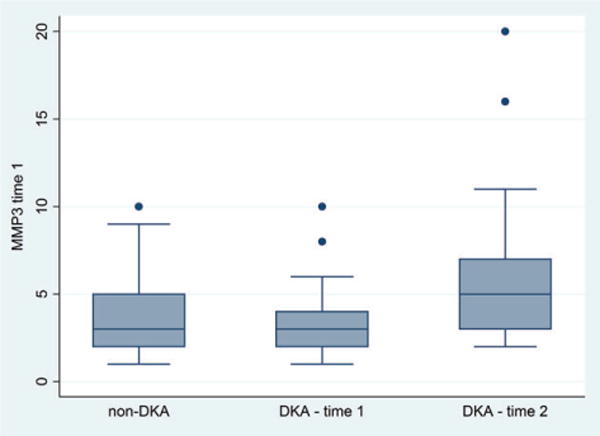
Plasma matrix metalloproteinase (MMP)-3 levels are presented for diabetes controls (n = 23), children with diabetic ketoacidosis (DKA) at presentation (time 1) (n = 34), and children with DKA during therapy (time 2) (n = 26). Box plots show the interquartile range (IQR) (shaded box), median (box center line) with fences extending to 1.5 times the IQR. Circulating MMP-3 levels were similar in children with DKA compared with diabetes controls at time 1 (mean 5 vs. 4 ng/mL, p = 0.81), but were significantly higher at time 2 (mean 8 vs. 4 ng/mL, p = 0.013).
Circulating MMP-9 levels were significantly higher in children with DKA compared with diabetes controls at time 1 (mean 67 vs. 25 ng/mL, p < 0.001, Fig. 5) and remained elevated at time 2 (mean = 59 ng/mL, p = 0.009). MMP-9 levels measured at time 1 and time 2 during DKA were not significantly different (p = 0.38). In children with DKA, there were no significant correlations between MMP-9 concentrations and DKA laboratory measures including serum pH, glucose, bicarbonate, sodium, BUN, or pCO2.
Fig. 5.
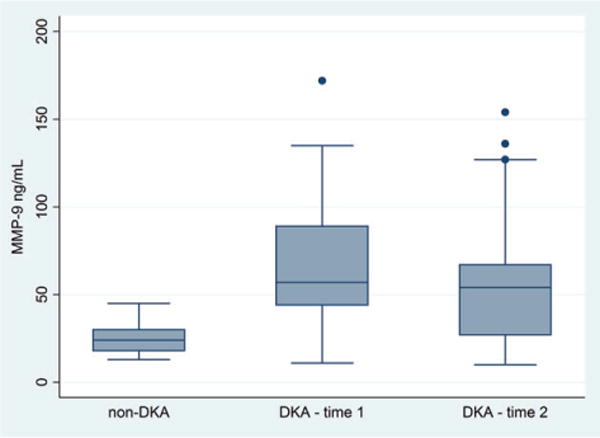
Plasma matrix metalloproteinase (MMP)-9 levels are presented for diabetes controls (n = 17), children with diabetic ketoacidosis (DKA) at presentation (time 1) (n = 19), and children with DKA during therapy (time 2) (n = 17). Box plots show the interquartile range (IQR) (shaded box), median (box center line), with fences extending to 1.5 times the IQR. Circulating MMP-9 levels were higher in children with DKA compared with diabetes controls at time 1 (mean 67 vs. 25 ng/mL, p < 0.001) and time 2 (mean = 59 ng/mL, p = 0.009).
Among children with DKA, there were no significant differences in circulating MMP levels at time 1 between children with new-onset diabetes (n = 12) and children with known diabetes [MMP-2 (mean 77 ± 40 vs. 78 ± 42 ng/mL; p = 0.91), MMP-3 (mean 3 ± 2 vs. 6 ± 9 ng/mL; p = 1.0), MMP-9 (mean 60 ± 29 vs. 71 ± 47; p = 0.90)]. There were also no significant differences at time 2 [MMP-2 (mean 111 ± 67 vs. 112 ± 68 ng/mL; p = 0.92), MMP-3 (mean 6 ± 3 vs. 10 ± 14 ng/mL; p = 0.73), MMP-9 (mean 54 ± 12 vs. 63 ± 58; p = 0.59)].
Time since diabetes diagnosis was similar in controls and children with DKA who had known diabetes (mean 5.2 vs. 4.5 yr, p = 0.93). Diabetes controls had lower hemoglobin A1c levels than children with DKA who had known diabetes [mean 9.3% (78.15 mmol/mol) vs. 10.6% (92.36 mmol/mol), p = 0.03]. MMP-9 levels at time 1 were correlated with hemoglobin A1c (r = 0.54, p = 0.003). There were no other significant correlations between MMP levels and hemoglobin A1c levels. In regression models controlling for hemoglobin A1c, the difference in MMP-9 levels at time 1 between children with DKA and diabetes controls remained significant (β = −31.3, p = 0.04).
Discussion
Circulating MMP-2 levels are reduced while MMP-9 levels are elevated in children with DKA compared with children with diabetes but without recent DKA. These changes in MMP expression during DKA could mediate BBB dysfunction, contributing to the vasogenic cerebral edema frequently observed during DKA treatment (5). The finding of decreased circulating MMP-2 levels is surprising as DKA is associated with systemic inflammation (28–30), and inflammatory diseases typically are associated with elevated levels of both MMP-2 and MMP-9 [including multiple sclerosis (13), rheumatoid arthritis (14), and systemic lupus erythematosus (14), bacterial meningitis (15), and Guillain–Barré syndrome (16)]. Patients with type 1 diabetes who are not in DKA have also been shown to have elevated circulating levels of MMP-2 and MMP-9 compared with individuals without diabetes (31). The underlying reason for MMP elevation in diabetes is not known, but may reflect the general state of inflammation in diabetes (28–30).
The mechanism underlying the reduction in circulating MMP-2 is unclear. Most inflammatory disease states do not demonstrate this pattern, but similar alterations have been described in ischemic stroke (32–34). During ischemic injury, cerebrovascular endothelial cells are the major source of circulating MMP-2 (35). Dynamic reductions in circulating MMP-2 often reflect either rapid degradation of circulating MMP-2, or distribution of MMP-2 to areas of active tissue remodeling (36). Prolonged hyperventilation has been hypothesized to be a factor causing cerebral hypoperfusion (37). Although we did not observe a significant correlation between MMP-2 levels and serum pCO2 levels, the small sample size may have limited the power to achieve statistical significance. Circulating MMP-2 concentrations, however, were positively correlated with serum pH. The correlation between circulating MMP-2 levels and initial pH was relatively constant at later time points during DKA therapy, indicating that the trigger for dynamic changes in MMP-2 concentrations precedes DKA therapy.
In contrast to circulating MMP-2 concentrations, circulating MMP-9 levels were higher in children with DKA compared with diabetes controls and remained elevated throughout the time period when DKA-related cerebral injury frequently occurs. Circulating MMP-9 concentrations frequently increase in systemic inflammatory conditions (38). Vascular MMP-9 production is increased in response to hyperglycemia (39). Hyperglycemia itself, however, does not appear to be a direct cause of BBB dysfunction because attenuation of hyperglycemia via insulin administration does not decrease BBB permeability (17). MMP-9 expression is increased in response to advanced glycation end products in states of chronic hyperglycemia (40), consistent with our subanalysis showing the associations between hemoglobin A1c and MMP-9 levels. Even after accounting for this association, however, children with DKA continued to have significantly higher levels of circulating MMP-9 when compared with diabetes controls. Systemic leukocytosis with neutrophil predominance can also result in high concentrations of circulating MMP-9 (41). Children in our study had substantial leukocytosis, similar to that documented previously in other studies of children with DKA (30) (Table 1).
Although circulating MMP-3 levels were statistically elevated in DKA patients at time 2 in comparison to diabetes controls, the clinical significance of this elevation is difficult to interpret given the overall low peak circulating levels in both groups. MMP-3 production is more often localized to sites of cerebral inflammation (specifically astrocytes) without concomitant increases in circulating concentrations (42, 43). The low levels of circulating MMP-3 do not preclude a role for MMP-3 in the pathophysiology of DKA-related cerebral injury, however, as other investigators have shown isolated intrathecal production of MMP-3 in cerebral injury without associated increases in circulating plasma MMP-3 levels (42).
Study limitations
The sample size in this exploratory study was relatively small and this limited our ability to detect significant correlations, particularly between metabolic alterations caused by DKA and MMP levels. In addition, the comparison between newly diagnosed patients and patients with known diabetes did not show any statistically significant differences in MMP levels; however, because of the small sample size, we cannot exclude those differences exists. The measurement of MMP activity in specific tissues is not feasible in human subjects, and thus, our studies cannot confirm causality of BBB dysfunction during DKA. Finally, because children present with variable durations of DKA (44), our data do not allow us to precisely time changes in MMP levels in relation to the onset of DKA.
Conclusion
Circulating MMP-2 levels are lower and MMP-9 levels are higher in children with DKA than in children with diabetes without DKA. Future studies are needed to examine associations between changes in MMP concentrations and neurological injury resulting from DKA in children, as well as to explore the potential role of MMP inhibitors in reducing such injury.
Acknowledgments
This study was supported by grant 1R01HD062417-01 from the Eunice Kennedy Shriver National Institute of Child Health & Human Development and by basic science grant 7-12-BS-060 from the American Diabetes Association. This project was also supported in part by the Health Resources and Services Administration (HRSA), Maternal and Child Health Bureau (MCHB), Emergency Medical Services for Children (EMSC) Network Development Demonstration Program under cooperative agreements U03MC00008, U03MC00001, U03MC00003, U03MC00006, U03MC00007, U03MC22684, and U03MC22685. The information or content and conclusions are those of the author and should not be construed as the official position or policy of, nor should any endorsements be inferred by HRSA, HHS or the U.S. Government.
We thank the PECARN Research Coordinators and clinicians around the PECARN Network who are enrolling children into this study. We also thank Julie Sarri for technical assistance in processing blood samples and assisting ELISA.
References
- 1.Dabelea D, Rewers A, Stafford JM, et al. Trends in the prevalence of ketoacidosis at diabetes diagnosis: the SEARCH for diabetes in youth study. Pediatrics. 2014;133:e938–e945. doi: 10.1542/peds.2013-2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ghetti S, Lee JK, Sims CE, Demaster DM, Glaser NS. Diabetic ketoacidosis and memory dysfunction in children with type 1 diabetes. J Pediatr. 2010;156:109–114. doi: 10.1016/j.jpeds.2009.07.054. [DOI] [PubMed] [Google Scholar]
- 3.Bello FA, Sotos JF. Cerebral oedema in diabetic ketoacidosis in children. Lancet. 1990;336:64. doi: 10.1016/0140-6736(90)91587-z. [DOI] [PubMed] [Google Scholar]
- 4.Duck SC, Wyatt DT. Factors associated with brain herniation in the treatment of diabetic ketoacidosis. J Pediatr. 1988;113:10–14. doi: 10.1016/s0022-3476(88)80521-3. [DOI] [PubMed] [Google Scholar]
- 5.Glaser NS, Wootton-Gorges SL, Buonocore MH, et al. Frequency of sub-clinical cerebral edema in children with diabetic ketoacidosis. Pediatr Diabetes. 2006;7:75–80. doi: 10.1111/j.1399-543X.2006.00156.x. [DOI] [PubMed] [Google Scholar]
- 6.Edge JA, Hawkins MM, Winter DL, Dunger DB. The risk and outcome of cerebral oedema developing during diabetic ketoacidosis. Arch Dis Child. 2001;85:16–22. doi: 10.1136/adc.85.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glaser N, Barnett P, McCaslin I, et al. Risk factors for cerebral edema in children with diabetic ketoacidosis. The Pediatric Emergency Medicine Collaborative Research Committee of the American Academy of Pediatrics. N Engl J Med. 2001;344:264–269. doi: 10.1056/NEJM200101253440404. [DOI] [PubMed] [Google Scholar]
- 8.Hoffman WH, Steinhart CM, el Gammal T, Steele S, Cuadrado AR, Morse PK. Cranial CT in children and adolescents with diabetic ketoacidosis. AJNR Am J Neuroradiol. 1988;9:733–739. [PMC free article] [PubMed] [Google Scholar]
- 9.Krane EJ, Rockoff MA, Wallman JK, Wolfsdorf JI. Subclinical brain swelling in children during treatment of diabetic ketoacidosis. N Engl J Med. 1985;312:1147–1151. doi: 10.1056/NEJM198505023121803. [DOI] [PubMed] [Google Scholar]
- 10.Hoffman WH, Casanova MF, Cudrici CD, et al. Neuroinflammatory response of the choroid plexus epithelium in fatal diabetic ketoacidosis. Exp Mol Pathol. 2007;83:65–72. doi: 10.1016/j.yexmp.2007.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoffman WH, Stamatovic SM, Andjelkovic AV. Inflammatory mediators and blood brain barrier disruption in fatal brain edema of diabetic ketoacidosis. Brain Res. 2009;1254:138–148. doi: 10.1016/j.brainres.2008.11.100. [DOI] [PubMed] [Google Scholar]
- 12.Gasche Y, Soccal PM, Kanemitsu M, Copin JC. Matrix metalloproteinases and diseases of the central nervous system with a special emphasis on ischemic brain. Front Biosci. 2006;11:1289–1301. doi: 10.2741/1883. [DOI] [PubMed] [Google Scholar]
- 13.Avolio C, Ruggieri M, Giuliani F, et al. Serum MMP-2 and MMP-9 are elevated in different multiple sclerosis subtypes. J Neuroimmunol. 2003;136:46–53. doi: 10.1016/s0165-5728(03)00006-7. [DOI] [PubMed] [Google Scholar]
- 14.Chang YH, Lin IL, Tsay GJ, et al. Elevated circulatory MMP-2 and MMP-9 levels and activities in patients with rheumatoid arthritis and systemic lupus erythematosus. Clin Biochem. 2008;41:955–959. doi: 10.1016/j.clinbiochem.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 15.Kanoh Y, Ohara T, Kanoh M, Akahoshi T. Serum matrix metalloproteinase-2 levels indicate blood-CSF barrier damage in patients with infectious meningitis. Inflammation. 2008;31:99–104. doi: 10.1007/s10753-007-9054-y. [DOI] [PubMed] [Google Scholar]
- 16.Nyati KK, Prasad KN, Verma A, Paliwal VK. Correlation of matrix metalloproteinases-2 and -9 with proinflammatory cytokines in Guillain-Barre syndrome. J Neurosci Res. 2010;88:3540–3546. doi: 10.1002/jnr.22514. [DOI] [PubMed] [Google Scholar]
- 17.Hawkins BT, Lundeen TF, Norwood KM, Brooks HL, Egleton RD. Increased blood-brain barrier permeability and altered tight junctions in experimental diabetes in the rat: contribution of hyperglycaemia and matrix metalloproteinases. Diabetologia. 2007;50:202–211. doi: 10.1007/s00125-006-0485-z. [DOI] [PubMed] [Google Scholar]
- 18.Batra A, Latour LL, Ruetzler CA, et al. Increased plasma and tissue MMP levels are associated with BCSFB and BBB disruption evident on post-contrast FLAIR after experimental stroke. J Cereb Blood Flow Metab. 2010;30:1188–1199. doi: 10.1038/jcbfm.2010.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gurney KJ, Estrada EY, Rosenberg GA. Blood–brain barrier disruption by stromelysin-1 facilitates neutrophil infiltration in neuroinflammation. Neurobiol Dis. 2006;23:87–96. doi: 10.1016/j.nbd.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 20.Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev. 2004;84:869–901. doi: 10.1152/physrev.00035.2003. [DOI] [PubMed] [Google Scholar]
- 21.Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 22.Gidday JM, Gasche YG, Copin JC, et al. Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am J Physiol Heart Circ Physiol. 2005;289:H558–H568. doi: 10.1152/ajpheart.01275.2004. [DOI] [PubMed] [Google Scholar]
- 23.Justicia C, Panes J, Sole S, et al. Neutrophil infiltration increases matrix metalloproteinase-9 in the ischemic brain after occlusion/reperfusion of the middle cerebral artery in rats. J Cereb Blood Flow Metab. 2003;23:1430–1440. doi: 10.1097/01.WCB.0000090680.07515.C8. [DOI] [PubMed] [Google Scholar]
- 24.Rosenberg GA, Cunningham LA, Wallace J, et al. Immunohistochemistry of matrix metalloproteinases in reperfusion injury to rat brain: activation of MMP-9 linked to stromelysin-1 and microglia in cell cultures. Brain Res. 2001;893:104–112. doi: 10.1016/s0006-8993(00)03294-7. [DOI] [PubMed] [Google Scholar]
- 25.Rosenberg GA. Matrix metalloproteinases in neuroinflammation. Glia. 2002;39:279–291. doi: 10.1002/glia.10108. [DOI] [PubMed] [Google Scholar]
- 26.Glaser NS, Ghetti S, Casper TC, Dean JM, Kuppermann N, Pediatric Emergency Care Applied Research Network DKAFSG Pediatric diabetic ketoacidosis, fluid therapy, and cerebral injury: the design of a factorial randomized controlled trial. Pediatr Diabetes. 2013;14:435–446. doi: 10.1111/pedi.12027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rouy D, Ernens I, Jeanty C, Wagner DR. Plasma storage at −80 degrees C does not protect matrix metalloproteinase-9 from degradation. Anal Biochem. 2005;338:294–298. doi: 10.1016/j.ab.2004.10.052. [DOI] [PubMed] [Google Scholar]
- 28.Close TE, Cepinskas G, Omatsu T, et al. Diabetic ketoacidosis elicits systemic inflammation associated with cerebrovascular endothelial cell dysfunction. Microcirculation. 2013;20:534–543. doi: 10.1111/micc.12053. [DOI] [PubMed] [Google Scholar]
- 29.Hoffman WH, Burek CL, Waller JL, Fisher LE, Khichi M, Mellick LB. Cytokine response to diabetic ketoacidosis and its treatment. Clin Immunol. 2003;108:175–181. doi: 10.1016/s1521-6616(03)00144-x. [DOI] [PubMed] [Google Scholar]
- 30.Hoffman WH, Helman SW, Passmore G. Acute activation of peripheral lymphocytes during treatment of diabetic ketoacidosis. J Diabetes Complications. 2001;15:144–149. doi: 10.1016/s1056-8727(00)00142-2. [DOI] [PubMed] [Google Scholar]
- 31.Derosa G, Avanzini MA, Geroldi D, et al. Matrix metalloproteinase 2 may be a marker of microangiopathy in children and adolescents with type 1 diabetes mellitus. Diabetes Res Clin Pract. 2005;70:119–125. doi: 10.1016/j.diabres.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 32.Horstmann S, Kalb P, Koziol J, Gardner H, Wagner S. Profiles of matrix metalloproteinases, their inhibitors, and laminin in stroke patients: influence of different therapies. Stroke. 2003;34:2165–2170. doi: 10.1161/01.STR.0000088062.86084.F2. [DOI] [PubMed] [Google Scholar]
- 33.Kreisel SH, Stroick M, Reuter B, Senn E, Hennerici MG, Fatar M. MMP-2 concentrations in stroke according to etiology: adjusting for enzyme degradation in stored deep-frozen serum and other methodological pitfalls. J Clin Neurosci. 2012;19:1564–1567. doi: 10.1016/j.jocn.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 34.Xu YZ, Zhao KJ, Yang ZG, et al. Decreased plasma decorin levels following acute ischemic stroke: correlation with MMP-2 and differential expression in TOAST subtypes. Mol Med Rep. 2012;6:1319–1324. doi: 10.3892/mmr.2012.1108. [DOI] [PubMed] [Google Scholar]
- 35.Lischper M, Beuck S, Thanabalasundaram G, Pieper C, Galla HJ. Metalloproteinase mediated occludin cleavage in the cerebral microcapillary endothelium under pathological conditions. Brain Res. 2010;1326:114–127. doi: 10.1016/j.brainres.2010.02.054. [DOI] [PubMed] [Google Scholar]
- 36.Lalu MM, Csont T, Schulz R. Matrix metalloproteinase activities are altered in the heart and plasma during endotoxemia. Crit Care Med. 2004;32:1332–1337. doi: 10.1097/01.ccm.0000127778.16609.ec. [DOI] [PubMed] [Google Scholar]
- 37.Ito H, Ibaraki M, Kanno I, Fukuda H, Miura S. Changes in the arterial fraction of human cerebral blood volume during hypercapnia and hypocapnia measured by positron emission tomography. J Cereb Blood Flow Metab. 2005;25:852–857. doi: 10.1038/sj.jcbfm.9600076. [DOI] [PubMed] [Google Scholar]
- 38.Goetzl EJ, Banda MJ, Leppert D. Matrix metalloproteinases in immunity. J Immunol. 1996;156:1–4. [PubMed] [Google Scholar]
- 39.Uemura S, Matsushita H, Li W, et al. Diabetes mellitus enhances vascular matrix metalloproteinase activity: role of oxidative stress. Circ Res. 2001;88:1291–1298. doi: 10.1161/hh1201.092042. [DOI] [PubMed] [Google Scholar]
- 40.Zhang F, Banker G, Liu X, et al. The novel function of advanced glycation end products in regulation of MMP-9 production. J Surg Res. 2011;171:871–876. doi: 10.1016/j.jss.2010.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kjeldsen L, Johnsen AH, Sengelov H, Borregaard N. Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J Biol Chem. 1993;268:10425–10432. [PubMed] [Google Scholar]
- 42.Grossetete M, Phelps J, Arko L, Yonas H, Rosenberg GA. Elevation of matrix metalloproteinases 3 and 9 in cerebrospinal fluid and blood in patients with severe traumatic brain injury. Neurosurgery. 2009;65:702–708. doi: 10.1227/01.NEU.0000351768.11363.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Savarin C, Stohlman SA, Rietsch AM, Butchi N, Ransohoff RM, Bergmann CC. MMP9 deficiency does not decrease blood-brain barrier disruption, but increases astrocyte MMP3 expression during viral encephalomyelitis. Glia. 2011;59:1770–1781. doi: 10.1002/glia.21222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wolfsdorf JI, Allgrove J, Craig ME, Edge J, Glaser N, Jain V. Diabetic ketoacidosis and hyperglycemic hyperosmolar state. Pediatr Diabetes. 2014;15:154–179. doi: 10.1111/pedi.12165. [DOI] [PubMed] [Google Scholar]