

Abstract
Cancer causes significant morbidity and mortality worldwide, and is the area most targeted in precision medicine. Recent development of high-throughput methods enables detailed omics analysis of the molecular mechanisms underpinning tumor biology. These studies have identified clinically actionable mutations, gene and protein expression patterns associated with prognosis, and provided further insights into the molecular mechanisms indicative of cancer biology and new therapeutics strategies such as immunotherapy. In this review, we summarize the techniques used for tumor omics analysis, recapitulate the key findings in cancer omics studies, and point to areas requiring further research on precision oncology.
In 2012, cancer was estimated to cause more than 8.2 million deaths worldwide, with 14.1 million new cases (1). It is expected to surpass heart diseases as the leading cause of death in the next few years in the United States (2). This heavy disease burden causes significant healthcare cost and diminished quality of life in both developed countries and developing economies alike (1). In vitro and in vivo studies identified a number of hallmarks of cancer, including self-sufficiency in growth signals, insensitivity to anti-growth signals, evading apoptosis, limitless replicative potential, sustained angiogenesis, and tissue invasion and metastasis (3). These hallmarks are continuously being refined with evolving research evidence (4).
Precision medicine is an approach that takes into account individual differences to guide disease prevention and treatment (5, 6), and oncology, i.e. the study of cancer, is the most prominent field targeted for precision medicine (7). In particular, the Precision Medicine Initiative aims to revolutionize how we improve health and treat disease, with the goal to “deliver the right treatment at the right time, every time, to the right person” (7, 8). Medical diagnostics and treatments have long been focused on the general principles that work for the majority of patients. However, the inter-individual differences are not addressed adequately for most diseases. For instance, cancer types are mostly defined by histopathology analysis, but patients suffering from the same type of cancer may have very different cancer driver mutations or tumor proteomic profiles, which lead to diverse response to chemotherapy and different prognosis (i.e. clinical course and outcome of disease). Without proper subtyping, these patients might be pooled together for clinical treatments without any consideration of their underlying causes of their particular diseases, resulting in potentially suboptimal choice of treatments. Precision oncology intended to better identify the inter-individual differences and to provide a better understanding of disease phenotypes and guide personalized treatment plans.
With the advent of omics technology and big data analytics, we can now gather detailed molecular information on the diseased cells, identify obscure patterns from the data effectively, and gather further insights into the biology of diseases and health states of individual patients (9). The recent availability of cancer “omics” data has created unique opportunities for characterizing the biological processes correlated with clinical phenotypes. Consortiums like The Cancer Genome Atlas (TCGA) (10) and International Cancer Genome Consortium (ICGC) (11) have profiled the genomic variation, DNA methylation landscapes (epigenomics), gene expression (transcriptomics), and protein expression as well as modification status (proteomics) by next-generation sequencing, protein arrays, mass spectrometry, and other high-throughput modalities for large numbers of patients (typically a hundred (proteomics) to several thousand (genomic and transcriptomic data)). Leveraging machine-learning methods, researchers are able to associate terabytes of data generated from high-throughput methods with clinically important phenotypes (12), such as drug responses or survival outcomes. The development of big data analytics methods and data integration frameworks would enable medical researchers to draw inferences from diverse types of omics information and to make accurate clinical predictions, which contributes to formulating personalized treatment plans for each patient (13).
A number of research articles have shown the potential utility of omics profiling in precision oncology (14). Although histopathology evaluation still serves as the backbone of most oncological diagnosis (15), recent research has indicated that omics information has the potential to complement and enhance pathology diagnosis. In particular, molecular profiles could provide additional information for tumor subtyping and identify previously unknown molecular aberrations of clinical importance (16). Thus, omics profiling holds the promise of augmenting cancer diagnosis and facilitating the development of personalized cancer management.
In this review, we summarize the utility of conventional clinical and pathology evaluations, illustrate the utility of omics profiling on precision oncology, and identify future research directions to better understand malignancies.
Conventional Oncology Assessments: Clinical and Pathology Evaluations
Clinical and pathology evaluation of tumor is indispensible to cancer detection, diagnosis, and formulating treatment plans. These assessments are part of the state-of-the-art practice (15): clinicians stage patients through medical imaging and tumor biopsy, and pathologists prepare microscopic slides from tissue samples obtained through surgery or biopsy, stain them with appropriate chemicals, review them under the microscope in detail, and describe their findings in pathology reports (17). For malignant cases, detailed microscopic evaluation is generally needed to assess the extent of tumor (18, 19) and the type of tumor (e.g. adenocarcinoma versus squamous cell carcinoma) (20), as well as to ascertain that the tumor is adequately removed during surgical excision (21).
Several qualitative annotations, such as tumor stage and grade, have clear clinical implications. Tumor stage is the evaluation of tumor spread, and the TNM staging system is the most widely used system for most cancers, such as breast cancer, prostate cancer, lung cancer, colorectal cancer, bladder cancer, and pancreatic cancer (22). There are three major components in the TNM system: tumor extent (T), lymph node involvement (N), and distant metastasis (M). An example of TNM staging criteria for non-small cell lung cancer is shown in Table I, and the mapping from T, N, and M status to stages is described in Table II (23). Note that different types of malignancy may have different staging systems (24), and there is no well-established TNM staging for brain cancer or malignancies of the spinal cord. The lack of staging for central nervous system (CNS)1 cancer is due to the fact that tumor histology and location of CNS tumor is a better prognostic predictor than tumor size; in addition, the CNS has no lymphatics, and most CNS tumor patients do not survive long enough to develop metastatic diseases (25). Tumor grade evaluates the level of tumor differentiation. Patients with higher-grade (less signs of differentiation) tumors often have worse survival outcomes (26, 27).
Table I. TNM staging of non-small cell lung cancer.
| T | ||
|---|---|---|
| T0 | No evidence of primary tumor. | |
| Tis | Carcinoma in situ. | |
| T1 | Tumor that is ≤3 cm in its greatest dimension, does not invade the visceral pleura, and is without bronchoscopic evidence of invasion more proximal than a lobar bronchus. The uncommon superficial spreading tumor of any size with its invasive component limited to the bronchial wall, which may extend proximal to the main bronchus, is classified as a T1a. | |
| T1a | Tumor is ≤2 cm in its greatest dimension. | |
| T1b | Tumor is >2 cm, but ≤3 cm, in its greatest dimension. | |
| T2 | Tumor with any of the following characteristics: >3 cm but ≤7 cm in its greatest dimension, invades a mainstem bronchus with its proximal extent at least 2 cm from the carina, invades the visceral pleura, or is associated with either atelectasis or obstructive pneumonitis that extends to the hilar region without involving the entire lung. | |
| T2a | Tumor is >3 cm, but ≤5 cm, in its greatest dimension. | |
| T2b | Tumor is >5 cm, but ≤7 cm, in its greatest dimension. | |
| T3 | Tumor with any of the following characteristics: >7 cm in its greatest dimension; invades the chest wall (including superior sulcus tumors), diaphragm, phrenic nerve, mediastinal pleura, parietal pericardium, or a mainstem bronchus less than 2 cm from the carina without invasion of the carina; is associated with either atelectasis or obstructive pneumonitis of the entire lung; or separate tumor nodule(s) are located in the same lung lobe as the primary tumor. | |
| T4 | Tumor of any size that invades the mediastinum, heart, great vessels, trachea, recurrent laryngeal nerve, esophagus, vertebral body, or carina; or separate tumor nodule(s) located in a different lobe of the ipsilateral lung. | |
| N | ||
| N0 | No regional lymph node involvement. | |
| N1 | Involvement of ipsilateral intrapulmonary, peribronchial, or hilar lymph nodes. | |
| N2 | Involvement of ipsilateral mediastinal or subcarinal lymph nodes. | |
| N3 | Involvement of contralateral mediastinal or hilar lymph nodes. Alternatively, involvement of either ipsilateral or contralateral scalene or supraclavicular lymph nodes. | |
| M | ||
| M0 | No distant metastasis. | |
| M1 | Metastasis. | |
| M1a | Malignant pleural effusion, pericardial effusion, pleural nodules, or metastatic nodules in the contralateral lung. | |
| M1b | Distant (extrathoracic) metastasis. |
Table II. Mapping T, N, and M status to non-small cell lung cancer stages.
| Tumor Stages | TNM |
|---|---|
| Stage 0 | TisN0M0 |
| Stage I | |
| IA | T1a-1bN0M0 |
| IB | T2aN0M0 |
| Stage II | |
| IIA | T1a-2aN1M0 or T2bN0M0 |
| IIB | T2bN1M0 or T3N0M0 |
| Stage III | |
| IIIA | T3N1M0 or T1a-3N2M0 or T4N0-1M0 |
| IIIB | T4N2M0 or T1a-4N3M0 |
| Stage IV | Any T Any N M1a-1b |
In addition to hematoxylin and eosin (H&E) stained slides, pathologists also use immunohistochemistry (IHC) to detect the presence of proteins and to semi-quantify protein expression levels (28). Previous research showed that it is possible to prioritize cancer marker candidates through the IHC semi-quantified protein levels (29).
Overall, histopathology evaluation defined cancer types and subtypes, and assessments on tumor grade and stage can stratify patients with different survival outcomes. However, these evaluations can be subjective (30, 31) and the results may not capture all of the clinically relevant inter-individual differences (32).
Omics Studies
The recent “omics revolution” provides great opportunities to link biological pathways to clinical phenotypes (33, 34). Advancements in omics profiling techniques enable researchers to view the panorama of the biological processes underpinning diseases and health status, which not only renders further insights into disease pathology (35), but also identifies biomarkers for clinical predictions (36). Discovering robust links between important clinical variables and their predictive features is the key to precision medicine (37). Here we discuss the clinical implications of genomics, epigenomics, transcriptomics, proteomics, and metabolomics information (Fig. 1), and illustrate how these findings could guide precision oncology.
Fig. 1.
Schematic diagram of omics modalities in precision oncology. Genomics, epigenomics, transcriptomics, proteomics, and metabolomics methods provide complementary information on the biology of tumorigenesis and cancer development.
Genomics
Genome sequencing provides the panorama of the DNA sequence changes of tumor tissues at single base pair resolution. By comparing tumor genome with a patient's germline sequence or a reference genome, researchers can identify genetic aberrations as well as their clinical implications (34, 38). Many of these variations are associated with clinically important phenotypes, such as response to targeted therapeutics (39) or survival outcomes (40).
As an illustration, in the past several years many drugs have been designed to target the proteins expressed from mutated genes in non-small cell lung tumors. For example, the therapeutic agents that target the effects of EGFR mutation (41), BRAF mutation (42), and MET amplification (43) have been designed—many initially for these mutations in other cancers (Table III) (44–50). As a result, numerous cancer patients are tested for their tumor genotypes before receiving targeted therapy (39), and the prognostic markers guide physicians in formulating treatment plans for individual patients (51). These advancements altered the clinical managements of malignancies tremendously (52).
Table III. Examples of genetic variations and associated targeted therapy agents for non-small cell lung cancer.
| Genes | Type of Variations | Estimated Prevalence in Lung Adenocarcinoma | Estimated Prevalence in Lung Squamous Cell Carcinoma | Targeted Therapy Agents |
|---|---|---|---|---|
| EGFR | Point mutations | 5–15% | <5% | Gefitinib, erlotinib, afatinib |
| ALK | Rearrangements | 5–15% | <5% | Crizotinib |
| MET | Amplification | 5% | 5% | Crizotinib |
| RET | Rearrangements | 1.8% | <1% | Cabozantinib |
| ROS1 | Rearrangements | 1.8% | 0% | Crizotinib |
| HER2 | Point mutations | <5% | 0% | Trastuzumab, afatinib |
| BRAF | Point mutations | <5% | 0% | Vemurafenib, dabrafenib |
In addition, genomics profiling rendered a systematic way toward understanding the biological processes underpinning important clinical phenotypes (53). Because tumor cells harbor many genetic variations, hundreds of genes can be associated with a phenotype by genomic analysis. Recent developments in pathway analysis provide effective ways to gather insights into the biology of the identified genes and proteins in cancer patients (54). As an illustration, pathway analysis of genes with recurrent somatic mutations revealed the role of Wnt/β-catenin signaling in carcinogenesis of hepatocellular carcinoma. For an individual tumor, through mapping a large number of altered genes or proteins into pathways, the dimensionality involved in the analyses can be reduced, which increases the explanatory power and facilitates biological interpretations. A few methods for conducting pathway analysis have been described (55). Researchers classify the most commonly used methods into three major categories: over-representation analysis, functional class scoring, and pathway topology. The design of effective pathway analysis algorithms is still an active area of research (55).
Epigenomics
Epigenomic changes, including DNA methylation and chromatin modifications, can affect the expression patterns of genes (56). DNA methylation is the reversible addition of a methyl group to DNA, which occurs most frequently on a cytosine adjacent to a guanine. DNA methylation profiles are heritable, and generally suppress gene expression if it occurred in the promoter regions (57). In addition to DNA methylation, there are a number of known chromatin modifications that result in epigenomic effects, including histone acetylation, methylation, phosphorylation, ubiquitination,SUMOylation, ADP-ribosylation, deimination, and proline isomerization (58). Depending on the particular histone modification, these alterations can have different effects on transcription. Cancer cells are known to exhibit many of these epigenomic changes in DNA methylation and chromatin modification (56), and profiling tools to investigate the status of many types of epigenetic modification are available (59).
Bisulfite treatment and sequencing is an effective method to identify the DNA methylation status at the single base pair resolution. This method works by using bisulfite to modify unmethylated cytosines to uracils, while sparing methylated cytosines (Fig. 2). By sequencing and comparing the sequences from bisulfite-treated as well as the untreated samples, researchers can identify the methylation status of each cytosine under study (60). A few large-scale studies used bisulfite-based methylation assay on human cancer samples. As an illustration, The Cancer Genome Atlas (TCGA) used Illumina's Infinium Human DNA Methylation 27 and Infinium Human DNA Methylation 450 platform to investigate the epigenomic landscape of more than 10 tumor types. These platforms can reveal the methylation status of 27,578 and more than 485,000 sites per sample at single-nucleotide resolution respectively (61).
Fig. 2.
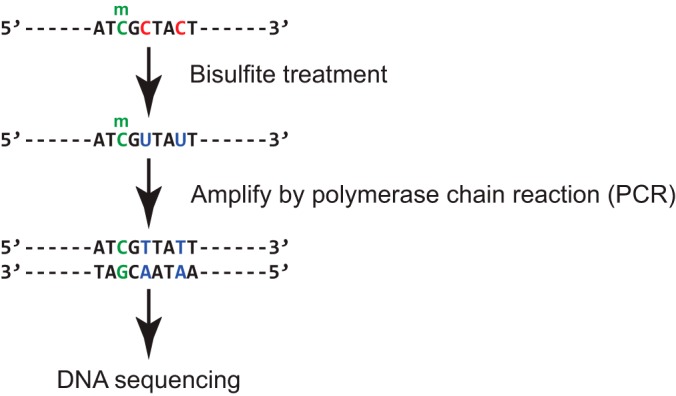
Bisulfite sequencing identifies cytosines with and without methylation at a single nucleotide resolution. Unmethylated cytosines (represented by “C” in the sequence) are converted to uracil (represented by “U”) by bisulfite treatment, which will be sequenced as thymine (represented by “T”). In contrast, methylated cytosines (5-methylcytosine; represented by “C” with a small “m” on the top) are resistant to bisulfite conversion, and will be sequenced as they are. By comparing the bisulfite treated and untreated samples, researchers can identify the methylation status and methylation rate of each cytosine at a single nucleotide resolution.
High-throughput DNA sequencing technologies coupled with chromatin immunoprecipitation (ChIP) methods are useful for identifying histone modifications (62). Using modification-specific antibodies, ChIP methods can immunoisolate DNA-histone complexes with desired histone modifications. The DNA sequences that interact with the modified histones can be identified through DNA microarrays (ChIP-chip) (63) or DNA sequencing (ChIP-seq) (64, 65).
DNA methylation patterns have been related to mutations in cancer driver genes in several cancer types, including colorectal cancer (66); indeed DNA methylation, demethylation and chromatin modification enzymes are often mutated in many types of cancer; for example, more than 17% of acute myeloid leukemia (AML) patients have mutations in DNA methyltransferase 3A (DNMT3A) gene (67). In addition, integrative studies on DNA methylation and gene expression data has revealed that CpG island methylation in promotors can explain the decreased gene expression patterns in a number of important genes (66). In some cancer types, the methylation signatures of selected genes were found to be prognostic and correlate with relapse-free survival of the patients (68, 69).
In addition, ChIP-seq methods revealed histone modification profiles in cancer, which can be linked to clinical phenotypes and inform tumor biology. As an illustration, ChIP-seq studies demonstrated that estrogen receptor, an important transcription factor affecting endocrine response and cell growth in breast cancer, has distinct binding patterns in breast cancer patients who are more likely to relapse (70). ChIP-chip analysis on cancer cell lines also shed light on the biological processes associated with tumor metastasis and aggressiveness (71, 72).
Transcriptomics
Contrasting with genomic and epigenomic studies, transcriptomic analyses focus on gene expression levels. Transcriptomics is the study of the complete set of mRNA transcripts in a cell and the quantity of each transcript (73). By assessing the amount of transcripts, researchers can estimate the gene expression levels in cells, which is a proxy of gene activity. Because of the good reproducibility of experiment modalities for transcriptomics analysis, it is a popular method to estimate gene activities in tumor cells (74).
RNA-sequencing (RNA-seq) is the current method of choice for profiling gene expression levels. It has several advantages over microarray studies: RNA-seq has low background noise, can identify a larger dynamic ranges of expression level, can distinguish among different isoforms and allelic expression, and is able to provide single base resolution and measurement of each transcript (73). The experimental procedure of a typical RNA-sequencing protocol involves the use of poly(T) magnetic beads to separate coding RNAs with poly(A) tails from noncoding RNAs, reverse transcription of RNAs to complementary DNAs (cDNAs), and sequencing of the resulting cDNAs (Fig. 3) (73). Developing different experimental protocols to profile RNAs with low-quantity or directly sequence RNAs without reverse transcription is still an active area of research (75).
Fig. 3.

A general workflow of RNA sequencing with reverse transcription to complementary DNA (cDNA). Coding mRNA molecules with poly-A tails are first isolated from the sample, and then reverse transcribed to cDNA. The cDNA is fragmented and sequenced, and the resulting sequence is mapped back onto the reference genome. The quantity of mapped sequences in each genic region is associated with the expression level of the gene. (Blue regions of the reference genome indicate introns, whereas red regions indicate exons.)
Before the advent of RNA-sequencing, microarrays were widely used to profile the transcriptomic landscape of cancerous tissues. One seminal study shows that the gene expression levels profiled by microarrays can distinguish different types of hematologic cancer (76). Because of the large quantities of DNA microarray data in the public domain, large repositories of microarray data still serve as important databases for research on drug repurposing (77) as well as disease re-classification (78, 79), although these repositories are becoming rapidly populated with RNA-Sequencing data sets.
Many reports demonstrate the utility of gene expression profiles for prognosis. Machine learning methods are the cornerstone of identifying nonobvious gene expression patterns associated with clinical phenotypes (Fig. 4). As an illustration, Beer et al. used gene expression patterns profiled by microarray to identify lung adenocarcinoma patients with different prognoses. They came up with a statistical model that predicts patient survival with gene expression features, which provides additional information for clinical managements (80). In addition, US patent 7,914,988 describes a 21-gene panel expression test for prostate cancer relapse prediction (81). Moreover, RNA-seq studies also reveal alternative splicing and fusion transcripts likely contributing to carcinogenesis in a number of cancers, including melanoma (82), breast cancer (83), and prostate adenocarcinoma (84).
Fig. 4.

A schematic diagram of using machine learning methods to predict clinical phenotypes. First, a training data set is collected, subsets of features associated with the phenotype of interest are selected, and a statistical model is built by the training data. A previously untouched test set using the same omics profiling methods is collected and treated as new input to the established machine learning model. The model provides predictions on the test input. By comparing the model output and the actual clinical phenotypes of the patients in the test set, researchers can estimate the performance of the prediction model.
Proteomics
Proteins are important building blocks of cells and they carry out essential functions in organisms. As malignant cells have distinct replication and metabolic processes, their protein quantities and activities are affected. Quantifying proteins and their modifications can determine different health and disease states. A number of high-throughput experimental methods are used to analyze the proteomic profiles of cancer, including mass spectrometry, protein arrays and antibody based-detection methods (85).
Mass spectrometry (MS) is a sensitive and robust method that quantifies peptide by their mass-to-charge (m/z) ratio (86). Companies have developed different types of mass spectrometers with different resolving power, sensitivity, dynamic range, throughput, and the ability to detect post-translational modifications for proteomics studies (87). The MS approach has many applications in cancer studies. As an illustration, MS studies reveal activated oncogenic kinases in non-small cell lung cancer samples and identify novel fusion proteins, such as ALK (88). Leveraging these crucial findings, researchers further demonstrate the effectiveness of Crizotinib, a tyrosine kinase inhibitor targeting ALK, MET, and ROS1 tyrosine kinases, in treating non-small cell lung cancer patients with ALK rearrangements (89). Protein profiling of TCGA cancer samples has stratified different colorectal subtypes that are overlapping but distinct from those identified by RNA-sequencing studies (90). Thus, new information can be obtained from global analyses of proteins.
Protein microarrays are another widely used analytical method for proteomics. There are two types of abundance-based protein arrays: capture arrays and reverse phase protein arrays (Fig. 5) (91). Capture arrays can be further classified into direct labeling and sandwich immunoassay. Direct labeling method labels proteins of interest with detectable markers, such as fluorescent probes, and captures the labeled proteins with antibodies fixed on a solid surface. This method can assay multiple samples at the same time, but requires chemically modifying the proteins (92). In addition, cross-reactive antibodies can cause false positives, which lower the specificity of the analysis. Sandwich immunoassay used two types of antibodies, one captures the proteins and the other carries the fluorescent molecule and binds to another epitope of the protein. This approach avoids labeling the proteins directly and has higher specificity; however, it requires two distinct of antibodies to profile each protein (91).
Fig. 5.
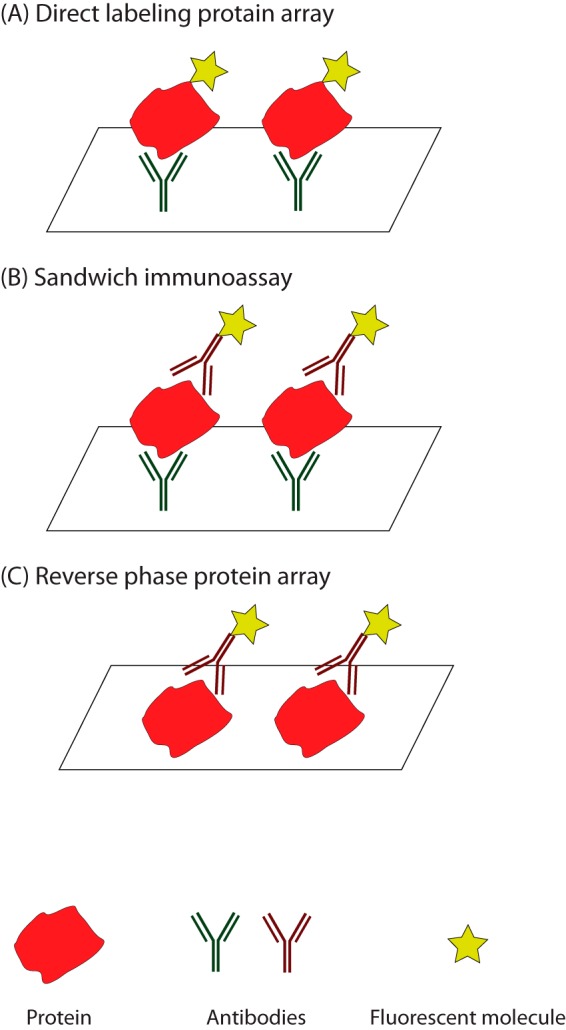
Protein array methods for proteomic profiling. A, Direct labeling method adds detectable markers to the proteins, and uses antibodies fixed on a solid surface to capture the proteins of interest. B, Sandwich method utilizes two types of antibodies to capture the proteins and to tag on the fluorescent molecules respectively. C. Reverse phase protein array method first prints protein lysate on a solid surface, and then uses antibodies to quantify the proteins of interest.
Reverse-phased protein array prints protein lysate to a solid surface, and introduces primary and secondary antibodies to quantify the proteins of interest (93, 94). This method allows researchers to screen many samples efficiently, but has a narrower dynamic range of detectable protein abundance (91). These array-based methods are also proved useful for cancer biomarker discovery. As an illustration, antibody arrays analysis reveals that IL-8 and growth-related oncogene (GRO) cytokines levels are potential biomarkers for monitoring response to HER2-targeted therapy in breast cancer (95). Data gathered from reverse-phase protein array also suggests that subsets of ovarian cancer patients can benefit from a combination of KIT and cyclin E2 inhibitors or a combination of PI3K and MAPK inhibitors (96).
Metabolomics
Metabolomics is defined as the study of the collection of metabolites in a system (e.g. cell, tissue, or organism) under a given set of conditions (97). Cancer cells have different metabolism from normal cells and use different metabolic pathways than normal cells: it is well established that most cancer cells generate energy by glycolysis regardless of the availability of oxygen, instead of using mitochondrial oxidative phosphorylation that noncancer cells use (the Warburg Effect) (98). With the advancement in high-throughput profiling tools for metabolites, metabolomics studies are expected to bring in further insights into cancer biology and biomarker discovery (99).
For metabolomics studies researchers typically use two major technologies, MS and nuclear magnetic resonance (NMR) (100, 101). MS can perform both targeted and untargeted analyses. Targeted analyses follow known molecules (typically one to several hundred) and can provide very sensitive quantification of key known compounds. However, it will miss the many metabolites not targeted. Untargeted metabolomics profile many thousands of features (molecules of particular column retention times and molecular mass) globally and can discover novel biomarkers found in specific conditions and thus identify new targets (100, 102). One-dimensional (1D) NMR can also profile the metabolites from blood plasma, urine, saliva, and tissue extracts (103, 104). NMR in two-dimensional (2D) mode can elucidate the molecular structure and facilitate molecule identification with increased signal dispersion (103). Recent advancement in the NMR technology has improved its detection sensitivity (105) and the availability of extensive NMR spectral databases has facilitated the identification of molecules (106). This fast and automated approach can be useful for clinical diagnosis and toxicological studies (103).
Metabolomics analyses can reveal cancer biology and detect cancer in a noninvasive fashion. For instance, metabolomics assays have identified the role of serine consumption in nucleotide synthesis, one-carbon metabolism, and cell proliferation in cancer cell lines (107). Another study shows that the serum concentration of a number of free fatty acids is different between breast invasive ductal carcinoma patients and healthy controls. These results not only provide potential biomarkers for cancer diagnosis, but also point to metabolic alterations associated with cancer development (108).
Integrative Omics Studies for Precision Oncology
The different omics described above characterize biomolecules at different levels. With an aim to incorporate information from different omics studies, integrative omics analyses account for various omics information to provide a more holistic view of cancer biology, as well as to generate better predictions for clinical phenotypes. A number of omics integration algorithms and tools are available for data exploration, analysis, and integration (109, 110).
For instance, one study investigated the somatic mutations from whole-exome sequencing, copy number alterations, DNA methylation, and mRNA levels quantified by RNA-sequencing of 3 299 tumor samples from 12 cancer types (111). In this analysis, 479 genetic and epigenetic alterations with concordant changes in gene expression are identified. Hierarchical classification shows that the majority of these tumors are either driven by somatic mutations or copy number variations. In addition, a number of genes in cell cycle signaling pathways, including TP53 and PIK3CA, have both mutations and copy number aberrations. These results characterize the potential driving events in cancer, and portray the global molecular aberrations in malignancy across tumor types (112).
In addition, the multi-omics integration can better identify molecular patterns associated with important clinical phenotypes (14). As an illustration, through incorporating the genomic and transcriptomic profiles of breast cancer patients, researchers define a novel 10-subtype classification system for the tumor. Each subtype is associated with distinct clinical characteristics and survival outcomes (113). With omics integration, we can better understand the key molecules in cancer development, and provide better clinical predictions. Proteomics, which is only getting incorporated into these analyses now, has the potential to greatly expand these studies.
Omics in Cancer Immunotherapy
Cancer immunotherapy is a treatment that uses patients' immune system to control or eliminate malignant cells and involves multiple technologies (114). This type of treatment exploits the fact that genetic aberrations in cancer cells can result in new peptides that are not normally expressed in benign cells (115). Some of the new peptides are transported to the cell surface, and the immune system is able to recognize these cancer cell-specific antigens (Fig. 6). Because cancer immunotherapy acts specifically on malignant cells, the side effects of immunotherapy are less severe. With the recent successful clinical trial on late-stage metastatic melanoma patients with no other treatment options, immunotherapy has gained much attention in recent years (116).
Fig. 6.
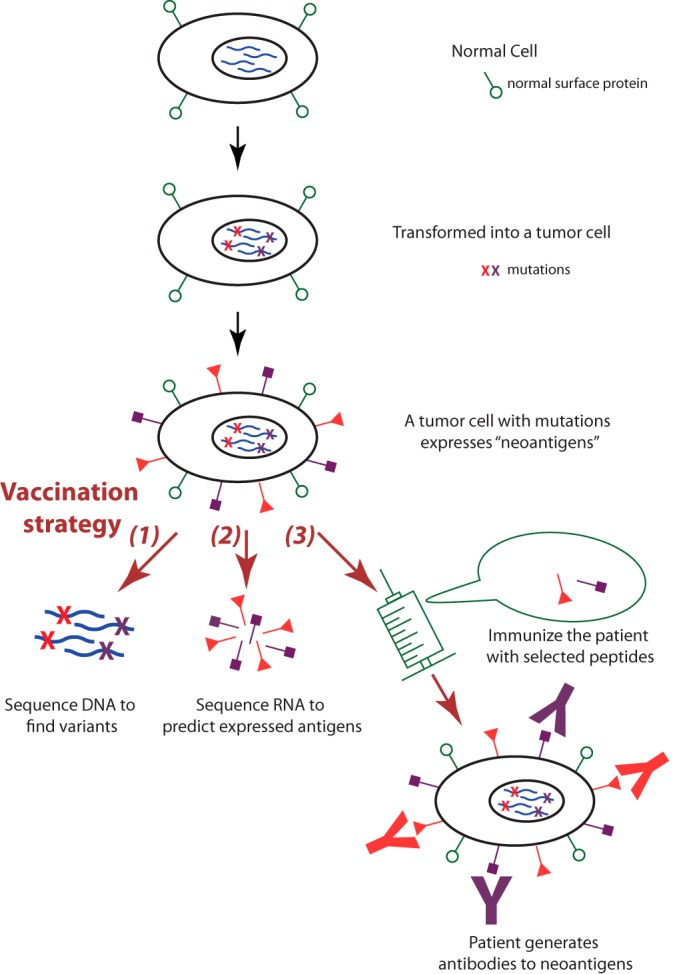
Omics applications in cancer immunotherapy. Cancer immunotherapy exploits the fact that genetic aberrations in the cancer genome can result in new antigens (neoantigens) not normally expressed in benign tissue. Researchers can sequence the tumor genome to identify potential neoantigens, use proteomic methods to characterize the expressed neoantigens, and design personalized cancer vaccines based on the identified neoantigens, which will elicit specific immune response against the tumor cells.
There are a few places where omics studies can facilitate the development of immunotherapy. First, genomic analysis is used to determine the genetic mutations leading to potentially actionable neoantigens. Second, proteomic methods can characterize neopeptides on the surface of the tumor cells. Identifying the expressed neoantigens using RNA-seq or proteomics methods is also useful for selecting immunotherapy regimens. Third, researchers can design personalized vaccines based on the identified neoantigens presented in the tumor. The introduced antigens in vaccines can trigger immune responses in the patients, prompting B cells to generate antibodies against the cancer cells (115).
In addition to providing insights into the design of immunotherapy, omics signatures can predict immunotherapy response as well. For instance, biomarkers in the peripheral blood can quantify the strength of immune response, identify the extent of epitope spreading, and detect autoimmunity. A number of biomarkers successfully predict the response to immunotherapy in clinical trials of many tumor types (117).
Challenges and Future Directions
Despite a considerable amount of research on cancer omics, our current knowledge of the molecular mechanisms of cancer biology is limited and the implementation of precision oncology is still far from perfect. As a first example, although mutations can be identified by genome sequencing, the driver mutations in a number of cancer patients are still unknown (118). As a second example, tumor tumors are heterogeneous (119) and constantly evolving (120), and the complete mutational spectrum of truncal and branch mutations in heterogeneous cancers are difficult to ascertain (121). Evolution of tumor cells can lead to acquired drug resistance and temporal variation of tumor omics (122). Heterogeneity may also account for different biomarker expression: for examples, some genes in the Oncotype DX® assay, a prognostic test for node-negative, estrogen receptor-positive breast cancer, showed variable expression levels in different tumor sections from the same patient (123). As a third example, some forms of chemotherapy and immunotherapy only work in a fraction of patients, and the biological mechanism underpinning treatment responses for many types of cancer remains largely unexplored (117, 124). It is possible to identify nonobvious omics patterns predictive of treatment efficacy, but it requires large cohorts to build and test the newly established omics signatures (125). Presently, the correlations between different omics modalities or between histopathology phenotypes and omics features are not systematically characterized, and it is possible that the integration of pathology and multi-omics can provide further information for precision oncology (126). Further research is needed to identify the additional driver mutations in cancers, to provide robust biomarkers for predicting treatment responses of different treatment modalities, and to show how different omics relate to one another. These integrative studies require scalable bioinformatics approaches to identify unrecognized genomic architectures and international collaborations to gather large patient cohorts that account for individual variations and population differences. With these studies, we can better translate biomedical discoveries from bench to bedside.
CONCLUSION
High throughput omics methods have greatly facilitated the development of precision oncology and are beginning to guide personalized cancer management. Here we summarize the key omics modalities useful for identifying clinical phenotypes, such as tumor types and subtypes, drug responses, and survival outcomes. Omics technology can complement current clinical and pathology evaluations by discovering previously unknown subtypes with clinical implications, identifying patients' prognoses, or predicting responses to treatments. Future studies on cancer mutations, functional aberrations, and omics integration have the potential to further improve the precision in precision medicine.
Footnotes
Author contributions: K.Y. designed research; K.Y. performed research; K.Y. analyzed data; K.Y. and M.S. wrote the paper.
* This work was supported by National Institutes of Health Grant 5U24CA160036-05. K.-H. Y. is a Howard Hughes Medical Institute International Student Research Fellow and Winston Chen Stanford Graduate Fellow. We thank the anonymous reviewers for their constructive comments. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
1 The abbreviations used are:
- CNS
- central nervous system
- ChIP
- chromatin immunoprecipitation
- RNA-seq
- RNA-sequencing
- NMR
- nuclear magnetic resonance
- FT-IR
- Fourier transform infrared spectroscopy
- GC-MS
- gas chromatography mass spectrometry
- LC-MS
- liquid chromatography mass spectrometry.
REFERENCES
- 1. Torre L. A., Bray F., Siegel R. L., Ferlay J., Lortet-Tieulent J., and Jemal A. (2015) Global cancer statistics, 2012. CA 65, 87–108 [DOI] [PubMed] [Google Scholar]
- 2. Siegel R. L., Miller K. D., and Jemal A. (2015) Cancer statistics, 2015. CA 65, 5–29 [DOI] [PubMed] [Google Scholar]
- 3. Hanahan D., and Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 4. Hanahan D., and Weinberg R. A. (2000) The hallmarks of cancer. Cell 100, 57–70 [DOI] [PubMed] [Google Scholar]
- 5. National Research Council Committee on A Framework for Developing a New Taxonomy of Disease (2011) Toward precision medicine: Building a knowledge network for biomedical research and a new taxonomy of disease, National Academies Press (US) [PubMed] [Google Scholar]
- 6. Snyder M. (2016) Genomics and Personalized Medicine: What Everyone Needs to Know, Oxford University Press [Google Scholar]
- 7. Collins F. S., and Varmus H. (2015) A new initiative on precision medicine. The New England journal of medicine 372, 793–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ashley E. A. (2015) The precision medicine initiative: a new national effort. JAMA 313, 2119–2120 [DOI] [PubMed] [Google Scholar]
- 9. Holzinger A., Dehmer M., and Jurisica I. (2014) Knowledge Discovery and interactive Data Mining in Bioinformatics–State-of-the-Art, future challenges and research directions. BMC bioinformatics 15, I1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cancer Genome Atlas Research Network, Weinstein J. N., Collisson E. A., Mills G. B., Shaw K. R., Ozenberger B. A., Ellrott K., Shmulevich I., Sander C., and Stuart J. M. (2013) The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genetics 45, 1113–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang J., Baran J., Cros A., Guberman J. M., Haider S., Hsu J., Liang Y., Rivkin E., Wang J., Whitty B., Wong-Erasmus M., Yao L., and Kasprzyk A. (2011) International Cancer Genome Consortium Data Portal–a one-stop shop for cancer genomics data. Database 2011, bar026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Larranaga P., Calvo B., Santana R., Bielza C., Galdiano J., Inza I., Lozano J. A., Armananzas R., Santafe G., Perez A., and Robles V. (2006) Machine learning in bioinformatics. Briefings Bioinformatics 7, 86–112 [DOI] [PubMed] [Google Scholar]
- 13. Bellazzi R., and Zupan B. (2008) Predictive data mining in clinical medicine: current issues and guidelines. Int. J. Medical Informatics 77, 81–97 [DOI] [PubMed] [Google Scholar]
- 14. Vucic E. A., Thu K. L., Robison K., Rybaczyk L. A., Chari R., Alvarez C. E., and Lam W. L. (2012) Translating cancer ‘omics’ to improved outcomes. Genome Res. 22, 188–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lakhani S. R., and Ashworth A. (2001) Microarray and histopathological analysis of tumours: the future and the past? Nat. Rev. Cancer 1, 151–157 [DOI] [PubMed] [Google Scholar]
- 16. Clinical Lung Cancer Genome, P., and Network Genomic, M. (2013) A genomics-based classification of human lung tumors. Sci. Translational Med. 5, 209ra153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Abeloff M., Armitage J., Niederhuber J., Kastan M., and Mckenna W. (2004) Clinical Oncology. Philadelphia, PA: Churchill Livingstone. Elsevier [Google Scholar]
- 18. Roberts T. E., Hasleton P. S., Musgrove C., Swindell R., and Lawson R. A. (1992) Vascular invasion in non-small cell lung carcinoma. J. Clin. Pathol. 45, 591–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ogawa J., Tsurumi T., Yamada S., Koide S., and Shohtsu A. (1994) Blood vessel invasion and expression of sialyl Lewisx and proliferating cell nuclear antigen in stage I non-small cell lung cancer. Relation to postoperative recurrence. Cancer 73, 1177–1183 [DOI] [PubMed] [Google Scholar]
- 20. Kumar V., Abbas A. K., Fausto N., and Aster J. C. (2014) Robbins and cotran pathologic basis of disease, Professional Edition: Expert Consult-Online, Elsevier Health Sciences [Google Scholar]
- 21. Baish J. W., and Jain R. K. (2000) Fractals and cancer. Cancer Res. 60, 3683–3688 [PubMed] [Google Scholar]
- 22. Sobin L. H., Gospodarowicz M. K., Wittekind C., and International Union against Cancer. (2010) TNM classification of malignant tumours, 7th Ed., Wiley-Blackwell, Chichester, West Sussex, UK; ; Hoboken, NJ [Google Scholar]
- 23. Goldstraw P., Crowley J., Chansky K., Giroux D. J., Groome P. A., Rami-Porta R., Postmus P. E., Rusch V., Sobin L., International Association for the Study of Lung Cancer International Staging, C., and Participating, I. (2007) The IASLC Lung Cancer Staging Project: proposals for the revision of the TNM stage groupings in the forthcoming (seventh) edition of the TNM Classification of malignant tumours. J. Thoracic Oncol. 2, 706–714 [DOI] [PubMed] [Google Scholar]
- 24. Greene F. L. (2002) AJCC cancer staging manual, Springer Science & Business Media [Google Scholar]
- 25. Edge S. B., and American Joint Committee on Cancer. (2010) AJCC cancer staging manual, 7th Ed., Springer, New York: [DOI] [PubMed] [Google Scholar]
- 26. Gronchi A., Miceli R., Shurell E., Eilber F. C., Eilber F. R., Anaya D. A., Kattan M. W., Honore C., Lev D. C., Colombo C., Bonvalot S., Mariani L., and Pollock R. E. (2013) Outcome prediction in primary resected retroperitoneal soft tissue sarcoma: histology-specific overall survival and disease-free survival nomograms built on major sarcoma center data sets. J. Clin. Oncol. 31, 1649–1655 [DOI] [PubMed] [Google Scholar]
- 27. Delahunt B., McKenney J. K., Lohse C. M., Leibovich B. C., Thompson R. H., Boorjian S. A., and Cheville J. C. (2013) A novel grading system for clear cell renal cell carcinoma incorporating tumor necrosis. Am. J. Surg. Pathol. 37, 311–322 [DOI] [PubMed] [Google Scholar]
- 28. Ramos-Vara J. A., and Miller M. A. (2014) When tissue antigens and antibodies get along: revisiting the technical aspects of immunohistochemistry–the red, brown, and blue technique. Veterinary Pathol. 51, 42–87 [DOI] [PubMed] [Google Scholar]
- 29. Chiang S. C., Han C. L., Yu K. H., Chen Y. J., and Wu K. P. (2013) Prioritization of cancer marker candidates based on the immunohistochemistry staining images deposited in the human protein atlas. PloS One 8, e81079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stang A., Pohlabeln H., Muller K. M., Jahn I., Giersiepen K., and Jockel K. H. (2006) Diagnostic agreement in the histopathological evaluation of lung cancer tissue in a population-based case-control study. Lung Cancer 52, 29–36 [DOI] [PubMed] [Google Scholar]
- 31. Grilley-Olson J. E., Hayes D. N., Moore D. T., Leslie K. O., Wilkerson M. D., Qaqish B. F., Hayward M. C., Cabanski C. R., Yin X., Socinski M. A., Stinchcombe T. E., Thorne L. B., Allen T. C., Banks P. M., Beasley M. B., Borczuk A. C., Cagle P. T., Christensen R., Colby T. V., Deblois G. G., Elmberger G., Graziano P., Hart C. F., Jones K. D., Maia D. M., Miller C. R., Nance K. V., Travis W. D., and Funkhouser W. K. (2013) Validation of interobserver agreement in lung cancer assessment: hematoxylin-eosin diagnostic reproducibility for non-small cell lung cancer: the 2004 World Health Organization classification and therapeutically relevant subsets. Arch. Pathol. Lab. Med. 137, 32–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wall D. P., and Tonellato P. J. (2012) The future of genomics in pathology. F1000 Med. Reports 4, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hayes D. F. (2013) OMICS-based personalized oncology: if it is worth doing, it is worth doing well! BMC Med. 11, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vogelstein B., Papadopoulos N., Velculescu V. E., Zhou S., Diaz L. A. Jr., and Kinzler K. W. (2013) Cancer genome landscapes. Science 339, 1546–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gehlenborg N., O'Donoghue S. I., Baliga N. S., Goesmann A., Hibbs M. A., Kitano H., Kohlbacher O., Neuweger H., Schneider R., Tenenbaum D., and Gavin A. C. (2010) Visualization of omics data for systems biology. Nat. Methods 7, S56–68 [DOI] [PubMed] [Google Scholar]
- 36. McShane L. M., Cavenagh M. M., Lively T. G., Eberhard D. A., Bigbee W. L., Williams P. M., Mesirov J. P., Polley M. Y., Kim K. Y., Tricoli J. V., Taylor J. M., Shuman D. J., Simon R. M., Doroshow J. H., and Conley B. A. (2013) Criteria for the use of omics-based predictors in clinical trials. Nature 502, 317–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gonzalez de Castro D., Clarke P. A., Al-Lazikani B., and Workman P. (2013) Personalized cancer medicine: molecular diagnostics, predictive biomarkers, and drug resistance. Clin. Pharmacol. Therapeutics 93, 252–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jones S., Anagnostou V., Lytle K., Parpart-Li S., Nesselbush M., Riley D. R., Shukla M., Chesnick B., Kadan M., Papp E., Galens K. G., Murphy D., Zhang T., Kann L., Sausen M., Angiuoli S. V., Diaz L. A. Jr., and Velculescu V. E. (2015) Personalized genomic analyses for cancer mutation discovery and interpretation. Sci. Translational Med. 7, 283ra253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sawyers C. (2004) Targeted cancer therapy. Nature 432, 294–297 [DOI] [PubMed] [Google Scholar]
- 40. Liu B., Yang L., Huang B., Cheng M., Wang H., Li Y., Huang D., Zheng J., Li Q., Zhang X., Ji W., Zhou Y., and Lu J. (2012) A functional copy-number variation in MAPKAPK2 predicts risk and prognosis of lung cancer. Am. J. Human Gen. 91, 384–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Paez J. G., Janne P. A., Lee J. C., Tracy S., Greulich H., Gabriel S., Herman P., Kaye F. J., Lindeman N., Boggon T. J., Naoki K., Sasaki H., Fujii Y., Eck M. J., Sellers W. R., Johnson B. E., and Meyerson M. (2004) EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304, 1497–1500 [DOI] [PubMed] [Google Scholar]
- 42. Bollag G., Tsai J., Zhang J., Zhang C., Ibrahim P., Nolop K., and Hirth P. (2012) Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discovery 11, 873–886 [DOI] [PubMed] [Google Scholar]
- 43. Tanizaki J., Okamoto I., Okamoto K., Takezawa K., Kuwata K., Yamaguchi H., and Nakagawa K. (2011) MET tyrosine kinase inhibitor crizotinib (PF-02341066) shows differential antitumor effects in non-small cell lung cancer according to MET alterations. J. Thoracic Oncol. 6, 1624–1631 [DOI] [PubMed] [Google Scholar]
- 44. Pao W., and Girard N. (2011) New driver mutations in non-small-cell lung cancer. Lancet. Oncol. 12, 175–180 [DOI] [PubMed] [Google Scholar]
- 45. Kawakami H., Okamoto I., Okamoto W., Tanizaki J., Nakagawa K., and Nishio K. (2014) Targeting MET amplification as a new oncogenic driver. Cancers 6, 1540–1552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Heist R. S., Sequist L. V., and Engelman J. A. (2012) Genetic changes in squamous cell lung cancer: a review. J. Thoracic Oncol. 7, 924–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kohno T., Tsuta K., Tsuchihara K., Nakaoku T., Yoh K., and Goto K. (2013) RET fusion gene: translation to personalized lung cancer therapy. Cancer Sci. 104, 1396–1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Scheffler M., Schultheis A., Teixido C., Michels S., Morales-Espinosa D., Viteri S., Hartmann W., Merkelbach-Bruse S., Fischer R., Schildhaus H. U., Fassunke J., Sebastian M., Serke M., Kaminsky B., Randerath W., Gerigk U., Ko Y. D., Kruger S., Schnell R., Rothe A., Kropf-Sanchen C., Heukamp L., Rosell R., Buttner R., and Wolf J. (2015) ROS1 rearrangements in lung adenocarcinoma: prognostic impact, therapeutic options and genetic variability. Oncotarget 6, 10577–10585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhao W., Choi Y.-L., Song J.-Y., Zhu Y., Xu Q., Zhang F., Jiang L., Cheng J., Zheng G., and Mao M. (2016) ALK, ROS1 and RET Rearrangements in Lung Squamous Cell Carcinoma Are Very Rare. Lung Cancer 94, 22–27 [DOI] [PubMed] [Google Scholar]
- 50. National Comprehensive Cancer Network (2016) NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines) Non-Small Cell Lung Cancer. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines) National Comprehensive Cancer Network [Google Scholar]
- 51. Singhal S., Vachani A., Antin-Ozerkis D., Kaiser L. R., and Albelda S. M. (2005) Prognostic implications of cell cycle, apoptosis, and angiogenesis biomarkers in non-small cell lung cancer: a review. Clin. Cancer Res. 11, 3974–3986 [DOI] [PubMed] [Google Scholar]
- 52. Chin L., and Gray J. W. (2008) Translating insights from the cancer genome into clinical practice. Nature 452, 553–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen R., Mias G. I., Li-Pook-Than J., Jiang L., Lam H. Y., Chen R., Miriami E., Karczewski K. J., Hariharan M., Dewey F. E., Cheng Y., Clark M. J., Im H., Habegger L., Balasubramanian S., O'Huallachain M., Dudley J. T., Hillenmeyer S., Haraksingh R., Sharon D., Euskirchen G., Lacroute P., Bettinger K., Boyle A. P., Kasowski M., Grubert F., Seki S., Garcia M., Whirl-Carrillo M., Gallardo M., Blasco M. A., Greenberg P. L., Snyder P., Klein T. E., Altman R. B., Butte A. J., Ashley E. A., Gerstein M., Nadeau K. C., Tang H., and Snyder M. (2012) Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell 148, 1293–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Papin J. A., Stelling J., Price N. D., Klamt S., Schuster S., and Palsson B. O. (2004) Comparison of network-based pathway analysis methods. Trends Biotechnol. 22, 400–405 [DOI] [PubMed] [Google Scholar]
- 55. Khatri P., Sirota M., and Butte A. J. (2012) Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Computational Biol. 8, e1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Iacobuzio-Donahue C. A. (2009) Epigenetic changes in cancer. Ann. Rev. Pathol. 4, 229–249 [DOI] [PubMed] [Google Scholar]
- 57. Jones P. A., and Takai D. (2001) The role of DNA methylation in mammalian epigenetics. Science 293, 1068–1070 [DOI] [PubMed] [Google Scholar]
- 58. Kouzarides T. (2007) Chromatin modifications and their function. Cell 128, 693–705 [DOI] [PubMed] [Google Scholar]
- 59. Hyun B. R., McElwee J. L., and Soloway P. D. (2015) Single molecule and single cell epigenomics. Methods 72, 41–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Frommer M., McDonald L. E., Millar D. S., Collis C. M., Watt F., Grigg G. W., Molloy P. L., and Paul C. L. (1992) A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. U.S.A. 89, 1827–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. National Cancer Institute TCGA Wiki. (2015) DNA methylation. Retrieved April 15, 2016, from https://wiki.nci.nih.gov/display/TCGA/DNA+methylation
- 62. Collas P. (2010) The current state of chromatin immunoprecipitation. Mol. Biotechnol. 45, 87–100 [DOI] [PubMed] [Google Scholar]
- 63. Buck M. J., and Lieb J. D. (2004) ChIP-chip: considerations for the design, analysis, and application of genome-wide chromatin immunoprecipitation experiments. Genomics 83, 349–360 [DOI] [PubMed] [Google Scholar]
- 64. Park P. J. (2009) ChIP-seq: advantages and challenges of a maturing technology. Nature reviews. Genetics 10, 669–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Euskirchen G. M., Rozowsky J. S., Wei C. L., Lee W. H., Zhang Z. D., Hartman S., Emanuelsson O., Stolc V., Weissman S., Gerstein M. B., Ruan Y., and Snyder M. (2007) Mapping of transcription factor binding regions in mammalian cells by ChIP: comparison of array- and sequencing-based technologies. Genome Res. 17, 898–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hinoue T., Weisenberger D. J., Lange C. P., Shen H., Byun H. M., Van Den Berg D., Malik S., Pan F., Noushmehr H., van Dijk C. M., Tollenaar R. A., and Laird P. W. (2012) Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 22, 271–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Thol F., Damm F., Ludeking A., Winschel C., Wagner K., Morgan M., Yun H., Gohring G., Schlegelberger B., Hoelzer D., Lubbert M., Kanz L., Fiedler W., Kirchner H., Heil G., Krauter J., Ganser A., and Heuser M. (2011) Incidence and prognostic influence of DNMT3A mutations in acute myeloid leukemia. J. Clin. Oncol. 29, 2889–2896 [DOI] [PubMed] [Google Scholar]
- 68. Sandoval J., Mendez-Gonzalez J., Nadal E., Chen G., Carmona F. J., Sayols S., Moran S., Heyn H., Vizoso M., Gomez A., Sanchez-Cespedes M., Assenov Y., Muller F., Bock C., Taron M., Mora J., Muscarella L. A., Liloglou T., Davies M., Pollan M., Pajares M. J., Torre W., Montuenga L. M., Brambilla E., Field J. K., Roz L., Lo Iacono M., Scagliotti G. V., Rosell R., Beer D. G., and Esteller M. (2013) A prognostic DNA methylation signature for stage I non-small-cell lung cancer. J. Clin. Oncol. 31, 4140–4147 [DOI] [PubMed] [Google Scholar]
- 69. Maruyama R., Toyooka S., Toyooka K. O., Virmani A. K., Zochbauer-Muller S., Farinas A. J., Minna J. D., McConnell J., Frenkel E. P., and Gazdar A. F. (2002) Aberrant promoter methylation profile of prostate cancers and its relationship to clinicopathological features. Clin. Cancer Res. 8, 514–519 [PubMed] [Google Scholar]
- 70. Ross-Innes C. S., Stark R., Teschendorff A. E., Holmes K. A., Ali H. R., Dunning M. J., Brown G. D., Gojis O., Ellis I. O., Green A. R., Ali S., Chin S. F., Palmieri C., Caldas C., and Carroll J. S. (2012) Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Seligson D. B., Horvath S., McBrian M. A., Mah V., Yu H., Tze S., Wang Q., Chia D., Goodglick L., and Kurdistani S. K. (2009) Global levels of histone modifications predict prognosis in different cancers. Am. J. Pathol. 174, 1619–1628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gupta R. A., Shah N., Wang K. C., Kim J., Horlings H. M., Wong D. J., Tsai M. C., Hung T., Argani P., Rinn J. L., Wang Y., Brzoska P., Kong B., Li R., West R. B., van de Vijver M. J., Sukumar S., and Chang H. Y. (2010) Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, 1071–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wang Z., Gerstein M., and Snyder M. (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nature Rev. Genetics 10, 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Marioni J. C., Mason C. E., Mane S. M., Stephens M., and Gilad Y. (2008) RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 18, 1509–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ozsolak F., and Milos P. M. (2011) RNA sequencing: advances, challenges and opportunities. Nature Rev. Genetics 12, 87–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Golub T. R., Slonim D. K., Tamayo P., Huard C., Gaasenbeek M., Mesirov J. P., Coller H., Loh M. L., Downing J. R., Caligiuri M. A., Bloomfield C. D., and Lander E. S. (1999) Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 286, 531–537 [DOI] [PubMed] [Google Scholar]
- 77. Iorio F., Rittman T., Ge H., Menden M., and Saez-Rodriguez J. (2013) Transcriptional data: a new gateway to drug repositioning? Drug Discovery Today 18, 350–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Quackenbush J. (2006) Microarray analysis and tumor classification. New Engl. J. Med. 354, 2463–2472 [DOI] [PubMed] [Google Scholar]
- 79. Huang H., Liu C. C., and Zhou X. J. (2010) Bayesian approach to transforming public gene expression repositories into disease diagnosis databases. Proc. Natl. Acad. Sci. U.S.A. 107, 6823–6828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Beer D. G., Kardia S. L., Huang C. C., Giordano T. J., Levin A. M., Misek D. E., Lin L., Chen G., Gharib T. G., Thomas D. G., Lizyness M. L., Kuick R., Hayasaka S., Taylor J. M., Iannettoni M. D., Orringer M. B., and Hanash S. (2002) Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nature Med. 8, 816–824 [DOI] [PubMed] [Google Scholar]
- 81. Chudin E., Lozach J., Fan J.-B., and Bibikova M. (2011) Gene expression profiles to predict relapse of prostate cancer. In: U.S. Patent and Trademark Office, ed., US [Google Scholar]
- 82. Berger M. F., Levin J. Z., Vijayendran K., Sivachenko A., Adiconis X., Maguire J., Johnson L. A., Robinson J., Verhaak R. G., Sougnez C., Onofrio R. C., Ziaugra L., Cibulskis K., Laine E., Barretina J., Winckler W., Fisher D. E., Getz G., Meyerson M., Jaffe D. B., Gabriel S. B., Lander E. S., Dummer R., Gnirke A., Nusbaum C., and Garraway L. A. (2010) Integrative analysis of the melanoma transcriptome. Genome Res. 20, 413–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Edgren H., Murumagi A., Kangaspeska S., Nicorici D., Hongisto V., Kleivi K., Rye I. H., Nyberg S., Wolf M., Borresen-Dale A. L., and Kallioniemi O. (2011) Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome Biol. 12, R6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Nacu S., Yuan W., Kan Z., Bhatt D., Rivers C. S., Stinson J., Peters B. A., Modrusan Z., Jung K., Seshagiri S., and Wu T. D. (2011) Deep RNA sequencing analysis of readthrough gene fusions in human prostate adenocarcinoma and reference samples. BMC Med. Genomics 4, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Tyers M., and Mann M. (2003) From genomics to proteomics. Nature 422, 193–197 [DOI] [PubMed] [Google Scholar]
- 86. Aebersold R., and Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 [DOI] [PubMed] [Google Scholar]
- 87. Domon B., and Aebersold R. (2006) Mass spectrometry and protein analysis. Science 312, 212–217 [DOI] [PubMed] [Google Scholar]
- 88. Rikova K., Guo A., Zeng Q., Possemato A., Yu J., Haack H., Nardone J., Lee K., Reeves C., Li Y., Hu Y., Tan Z., Stokes M., Sullivan L., Mitchell J., Wetzel R., Macneill J., Ren J. M., Yuan J., Bakalarski C. E., Villen J., Kornhauser J. M., Smith B., Li D., Zhou X., Gygi S. P., Gu T. L., Polakiewicz R. D., Rush J., and Comb M. J. (2007) Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 131, 1190–1203 [DOI] [PubMed] [Google Scholar]
- 89. Shaw A. T., Kim D. W., Nakagawa K., Seto T., Crino L., Ahn M. J., De Pas T., Besse B., Solomon B. J., Blackhall F., Wu Y. L., Thomas M., O'Byrne K. J., Moro-Sibilot D., Camidge D. R., Mok T., Hirsh V., Riely G. J., Iyer S., Tassell V., Polli A., Wilner K. D., and Janne P. A. (2013) Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. New Engl. J. Med. 368, 2385–2394 [DOI] [PubMed] [Google Scholar]
- 90. Zhang B., Wang J., Wang X., Zhu J., Liu Q., Shi Z., Chambers M. C., Zimmerman L. J., Shaddox K. F., Kim S., Davies S. R., Wang S., Wang P., Kinsinger C. R., Rivers R. C., Rodriguez H., Townsend R. R., Ellis M. J., Carr S. A., Tabb D. L., Coffey R. J., Slebos R. J., Liebler D. C., and Nci C. (2014) Proteogenomic characterization of human colon and rectal cancer. Nature 513, 382–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. LaBaer J., and Ramachandran N. (2005) Protein microarrays as tools for functional proteomics. Current Opinion Chem. Biol. 9, 14–19 [DOI] [PubMed] [Google Scholar]
- 92. Haab B. B. (2003) Methods and applications of antibody microarrays in cancer research. Proteomics 3, 2116–2122 [DOI] [PubMed] [Google Scholar]
- 93. Brennan D. J., O'Connor D. P., Rexhepaj E., Ponten F., and Gallagher W. M. (2010) Antibody-based proteomics: fast-tracking molecular diagnostics in oncology. Nat. Rev. Cancer 10, 605–617 [DOI] [PubMed] [Google Scholar]
- 94. Spurrier B., Ramalingam S., and Nishizuka S. (2008) Reverse-phase protein lysate microarrays for cell signaling analysis. Nat. Protocols 3, 1796–1808 [DOI] [PubMed] [Google Scholar]
- 95. Vazquez-Martin A., Colomer R., and Menendez J. A. (2007) Protein array technology to detect HER2 (erbB-2)-induced ‘cytokine signature’ in breast cancer. Eur. J. Cancer 43, 1117–1124 [DOI] [PubMed] [Google Scholar]
- 96. Bast R. C. Jr., Hennessy B., and Mills G. B. (2009) The biology of ovarian cancer: new opportunities for translation. Nat. Rev. Cancer 9, 415–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Rochfort S. (2005) Metabolomics reviewed: a new “omics” platform technology for systems biology and implications for natural products research. J. Natural Products 68, 1813–1820 [DOI] [PubMed] [Google Scholar]
- 98. Vander Heiden M. G., Cantley L. C., and Thompson C. B. (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Claudino W. M., Quattrone A., Biganzoli L., Pestrin M., Bertini I., and Di Leo A. (2007) Metabolomics: available results, current research projects in breast cancer, and future applications. J. Clin. Oncol. 25, 2840–2846 [DOI] [PubMed] [Google Scholar]
- 100. Shulaev V. (2006) Metabolomics technology and bioinformatics. Briefings in Bioinformatics 7, 128–139 [DOI] [PubMed] [Google Scholar]
- 101. Chan E. C., Koh P. K., Mal M., Cheah P. Y., Eu K. W., Backshall A., Cavill R., Nicholson J. K., and Keun H. C. (2009) Metabolic profiling of human colorectal cancer using high-resolution magic angle spinning nuclear magnetic resonance (HR-MAS NMR) spectroscopy and gas chromatography mass spectrometry (GC/MS). J. Proteome Res. 8, 352–361 [DOI] [PubMed] [Google Scholar]
- 102. Blekherman G., Laubenbacher R., Cortes D. F., Mendes P., Torti F. M., Akman S., Torti S. V., and Shulaev V. (2011) Bioinformatics tools for cancer metabolomics. Metabolomics : Official J. Metabolomic Soc. 7, 329–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Beckonert O., Keun H. C., Ebbels T. M., Bundy J., Holmes E., Lindon J. C., and Nicholson J. K. (2007) Metabolic profiling, metabolomic and metabonomic procedures for NMR spectroscopy of urine, plasma, serum and tissue extracts. Nat. Protocols 2, 2692–2703 [DOI] [PubMed] [Google Scholar]
- 104. Bertram H. C., Eggers N., and Eller N. (2009) Potential of human saliva for nuclear magnetic resonance-based metabolomics and for health-related biomarker identification. Anal. Chem. 81, 9188–9193 [DOI] [PubMed] [Google Scholar]
- 105. Smolinska A., Blanchet L., Buydens L. M., and Wijmenga S. S. (2012) NMR and pattern recognition methods in metabolomics: from data acquisition to biomarker discovery: a review. Anal. Chim. Acta 750, 82–97 [DOI] [PubMed] [Google Scholar]
- 106. Wishart D. S., Jewison T., Guo A. C., Wilson M., Knox C., Liu Y., Djoumbou Y., Mandal R., Aziat F., Dong E., Bouatra S., Sinelnikov I., Arndt D., Xia J., Liu P., Yallou F., Bjorndahl T., Perez-Pineiro R., Eisner R., Allen F., Neveu V., Greiner R., and Scalbert A. (2013) HMDB 3.0–The Human Metabolome Database in 2013. Nucleic Acids Res. 41, D801–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Labuschagne C. F., van den Broek N. J., Mackay G. M., Vousden K. H., and Maddocks O. D. (2014) Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Reports 7, 1248–1258 [DOI] [PubMed] [Google Scholar]
- 108. Lv W., and Yang T. (2012) Identification of possible biomarkers for breast cancer from free fatty acid profiles determined by GC-MS and multivariate statistical analysis. Clin. Biochem. 45, 127–133 [DOI] [PubMed] [Google Scholar]
- 109. Gao J., Aksoy B. A., Dogrusoz U., Dresdner G., Gross B., Sumer S. O., Sun Y., Jacobsen A., Sinha R., Larsson E., Cerami E., Sander C., and Schultz N. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Nibbe R. K., Koyuturk M., and Chance M. R. (2010) An integrative -omics approach to identify functional sub-networks in human colorectal cancer. PLoS Computational Biol. 6, e1000639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Ciriello G., Miller M. L., Aksoy B. A., Senbabaoglu Y., Schultz N., and Sander C. (2013) Emerging landscape of oncogenic signatures across human cancers. Nat. Genetics 45, 1127–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Kristensen V. N., Lingjaerde O. C., Russnes H. G., Vollan H. K., Frigessi A., and Borresen-Dale A. L. (2014) Principles and methods of integrative genomic analyses in cancer. Nature Revi. Cancer 14, 299–313 [DOI] [PubMed] [Google Scholar]
- 113. Dawson S. J., Rueda O. M., Aparicio S., and Caldas C. (2013) A new genome-driven integrated classification of breast cancer and its implications. EMBO J. 32, 617–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Palucka K., and Banchereau J. (2012) Cancer immunotherapy via dendritic cells. Nature Rev. Cancer 12, 265–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Schumacher T. N., and Schreiber R. D. (2015) Neoantigens in cancer immunotherapy. Science 348, 69–74 [DOI] [PubMed] [Google Scholar]
- 116. Mellman I., Coukos G., and Dranoff G. (2011) Cancer immunotherapy comes of age. Nature 480, 480–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Disis M. L. (2011) Immunologic biomarkers as correlates of clinical response to cancer immunotherapy. Cancer Immunol., Immunotherapy 60, 433–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Pao W., and Hutchinson K. E. (2012) Chipping away at the lung cancer genome. Nature Med. 18, 349–351 [DOI] [PubMed] [Google Scholar]
- 119. Bedard P. L., Hansen A. R., Ratain M. J., and Siu L. L. (2013) Tumour heterogeneity in the clinic. Nature 501, 355–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Merlo L. M., Pepper J. W., Reid B. J., and Maley C. C. (2006) Cancer as an evolutionary and ecological process. Nature Rev. Cancer 6, 924–935 [DOI] [PubMed] [Google Scholar]
- 121. Gerlinger M., Horswell S., Larkin J., Rowan A. J., Salm M. P., Varela I., Fisher R., McGranahan N., Matthews N., Santos C. R., Martinez P., Phillimore B., Begum S., Rabinowitz A., Spencer-Dene B., Gulati S., Bates P. A., Stamp G., Pickering L., Gore M., Nicol D. L., Hazell S., Futreal P. A., Stewart A., and Swanton C. (2014) Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nature Genetics 46, 225–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Marusyk A., and Polyak K. (2010) Tumor heterogeneity: causes and consequences. Biochim. Biophys. Acta 1805, 105–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Kim C., and Paik S. (2010) Gene-expression-based prognostic assays for breast cancer. Nature reviews. Clinical Oncol. 7, 340–347 [DOI] [PubMed] [Google Scholar]
- 124. La Thangue N. B., and Kerr D. J. (2011) Predictive biomarkers: a paradigm shift towards personalized cancer medicine. Nature reviews. Clin. Oncol. 8, 587–596 [DOI] [PubMed] [Google Scholar]
- 125. Chao T. C., Hansmeier N., and Halden R. U. (2010) Towards proteome standards: the use of absolute quantitation in high-throughput biomarker discovery. J. Proteomics 73, 1641–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Moch H., Blank P. R., Dietel M., Elmberger G., Kerr K. M., Palacios J., Penault-Llorca F., Rossi G., and Szucs T. D. (2012) Personalized cancer medicine and the future of pathology. Virchows Archiv 460, 3–8 [DOI] [PubMed] [Google Scholar]