

Abstract
Oocytes undergo a range of complex processes via oogenesis, maturation, fertilization, and early embryonic development, eventually giving rise to a fully functioning organism. To understand proteome composition and diversity during maturation of human oocytes, here we have addressed crucial aspects of oocyte collection and proteome analysis, resulting in the first proteome and secretome maps of human oocytes. Starting from 100 oocytes collected via a novel serum-free hanging drop culture system, we identified 2,154 proteins, whose function indicate that oocytes are largely resting cells with a proteome that is tailored for homeostasis, cellular attachment, and interaction with its environment via secretory factors. In addition, we have identified 158 oocyte-enriched proteins (such as ECAT1, PIWIL3, NLRP7)1 not observed in high-coverage proteomics studies of other human cell lines or tissues. Exploiting SP3, a novel technology for proteomic sample preparation using magnetic beads, we scaled down proteome analysis to single cells. Despite the low protein content of only ∼100 ng per cell, we consistently identified ∼450 proteins from individual oocytes. When comparing individual oocytes at the germinal vesicle (GV) and metaphase II (MII) stage, we found that the Tudor and KH domain-containing protein (TDRKH) is preferentially expressed in immature oocytes, while Wee2, PCNA, and DNMT1 were enriched in mature cells, collectively indicating that maintenance of genome integrity is crucial during oocyte maturation. This study demonstrates that an innovative proteomics workflow facilitates analysis of single human oocytes to investigate human oocyte biology and preimplantation development. The approach presented here paves the way for quantitative proteomics in other quantity-limited tissues and cell types. Data associated with this study are available via ProteomeXchange with identifier PXD004142.
Mammalian oocytes are exceptional cells in that they are extremely long-lived and start the meiotic program during fetal development, arrest for a long period of time, and become recruited from the resting to the growing pool for final maturation and fertilization. Before resumption of maturation, oocytes undergo extensive growth, which is accompanied by high transcription rate, while fully matured oocytes become transcriptionally silenced (1, 2). Various studies have investigated gene expression programs in human oocytes (3–7), yet the characterization of translation into functional protein products at different stages of oocyte growth and maturation remains poorly understood. During oocyte growth, many maternally transcribed mRNAs can be stored dispersed throughout the cytoplasm or in localized messenger ribonucleoprotein complexes in a translationally silent state (8). Tight translational control of these transcripts is crucial for properly timed oocyte development and maturation (9) until zygotic genome activation (10). In general, this is achieved by modulating mRNA polyadenylation where translation is suppressed by poly-adenylation and activated by deadenylation to induce development and meiosis (11–13). Recent data have shown that translation of a subset of oocyte mRNAs that are critical for early embryo development are also under the control of surrounding follicular (granulosa) cell inputs (14), but the signals are still poorly understood.
Oocytes are among the largest cells in many animals, although their size differs between species. Oocytes in Xenopus laevis are 1–1.3 mm in diameter and are visible even by the naked eye, while mammalian oocytes are 1,000-fold smaller with diameters of 50–120 μm. Because of their size and accessibility, Xenopus oocytes have been an attractive model in developmental biology that is tractable by many techniques, including proteomics (15). Proteomics of mammalian oocytes is more challenging, requiring large numbers of cells as has been done for mouse, cow, and pig (16). For example, several thousands of oocytes were required for the profiling of mouse oocytes across several maturation stages (17, 18). Human oocytes have so far remained inaccessible for proteomic studies because of their small size compared with Xenopus and since they can only be collected in small numbers in a time-consuming process. This makes it practically impossible to collect the hundreds or thousands of oocytes that have been used to investigate similarly sized mouse oocytes. To solve this issue, the only possibility would be to increase sensitivity of the used technology to study these cells in smaller numbers or even individually. Assuming a sphere with a diameter of 100 μm and an intracellular protein concentration of 200g/l (19), a single human oocyte has an estimated protein content of 100 ng, a number that correlates well with previous estimates (20, 21). Such a low amount of starting material presents an enormous challenge for proteomics, considering that the analysis of complex proteomes by LC-MS typically requires 100–500 ng of protein to be loaded on column. Therefore, the true challenge of obtaining proteome data from a single human oocyte resides in the efficient extraction, purification, and digestion of the 100 ng protein it contains, with only minimal losses. Xenopus oocytes are three orders of magnitude larger than human oocytes containing ∼100 μg protein per oocyte (22), which is well within the manageable range of present-day proteomic technologies. This has enabled several groups to study frog oocyte proteomes in detail (15, 23, 24) even at the single-cell level (25, 26).
In the current study, we aimed to characterize the human oocyte proteome and secretome to identify proteins that underlie oocyte maturation. In addition, we aimed to investigate whether single-cell proteomics is possible for human oocytes, benefiting from a novel protocol for sample preparation, SP3, which we introduced recently for low-input proteomics (27). In an initial experiment from 100 oocytes collected via a newly developed hanging drop culture system we identified over 2,100 proteins and more than 300 in their secretome, including 158 that had not been identified in other cell types and thus may have an oocyte-specific function. Application of SP3, a recently developed sample preparation method for low-input proteomics, allowed us to scale down proteome analysis identifying ∼500 proteins in single oocytes. Comparative analysis of individual mature and immature cells showed differential expression of proteins involved in DNA replication and genome integrity. This demonstrates the feasibility of quantitative single-cell proteomics that should provide a unique opportunity to gain insight in human oocyte biology and fertility.
EXPERIMENTAL PROCEDURES
Collection of Oocytes
This research was approved by Slovenian Medical Ethical Committee (Ministry of Health, Republic of Slovenia) and the local Ethical Committee of the European Molecular Biology Laboratory, Heidelberg, Germany. Oocytes were collected from the in vitro fertilization program at the University Medical Centre Ljubljana with informed consent by donors. We included the oocytes that would be otherwise discarded in daily medical practice: immature (GV or MI) oocytes that cannot be fertilized and mature (MII) oocytes that did not fertilize in vitro. Immature oocytes were collected on days 0 or 1 (after ultrasound-guided aspiration of follicles and enzymatic denudation of cumulus cells using hyaluronidase or at checking for fertilization) and mature, nonfertilized oocytes on day 2 (one day after checking for fertilization) of in vitro fertilization procedure. All oocytes were collected without cumulus (granulosa) cells.
Culture of Oocytes
The oocytes were cultured in the conventional IVF culture system or in a hanging drop culture system. For conventional IVF culture, the oocytes were prepared in a closed system in covered plastic and sterile four well-dishes (Thermo Scientific, NuncTM4-Well Dish, cat. no. 144444) with 0.5 ml of Universal IVF Medium containing glucose, sodium pyruvate, calcium chloride, magnesium sulfate, potassium chloride, sodium chloride, sodium bicarbonate, sodium phosphate, gentamicin sulfate, human albumin solution, SSR® (Synthetic Serum Replacement), and Milli RX Water (Origio, Denmark) per well. For the hanging drop culture system, each oocyte was transferred into 8 μl droplet of sterile DMEM/F12 (Dulbecco's Modified Eagle's Medium/Nutrient Mixture F-12 Ham, Sigma-Aldrich, D8900) supplemented with NaHCO3: 0.74 g/200 ml and penicillin (2 ml/200 ml) and with pH adjusted to 7.4 using NaOH in the cover of a plastic and sterile Petri dish (diameter 35 × 10 mm; Thermo Scientific, Nunc TC Petri Dish, cat. no. 153066). The cover was then inverted and put on top of the dish itself that was filled with 3 ml of sterile PBS to prevent the droplet from evaporation. The oocyte was incubated overnight in this culture system in a CO2-incubator (37 °C and 6% CO2 in air). Up to five oocytes were cultured in one hanging droplet. Individual cells were monitored microscopically before and after the overnight incubation, and the cells that showed signs of degeneration (brown or degraded cytoplasm) were excluded (∼15%). For secretome analyses, the corresponding droplets of culture medium after hanging drop culture were collected and analyzed. Cells and their secretomes were stored at −20 °C until proteome analysis.
Sampling of Oocytes after Hanging Drop or Conventional IVF Culture
Each oocyte was washed four times and denuded by transfer from one droplet of PBS to another by a denudation pipette (diameter 0.134–0.145 mm; Swemed by Vitrolife, ref. 14301) and at the end transferred into a small microcentrifuge tube in a minimal volume of PBS; the tube was centrifuged in a microcentrifuge 3 min at 5,590 × g and then stored at −20 °C. This was continued until the desired number of cells had been collected. After oocyte culturing in a hanging drop culture system, each corresponding droplet of culture medium was directly transferred into a microcentrifuge tube and stored at −20 °C. All samples were collected step by step, and several microcentrifuge tubes were combined into a sample.
Oocyte Immunocytochemistry
Fresh immature and mature oocytes were fixed in 4% formaldehyde for 10 min, washed two times in PBS, permeabilized with 0.3% Triton X-100 for 10 min, washed two times in PBS, and incubated in 10% fetal bovine serum (FBS) for 20 min. Then the nonwashed oocytes were transferred into the solution of primary antibodies and were incubated for 1 h. Thereafter, the oocytes were washed five times in PBS and transferred into the solution of secondary antibodies and incubated for 30 min. The oocytes were then washed five times in PBS and transferred into a drop of Vectashield mounting medium (Vector Laboratories, HI-1200) with DAPI. After 15 min, cells were covered by a coverslip and monitored by fluorescence microscopy (Nikon DS-Fi1 equipped by Nikon Digital.Sight camera). For a negative control, the primary antibodies were omitted. The whole procedure was performed at room temperature, and during all incubations, the oocytes were in the dark. The primary mouse antibodies and dilutions were: mouse anti-PCNA (Abcam, ab29, 1:400), rabbit anti-DNMT1 (Abcam, ab19905, 1:400), and rabbit anti-ECAT1 (Abcam, ab126339, 1:200) and the secondary antibodies were: Alexa Fluor 488 goat anti-mouse IgG (Molecular Probes, A-11001, 1:200) and Cy3 goat anti-rabbit IgG (Molecular Probes, A10520, 1:200).
Proteomic Sample Preparation
Collections of 100 oocytes were lysed in 50 mm HEPES buffer at pH 8.5 (Sigma) containing 0.1% Rapigest (Waters), and proteins were TCA precipitated. After resolubilization, proteolysis was carried out overnight at 37 °C with a mixture of trypsin and rLysC (Promega) at an enzyme to substrate ratio of 1:25.
Samples containing 10, 5, or single oocytes were processed via SP3, a single-tube method utilizing magnetic beads for unbiased protein and peptide capture and recovery as described previously (27). Briefly, oocytes were lysed in 2% SDS (Bio-Rad), 1X cOmplete Protease Inhibitor Mixture-EDTA (Roche), 5 mm EDTA, 5 mm EGTA, 10 mM NaOH, and 10 mm DTT, in 10 mm HEPES buffer at pH 8.5 (Sigma). After heating at 99 °C for 10 min followed by another 10 min in the Biorupter (prewarmed, high level), proteins were reduced (adding 5 μl of 200 mm DTT (Bio-Rad) per 100 μl of lysate) and alkylated (addition of 10 μl of 400 mm iodoacetamide (IAA) per 100 μl of lysate). The sample was acidified with formic acid (pH 2), SeraMag paramagnetic beads (Thermo Scientific) were added (1:100 v/v), and protein binding was induced by addition of acetonitrile (50% final concentration). After incubation on a magnetic rack, the supernatant was discarded, and beads were sequentially rinsed with 200 μl of 70% absolute ethanol and 180 μl of 100% acetonitrile, thereby removing SDS. Rinsed beads were then eluted off the beads in 5 μl of 50 mm HEPES, pH 8. A mixture of trypsin and rLysC (Promega) was added (1:25 enzyme to substrate ratio) for digestion overnight at 37 °C. For subsequent peptide clean-up and recovery, 100% acetonitrile was added to achieve a final concentration >95%. While on the magnetic rack, the supernatant was discarded, and the beads rinsed with 180 μl of 100% acetonitrile. Peptides were eluted in water containing 2% dimethyl sulfoxide, acidified with formic acid, and analyzed by LC-MSMS. Optionally, peptides were isotope-labeled via reductive methylation within the SP3-workflow as previously described (27).
Liquid Chromatography-Tandem MS (LC-MSMS)
Peptides were analyzed by liquid chromatography (LC) coupled to an Orbitrap Velos Pro mass spectrometer (Thermo Fisher Scientific) using a Proxeon nanospray source. Reverse phase chromatography was performed with a nanoACQUITY Ultra-Performance LC system (Waters) fitted with a trapping column (nanoAcquity Symmetry C18, 5 μm, 180 μm × 20 mm) and an analytical column (nanoAcquity BEH C18, 1.7 μm, 75 μm × 200 mm) directly coupled to the ion source. The mobile phases for LC separation were 0.1% (v/v) formic acid in LC-MS-grade water (solvent A) and 0.1% (v/v) formic acid in can (solvent B). Peptides were separated at a constant flow rate of 300 nl/min with a linear gradient of solvent B from 3 to 40% for 145 min. The MS1 scan was acquired in the Orbitrap from m/z 300 to 1,700 at a maximum filling time of 500 ms and 106 ions. The resolution was set to 30,000. Fragmentation was performed in the LTQ by collision-induced dissociation, selecting up to 15 most intense ions (top15) at an isolation window of 2 Da. Target ions previously selected for fragmentation were dynamically excluded for 30 s with relative mass window of 10 ppm. MS/MS selection threshold was set to 2,000 ion counts. A lock mass correction was applied using a background ion (m/z 445.12003). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD004142.
Experimental Design and Statistical Rationale
Oocytes were collected and classified by maturation stage (GV or MII) and prepared for proteome analysis in pools of 100, 10, 5, or single cells. Comparison of mature and immature cells was performed both with and without the use of stable isotopes for protein quantification, depending on the experiment as described in the results section. In both cases, raw LC-MS files were processed with MaxQuant (28) version 1.3.0.5 and the Andromeda search engine (29). The MS/MS spectra were searched against the Human UniProt database (downloaded on June 21, 2011) containing 69,906 forward sequences that was appended to the same number of reverse sequences and 265 common contaminants. The precursor mass tolerance was set to 20 ppm for the first pass and 6 ppm for the second pass. The fragment mass tolerance was 0.5 Da. Unique- and razor peptides were used for quantification, using only unmodified peptides. Cysteine carbamidomethylation and methionine oxidation were set as fixed and variable modification, respectively. The minimum peptide length was set to six amino acids, the enzyme specificity was set to trypsin/P, the maximum allowed miss-cleavage was set to 2, and the false discovery rate was set to 0.01 for both peptide and protein identifications. Requantification and match between runs were also performed unless stated otherwise. The protein identification was reported as a “protein group” if no unique peptide sequence to a single database entry was identified. Protein abundance was estimated from iBAQ values (30) and label-free quantification (LFQ) (31), performed within MaxQuant. Proteins were considered differentially expressed when showing a LFQ fold change larger than 2.5 times the standard deviation. Prior secretion evidence was considered confirmed for proteins with the UniProt keyword “Signal” or “Secreted” or reaching the significance threshold in Signal P4.0 (32). Protein functional annotation and pathway analysis was performed using the database for annotation, visualization and integrated discovery (DAVID) Bioinformatics Resources 6.7 (33) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (34). To elucidate oocyte-specific proteins, the human oocyte proteome retrieved in this study was compared with exhaustive proteomes of other cell types reported in the literature.
RESULTS
Proteome Analysis of Human Oocytes from a Hanging Drop Culture System
For an initial proteome analysis, we collected 100 mature (MII stage) and 100 immature (GV stage) human oocytes that would otherwise be discarded in the in vitro fertilization program. Oocytes were incubated in IVF media as is usually done for in vitro fertilization (Fig. 1A), washed, and processed for analysis by LC-MSMS, identifying ∼1,100 proteins in both maturation states (Table S1). To increase proteome coverage and to facilitate future secretome analysis, we utilized a hanging drop culture system incubating oocytes in a small volume (8 μl) of DMEM/F12 medium devoid of albumin or other proteinaceous constituents (Fig. 1B). From 100 mature and 100 immature cells (Fig. S1) analyzed in technical replicate LC-MSMS runs, greater than 2,100 proteins were identified (Table S1) the majority of which are contained in the data set obtained under IVF conditions (Fig. S2). The proteins exclusively identified from hanging drops are present at a lower abundance as judged from iBAQ values, (Fig. 1C), indicating an increased sampling depth collectively spanning six orders of magnitude.
Fig. 1.

Proteome analysis of oocytes from two different culture systems. (A) Conventional IVF culture system with oocyte culturing in a closed system–conventional IVF medium in covered four-well dishes (0.5 ml of medium per well). (B) New hanging drop culture system culturing oocytes in small, 8 μl droplets of DMEM/F12 medium. After culturing, both oocytes and droplets of corresponding medium were sampled to analyze the proteome and secretome. (C) Estimated abundance by iBAQ value of proteins identified in oocytes grown in IVF media and in hanging drops. Blue symbols: proteins identified both in IVF and hanging drops. Red symbols: proteins identified in hanging drops only.
Functional Annotation of the Human Oocyte Proteome in Comparison to Cell Lines from Different Human Tissues and Organs
In the combined data sets obtained from oocytes grown in IVF and hanging drop conditions, we identified 2,154 proteins (Table S1) that we used collectively for a more detailed exploration of the oocyte proteome. Beyond performing a gene ontology enrichment analysis, we compared our data to proteomic data sets of various other cell types to gain insight in 1) biological processes that distinguish oocytes from other cells and 2) individual proteins that are uniquely or preferentially expressed in oocytes.
To answer the first question, we realized that the 2,154 identified proteins represent a reasonable fraction of the oocyte proteome but also that its overall depth is limited compared with other proteomes obtained from cell types that can be collected in much larger quantities. We reasoned that a comparison between such disparate data sets may still be informative by exploiting the relative protein abundance in the respective data sets, estimated from iBAQ values. Specifically, the comparison of the 2,154 oocyte proteins (representing the most abundant proteins of the proteome) to the same number of the most highly expressed proteins in another cell type should reveal the biological functionalities that are most critical for each of the cell types. To investigate this, we compiled a reference proteome obtained from an exhaustive proteomic data set (11,731 proteins) accrued from 11 distinct cell lines of various origins (35). Specifically, we selected the 2,154 most abundant proteins estimated from the summed iBAQ intensities across these cell lines, thus creating an “average” cellular proteome that we refer to here as the reference proteome (Table S2). Strikingly, only 1,105 proteins were shared between the oocyte and the reference, while ∼1,000 were unique for each of them (Table S2), indicating a drastic difference in the protein composition in the upper segments of the respective proteomes. Interestingly, of the oocyte proteins falling outside the top 2,154 in the reference proteome, 784 proteins ranked between position 2,200 and 9,800 based on iBAQ values, while 284 were not identified at all in the reference proteome (Fig. S3). In a comparative gene ontology (GO)-analysis of the proteins that were specific for either proteome subset, several functionalities stood out that were enriched in oocytes (Fig. 2, Table S3). This included vesicle-mediated transport (68 proteins) and 135 proteins classified in the extracellular region, suggesting that secretion and communication with the cellular environment is of crucial importance for oocytes. This is further hinted by the expression of 48 cell adhesion proteins that were absent or lowly expressed in the 11 cell lines, including various extracellular matrix proteins (e.g. JUP, LOXL2, ICAM1, ACE, CDH2, CD99), integrins (ITGA1, ITGAV, ITGAE, ITGA9) and focal adhesion proteins (e.g. LIMA1, PAK1, TES). It was striking to note that several ontology groups related to cellular defense and homeostasis were enriched in oocytes (“homeostatic process,” “response to wounding,” “response to organic substance,” “defense response”), reflecting a reactive cellular phenotype and suggesting a main function in maintaining a steady state.
Fig. 2.
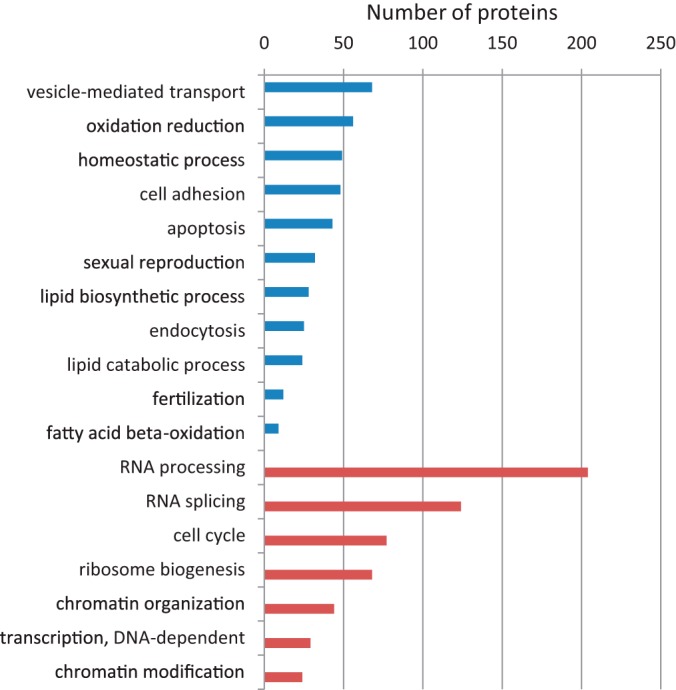
Gene ontology terms of proteins exclusively identified in oocytes (blue) and in the reference proteome obtained from 11 cell lines and embryonic stem cells (red).
Several functional classes related to lipid transport and metabolism were enriched in oocytes, collectively including 41 lipid-binding proteins that were not present in the reference proteome (Table S3). Specifically, these comprised 28 and 24 proteins in lipid biosynthetic and catabolic processes, respectively, in line with the recognized role of fatty acid utilization for the promotion of oocyte quality (36, 37).
In addition, 32 oocyte-enriched proteins were grouped in the GO-classes “sexual reproduction” and “fertilization” that were not identified in the reference proteome, including ZP1-ZP4 (constituting the zona pellucida), CD9 (involved in sperm–egg recognition and fusion), BMP15 and GDF9 (oocyte-specific growth factors required for follicle development), HENMT1 (methyltransferase to stabilize piRNAs, essential for gametogenesis), BCL2L10 (suppressing apoptosis), CHEK1 (required for cell cycle arrest), and PTTG1 (preventing chromosome segregation). Many of these proteins are well-known for their roles in gamete development and maturation.
Of equal interest was the large number (∼1,000) of proteins in the reference proteome that were not identified in oocytes (Table S2). Interestingly, these comprised large groups of proteins involved in RNA processing (204 proteins), transcription (29 proteins), splicing (124 proteins), ribosome biogenesis (68 proteins), cell cycle (77 proteins), and chromatin organization (44 proteins) (Table S3). Among the cellular components gene ontology cellular component (GO-CC), molecular machineries corresponding with these functionalities were missing in oocytes, including the spliceosome (65 proteins) and the ribosome (74 proteins) as well as usually abundant proteins that are crucial for transcription (e.g. RNA polymerases RNA polymerase (POLR) POLR2A, 2B, 2D, and 2L, as well as several GTF2 general transcription factors). Much of this can be attributed to the fact that oocytes do not have a visible nucleus at this stage of maturation (GV, MII).
Taking these findings together, this analysis shows that oocytes are largely resting cells with a proteome that is tailored to homeostasis, cellular attachment, and interaction via secretory factors. This is in stark contrast to dividing cells represented in the reference panel of 11 cell lines with a proteome targeted at growth and proliferation, where processes like chromatin organization, transcription, splicing, and translation are prominently represented among the most highly expressed proteins while these have remained invisible in oocytes.
Identification of Oocyte-Specific Proteins
Beyond a global description of oocyte-enriched GO-terms, we next aimed to identify individual proteins that are uniquely or preferentially expressed in oocytes. Therefore, we selected several data sets from the literature that have exhaustively characterized the proteomes of cell types distantly or more closely related to oocytes. In addition to the 11 human cancer cell lines mentioned before (11,731 proteins) (35), these included data on human embryonic stem cells (7,952 proteins) (38), human sperm (6,198 proteins) (39), and human blastocysts (1,307 proteins) (40) (Table S4). We argued that any protein that was identified in our (still rather superficial) analysis (2,154 proteins) but that was not detected in these in-depth studies will make a good candidate to be uniquely or preferentially expressed in oocytes.
When comparing our oocyte proteome to the two largest reference data sets (obtained from 11 cell lines and from embryonic stem cells), 158 proteins were exclusively identified in oocytes (Table S4). A gene ontology analysis of the ∼80 proteins within this group that could be mapped to GO terms returned highly similar biological processes as in the previous analysis above (several processes related to reproduction, defense response, response to wound healing, and lipid transport) (Table S5). This indicates that even in this much-stricter selection, biological functionalities emerge that are highly intrinsic to and specific for oocytes. In addition, extracellular proteins constitute the only cellular localization that was enriched in this oocyte-specific proteome (46 out of 89 mapped proteins), again emphasizing the importance of secretory proteins for oocytes. These include extracellular matrix proteins (e.g. ZP1–4, ACAN, KAL1), proteases (e.g. ACE, OVCH, ASTL, HTRA1) and protease inhibitors (several SERPINs, ITIH1), likely reflecting their intricate interaction in regulating shedding of cell-surface proteins.
Only half of the 158 proteins in the oocyte-specific proteome produced a sufficiently large group-size to result in an enriched GO-term (Table S5), thus still masking an equally sized group of individual proteins potentially representing an important biological functionality. This includes many proteins that were identified before in blastocysts and/or mouse oocytes, adding confidence that these indeed have a role to play in human oocytes. For instance, this applies to several maternal effect proteins such as ZAR1 and its paralog ZAR1L (critical for the oocyte-to-embryo transition (41); DPPA3 (required for normal preimplantation development, involved in transcriptional repression, and epigenetic chromatin reprogramming in the zygote (42)); DNMT1 (responsible for maintaining methylation patterns in development (43); TLE6 (important for oocyte developmental competence and embryogenesis (44)); NLRP, OOEP, and PADI6. Other proteins that have only been identified in oocytes are the hormone GPHA2 and the oxytocin carrier protein neurophysin (OXT), as well as KPNA7, an oocyte-specific nuclear importin required for fertility in mice (45, 46). The most salient findings of these analyses are summarized in Fig. 3. Several of these have also been detected in blastocysts but were absent in the sperm proteome (Table S4), supporting their candidacy as oocyte-specific proteins. Further evidence for this is provided by Human Proteome Map (www.humanproteomemap.org) where several proteins were not identified in any tissue (e.g. BCL2L10, FOLR4, GPHA2, MUC20, OOEP, OVCH1, OXT, OOSP2, ZP1. ZP3, ZP4), while several others were reported only in a limited number of organs (Fig. S4). The latter may still be an overestimation since most of these proteins are reported as single-peptide identifications, emphasizing that these large-scale data should be used with caution (47, 48) and thus hampering a more systematic analysis of our data based on this resource.
Fig. 3.

Proteins and their gene ontology classification identified in human oocytes but absent in the proteomes of 11 human cell lines, human embryonic stem cells, and sperm.
Comparative Proteome Analysis Reveals Human Disease-Related Oocyte-Enriched Proteins
The oocyte proteome has been best characterized in mice, identifying ∼2,700 (17) and ∼3,600 proteins (18) in GV and MII-oocytes. Strikingly, ∼600 and 770 of the proteins in our data set had not been identified previously in these respective studies, 470 of which were not found in either of them (Table S4), suggesting a profound difference in the proteome composition of mouse and human oocytes. A closer inspection revealed that several of the genes encoding these proteins are absent in the mouse genome (Table S6), demonstrating the importance of the availability of proteome composition of human oocytes. For instance, NLRP7, NLRP11, NLRP13, ECAT1, OVCH1, and even ZP4, a major constituent of the zona pellucida of human oocytes, have no orthologs in mice. Importantly, several of these have been causatively linked to disorders in oocyte development or pregnancy (Table S6). This extends to other oocyte-specific proteins, either with (e.g. NLRP5, GDF9, BMP15, ANXA5, DIAPH2, HADHA) or without mouse orthologs (e.g. ATAD3B, ATAD3C, CCNI2), and that have or have not been linked to disease before (Table S6). Finally, our analyses identified two proteins, LDHAL6A and PIWIL3, that have no mouse ortholog and whose expression is thought to be confined to male germ cells (Table S6). While not much is known about LDHAL6A, PIWIL3, a protein involved in piRNA biogenesis, was identified in cows only recently as the first demonstration that PIWIL3 is expressed in oocytes (49). Therefore, our data are the first to show that PIWIL3 is also present in human oocytes. Identification of PIWIL3 did not result from sperm because it was also identified in immature (GV) oocytes, which have not been exposed to sperm in any way. In addition, the identification of the piRNA methyltransferase HENMT1 seems to suggest that piRNAs in oocytes are stabilized via 3′-methylation.
ECAT1 (KHDC3L), the epigenetics-related ES cell-associated transcript 1, was only observed in blastocysts (Table S4), and our data demonstrate the expression of this protein in oocytes for the first time. To further corroborate this, we performed immuno-cytochemistry showing that ECAT1 was concentrated as a ring at the oocyte cortex with a nonhomogenous distribution regardless of oocyte maturity (Fig. S5). In mature oocytes, it was also concentrated in the region of polar body while in others it was protruded into the zona pellucida. ECAT1 was also expressed in fertilized oocytes (zygotes) not cleaving after an in vitro fertilization procedure (Fig. S5). These observations complement previous findings associating ECAT1 with biparental complete hydatidiform mole in humans, an abnormal form of pregnancy in which a nonviable fertilized oocyte implants in the uterus and converts a normal pregnancy into an abnormal one and does not come to the term (50–52).
Protein Decretion by Oocytes
Our proteome data indicated that protein secretion is one of the most prominently represented processes in oocytes. This is revealed most strikingly by the observation that 424 out of the 2,154 proteins (20%) detected in oocytes can be designated as secretory proteins (i.e. with prior secretion evidence based on the presence of a signal peptide and gene ontology) (Table S1). To investigate if any of these could indeed be detected in conditioned media of oocytes, we performed a secretome analysis of 100 oocytes via hanging drops in the absence of serum albumin in the medium. In this oocyte secretome, we identified 383 proteins, 338 of which were not present in the medium prepared in the absence of oocytes (Table S7). Of these, 299 were identified both in the oocyte proteome and secretome, while 39 proteins were unique to the secretome (Table S8). The secretome comprised 97 proteins with prior evidence of being secreted, such as GDF9, endoplasmin, CGREF1, ITIH1, ICAM1, and MIF.
Proteins in the secretome span a variety of functionalities (Table S9) featuring “proteolysis” as the largest group (39 proteins). Other functionalities among the top-enriched processes reflect functional groups also observed for the total proteome, e.g. cellular homeostasis and several processes related to response and defense to environment. Other interesting proteins in the oocyte secretome include WFDC2 (HE4), a small secretory protein, which has been implied in sperm maturation (53) and which is overexpressed in ovarian cancer (54). GNPDA1 (OSCILLIN) (55) is involved in oocyte activation and early development of the embryo. Collectively, these data are the first to give direct insight in the proteins secreted by human oocytes, providing a lead to investigate their potential role in the communication with other follicular cells (Fig. S6).
Proteome Analysis of 10, 5, and Single Oocytes
Given the large effort that is required to collect 100 oocytes for proteome analysis, we wondered if better sensitivity may be obtained benefiting from a novel method for sample preparation that we developed recently (27). The approach, termed SP3, follows a single-tube protocol using paramagnetic beads and has been proven highly suitable for quantity-limited samples. Therefore, we collected 10, 5, and single oocytes and processed them via SP3 prior to analysis by LC-MSMS.
From 10- and 5-cell pools, we identified 887 and 713 proteins, respectively. Even starting from a single oocyte, we identified 445 proteins (Table S10). To our knowledge, this represents the first proteome analysis of a single human oocyte or even of any human cell type. Moreover, we believe that the number of identified proteins is surprisingly large considering the very low amount of starting material. When extending this analysis to include data from five additional single oocytes we consistently identified 450–500 proteins per cell (Table S11). Even when deselecting the “match between runs” option in MaxQuant, meaning that each sample was treated individually without transfer of peptide identifications, we identified 400–450 proteins per oocyte (Table S11). This implies that proteomes of single oocytes can be obtained independent of any reference but that the number of identifications may benefit from analyzing several in parallel. Importantly, in these samples we were able to identify several proteins related to human reproduction (Tables S10 and S11): steroid biosynthetic process (CYB5R3, FDXR, ACAA2), fertilization (NLRP5, MFGE8, NPM2, OOEP, ZP1, ZP2, ZP3, ZP4), embryo implantation and pregnancy (FKBP4, BSG (Basigin), SOD1), cell cycle and meiosis proteins (SKP1, WEE2, PCNA, CALM2, CUL1, ITPR1, YWHAE, YWHAB, YWHAH, YWHAG, YWHAQ, YWHAZ), and a range of maternal effect proteins (OOEP, TLE6, NLRP5, NPM2, DNMT1). The latter group also included ECAT1, which was identified by multiple peptides even in single oocytes. The fact that many of these proteins are oocyte-enriched indicates that these highly relevant proteins are accessible by single-cell proteomics, afforded by efficient sample preparation via SP3. This may have important implications when applying this technology e.g. for diagnostic purposes.
Comparison of Immature and Mature Oocytes
We examined whether our data could be used to identify differences in protein expression between immature (GV state) and mature oocytes (MII state), either from analysis in bulk or from single oocytes. First, we used label-free quantification (LFQ) values of proteins identified from 100 mature and 100 immature cells (Table S1), obtaining very high correlation both in those cultured in IVF and hanging drop conditions (R2 = 0.86 and 0.89, respectively) (Figs. 4A and 4B). This indicates the robustness of our approach and suggests that the proteomes of both cell states are very similar. Since the scarcity of samples did not permit us to run biological replicates of 100 cells, we considered the proteins showing a LFQ fold change larger than 2.5 times the standard deviation of the mean both in IVF culture media and in hanging drops, identifying just three proteins that met these criteria. The protein expressed most prominently in MII cells was the oocyte-specific kinase Wee2 that functions to maintain meiotic arrest in GV oocytes and is required for exit from metaphase II. In GV cells the most enriched protein was Tudor and KH domain-containing protein (TDRKH) (Fig. 4) whose primary function is in piRNA biogenesis in germ cells. The other was caprin-2, an RNA-binding protein that was slightly enriched in GV cells.
Fig. 4.

Proteome comparison of mature and immature oocytes. (A) Label-free quantification (LFQ) of proteins in pools of 100 mature (MII) and 100 immature (GV) oocytes maintained in hanging drops. (B) Same as in A but oocytes maintained in IVF media. (C) Label-free quantification of proteins in single oocytes. The absence of red or blue bars indicates that the respective proteins were not identified in MII oocytes (TDRKH) or GV oocytes (DNMT1, Wee2, PCNA). (D) Protein quantification by stable-isotope-containing dimethyl labeling in pools of 10 mature (MII) and immature (GV) oocytes.
Next, to estimate differential protein expression between the six proteomes of single GV and MII oocytes (Table S11), we selected proteins that were identified by maximally one peptide across all three cells in one maturation stage, while they were identified by at least two peptides in each of the three cells of the other maturation stage. This selection resulted in TDRKH as the only protein that was increased in GV oocytes and 18 proteins that were higher in the MII stage (Table S12). Interestingly, the latter included Wee2, as before, but also PCNA, a critical regulator of DNA replication, and its interaction partner DNMT1, the DNA methyltransferase responsible for maintaining DNA methylation during the cell cycle (Fig 4C). In addition, the interaction partners CKAP5, BUB1B and TACC3, regulating spindle formation and mitotic checkpoint control, were consistently identified in MII oocytes, while they were absent in the GV stage (Table S12), indicating strict cell cycle control in mature oocytes.
To corroborate these label-free quantification data, we performed proteome quantification by stable isotope labeling. Specifically, we collected three groups of 10 cells each of GV and MII oocytes, which were isotope-labeled via reductive methylation as an integrated step during SP3 sample preparation. Pair-wise comparisons of GV versus MII oocytes resulted in 757 proteins that were identified and quantified in all three biological replicates (Table S13). Interestingly, Wee2, PCNA, and DNMT1 were among the most enriched proteins in MII oocytes (Fig. 4D). Again as before, TDRKH was strongly increased in GV cells (Fig. 4D). Thus, stable isotope labeling as the more robust quantification method validates our single-cell data obtained via label-free quantification. To further investigate these finding we performed immuno-cytochemistry for PCNA and DNMT1 in GV and MII oocytes. PCNA was clearly visible across the entire oocyte in both maturation stages (Fig. 5A). DNMT1 localizes tightly around the nucleus in GV oocytes, while it evenly spreads throughout the cytoplasm in the MII stage (Fig. 5B). Although protein expression levels cannot be estimated from these experiments, the redistribution of DNMT1 suggests its dynamic involvement in the maturation process. When analyzing parthenogenetic embryos, representing the next maturation stage, we observed strong staining of DNMT1 with the highest density around the nucleus (Fig. 5B). This is in line with the increased expression of DNMT1 during maturation as suggested by single-oocyte proteomics. Collectively, our data identify TDRKH, Wee2, PCNA, and DNMT1 as maturation-specific proteins that can be identified and quantified in single oocytes due to highly efficient sample preparation via SP3.
Fig. 5.
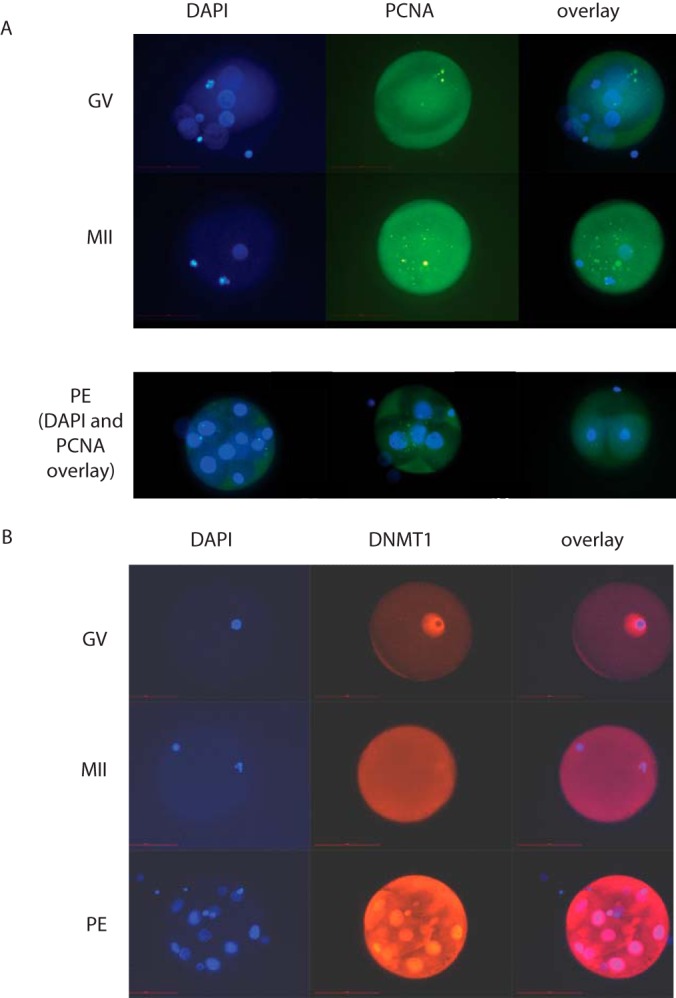
Immunofluorescence microscopy of PCNA (A) and DNMT1 (B) in oocytes at the germinal vesicle (GV) state, in metaphase II (MII) and in parthenogenetic embryos (PE).
DISCUSSION
In this study, we optimized the procedure for collecting human oocytes for proteome analysis and used SP3 as a novel sample preparation method to scale down proteome analysis to single oocytes. This not only generated the first single-cell proteome of a human cell but also identified proteins that are differentially expressed in mature and immature oocytes. In addition, we analyzed the secretome of human oocytes for the first time, extending our proteome data that indicated protein secretion as a prominent biological process in oocytes.
Using the hanging drop system, we identified 2,154 different proteins from pools of 100 human oocytes. Global gene ontology analysis revealed that oocytes express a repertoire of proteins focusing on cellular homeostasis and secretion, along with a specific subproteome involved in fertilization and reproduction. Of particular interest is the relatively large proportion of 158 oocyte-enriched proteins that had not been observed in other proteomes and that may thus fulfill important roles in sustaining oocyte functionality. For example, the epigenetics-related protein ES cell-associated transcript 1 (ECAT1/KHDC3L) identified here is known to be expressed in blastocysts (40) and thus supports the sole report on the expression of gene ECAT1 (C6orf221) in human oocytes (51). Its high importance for early human development is indicated by the association of mutated ECAT1 with recurrent biparental complete hydatidiform mole, an abnormal form of pregnancy where a nonviable fertilized oocyte implants in the uterus that does not come to the term (50–52). Other functional groups that likely sustain oocyte-specific processes include proteins involved in lipid metabolism, in line with the recognition that fatty acid utilization promotes oocyte quality (36, 37). The extended repertoire of oocyte-enriched proteins identified here may provide a lead toward a panel of markers indicating oocyte quality.
We consistently identified ∼450 proteins in single oocytes, which in itself reflects a significant technological advancement considering the very small amount of starting material. Oocytes have a diameter of 100 μm, corresponding to a volume of 0.5E6 μm3 and an estimated protein content of 100 ng (20, 21). By comparison, the volume of a HeLa cell has been estimated between 1,000–5,000 μm3 (56, 57), i.e. one oocyte corresponds to 100–500 HeLa cells. In contrast, Xenopus oocytes are 1 mm in diameter and are thus 1,000x bigger in size and protein content than human oocytes. Therefore, previous proteomic studies on Xenopus oocytes have benefited from the large cell size to operate at the single-cell level (25, 26) without being severely limited by protein amount.
As a result, our study marks an important step forward in oocyte biology. Especially the identification of many oocyte-specific and proteins that have been associated with fertility-related disorders implies that single-cell proteomics may be highly informative to monitor oocyte quality in a personalized manner. In addition, by label-free quantification, we identified Wee2, DNMT1, and PCNA to be preferentially expressed in MII-stage oocytes. The close functional and physical interaction between these proteins to control DNA replication strongly suggests that maintaining genome integrity is crucial in MII oocytes. The finding that TDRKH was highly expressed in GV oocytes is interesting from the perspective that PIWIL3, another protein in the piRNA biogenesis pathway, was identified in human oocytes for the first time. This not only indicates that piRNA biogenesis is operational in human oocytes but also that this process is regulated especially at the GV stage. Added to the notion that piRNAs have an established role in the protection against transposon-mediated genomic rearrangements (58), this may spark future studies focusing on the exact timing and mechanism of this process in oocytes.
At the same time, the wider implication of our study is that many other rare biological specimen may now come within reach for effective proteomic interrogation. Many examples exist, ranging from FACS-sorted cell populations to a large diversity of clinical samples. Importantly, the message of our study is that one of the main bottle necks in analyzing such samples is in sample preparation. Among the key characteristics contributing to the power of SP3 are the compatibility with buffers for protein extraction, efficiency in capturing proteins and peptides, and short incubation steps all in a single vessel. SP3-mediated sample preparation may not only be beneficial for proteomic approaches using liquid chromatography for peptide separation but also for capillary electrophoresis-based platforms that have shown excellent performance in subsequent MS-detection of samples in the ∼20 ng range (59). We wish to emphasize that after SP3 we analyzed samples via LC-MS using modern (Orbitrap velos, Q-Exactive “classic”) but not latest-generation mass spectrometers (e.g. Orbitrap Fusion, Q-Exactive-HF). Since these or similar instruments are widely used in the proteomics community, implementation of SP3 should empower many labs to handle quantity-limited samples in a routine fashion.
We conclude that the hanging drop culture system in combination with efficient sample preparation via SP3 represents an exciting new technology to study the human oocyte proteome and secretome. The ability to do so even for single cells opens the way to gain better insight into oocyte biology, female (in)fertility, and human preimplantation development in the future.
Supplementary Material
Acknowledgments
We would like to thank all patients who kindly donated their oocytes for this research and the staff of the Reproductive Unit, Department of Obstetrics and Gynaecology, University Medical Centre Ljubljana for their support. The EMBL Proteomics Core Facility is gratefully acknowledged for their expert support.
Footnotes
Author contributions: I.V.K., C.H., and J.K. designed the research; I.V.K., and S.L. performed the research; I.V.K., S.L., C.H., and J.K. analyzed data; and I.V.K. and J.K. wrote the paper.
 This article contains supplemental material.
This article contains supplemental material.
The authors declare no conflict of interest.
1 The abbreviations used are:
- ECAT
- ES cell-associated transcript 1 protein
- PIWIL
- Piwi-like protein
- NLRP
- NACHT, LRR and PYD domains-containing protein
- FBS
- fetal bovine serum
- GV
- germinal vesicle
- IVF
- in vitro fertilization
- LC-MSMS
- liquid chromatography coupled to tandem mass spectrometry
- LFQ
- label-free quantification
- MII
- metaphase II
- PBS
- phosphate-buffered saline
- SP3
- single-pot solid-phase-enhanced sample preparation
- ZP
- zona pellucida
- JUP
- Junction plakoglobin
- LOXL
- Lysyl oxidase-like protein
- ICAM
- Intercellular adhesion molecule
- ACE
- Angiotensin-converting enzyme
- CDH
- Cadherin
- CD
- cluster of differentiation
- ITGA
- integrin alpha
- ITGAV
- integrin alpha five
- ITGE
- integrin epsilon
- LIMA
- LIM domain and actin-binding protein
- PAK
- Serine/threonine-protein kinase
- TES
- testin
- ZP
- Zona pellucida sperm-binding protein
- BMP
- Bone morphogenetic protein
- GDF9
- Growth/differentiation factor 9
- HENMT
- HEN1 methyltransferase homolog
- BCL2L
- Bcl-2-like protein
- CHEK
- Checkpoint kinase
- PTTG1
- Pituitary tumor-transforming gene 1 protein
- ACAN
- Aggrecan core protein
- KAL
- Kallmann syndrome protein
- ACE
- Angiotensin-converting enzyme
- OVCH
- ovochymase
- ASTL
- Ovastacin
- HTRA1
- Serine protease HTRA1
- SERPIN
- serine protease inhibitor
- ITIH
- Inter-alpha-trypsin inhibitor heavy chain
- ZAR
- Zygote arrest protein
- DPPA
- Developmental pluripotency-associated protein
- DNMT
- DNA (cytosine-5)-methyltransferase 3A
- TLE
- Transducin-like enhancer protein
- KPNA
- Karyopherin subunit alpha
- NLRP
- NACHT, LRR and PYD domains-containing protein
- OOEP
- Oocyte-expressed protein
- PADI
- Protein-arginine deiminase
- GPHA
- Glycoprotein hormone alpha
- BCL2L
- BCL2L: Bcl-2-like protein
- MUC
- mucin
- OOSP
- Oocyte-secreted protein
- OVCH
- ovochymase
- ANXA
- annexin alpha
- DIAPH
- Protein diaphanous homolog
- HADHA
- Trifunctional enzyme subunit alpha, mitochondrial
- ATAD
- ATPase family AAA domain-containing protein
- CCNI
- Cyclin-I
- LDHAL
- L-lactate dehydrogenase A-like
- PIWIL
- Piwi-like protein
- CGREF
- Cell growth regulator with EF hand domain protein
- ICAM1
- Intercellular adhesion molecule 1
- MIF
- Macrophage migration inhibitory factor
- WFDC
- WAP four-disulfide core domain protein
- CGREF
- Cell growth regulator with EF hand domain protein
- ICAM1
- Intercellular adhesion molecule 1
- MIF
- Macrophage migration inhibitory factor
- WFDC
- WAP four-disulfide core domain protein
- CYB
- Cytochrome b5 reductase
- FDXR
- Ferredoxin reductase
- ACAA
- Acetyl-CoA acyltransferase
- MFGE
- Lactadherin
- FKBP
- FK506-binding protein
- SOD1
- Superoxide dismutase 1
- SKP
- S-phase kinase-associated protein
- CALM
- calmodulin
- CUL
- cullin
- ITPR
- Inositol 1,4,5-trisphosphate receptor
- YWHAE
- YWHAB, YWHAH, YWHAG, YWHAQ, and YWHAZ: 14–3-3 proteins epsilon, beta, eta, gamma, theta and zeta, respectively
- CKAP
- Cytoskeleton-associated protein
- BUB
- Mitotic checkpoint serine/threonine-protein kinase BUB
- TACC
- Transforming acidic coiled-coil-containing protein
- FACS
- Fluorescence-activated cell-sorting.
REFERENCES
- 1. De La Fuente R., Viveiros M. M., Burns K. H., Adashi E. Y., Matzuk M. M., and Eppig J. J. (2004) Major chromatin remodeling in the germinal vesicle (GV) of mammalian oocytes is dispensable for global transcriptional silencing but required for centromeric heterochromatin function. Dev. Biol. 275, 447–458 [DOI] [PubMed] [Google Scholar]
- 2. De La Fuente R., and Eppig J. J. (2001) Transcriptional activity of the mouse oocyte genome: Companion granulosa cells modulate transcription and chromatin remodeling. Dev. Biol. 229, 224–236 [DOI] [PubMed] [Google Scholar]
- 3. Virant-Klun I., Knez K., Tomazevic T., and Skutella T. (2013) Gene expression profiling of human oocytes developed and matured in vivo or in vitro. BioMed Res. Intl. 2013, 879489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gasca S., Pellestor F., Assou S., Loup V., Anahory T., Dechaud H., De Vos J., and Hamamah S. (2007) Identifying new human oocyte marker genes: A microarray approach. Reprod. Biomed. Online 14, 175–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kocabas A. M., Crosby J., Ross P. J., Otu H. H., Beyhan Z., Can H., Tam W. L., Rosa G. J., Halgren R. G., Lim B., Fernandez E., and Cibelli J. B. (2006) The transcriptome of human oocytes. Proc. Natl. Acad. Sci. U.S.A. 103, 14027–14032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Labrecque R., and Sirard M. A. (2014) The study of mammalian oocyte competence by transcriptome analysis: Progress and challenges. Mol. Human Reprod. 20, 103–116 [DOI] [PubMed] [Google Scholar]
- 7. O'Shea L. C., Mehta J., Lonergan P., Hensey C., and Fair T. (2012) Developmental competence in oocytes and cumulus cells: Candidate genes and networks. Syst. Biol. Reprod. Med. 58, 88–101 [DOI] [PubMed] [Google Scholar]
- 8. Hafidh S., Capková V., and Honys D. (2011) Safe keeping the message: mRNP complexes tweaking after transcription. Adv. Exp. Med. Biol. 722, 118–136 [DOI] [PubMed] [Google Scholar]
- 9. MacNicol M. C., and MacNicol A. M. (2010) Developmental timing of mRNA translation–integration of distinct regulatory elements. Mol. Reprod. Dev. 77, 662–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guzeloglu-Kayisli O., Lalioti M. D., Babayev E., Torrealday S., Karakaya C., and Seli E. (2014) Human embryonic poly(A)-binding protein (EPAB) alternative splicing is differentially regulated in human oocytes and embryos. Mol. Human Reprod. 20, 59–65 [DOI] [PubMed] [Google Scholar]
- 11. Tay J., Hodgman R., and Richter J. D. (2000) The control of cyclin B1 mRNA translation during mouse oocyte maturation. Dev. Biol. 221, 1–9 [DOI] [PubMed] [Google Scholar]
- 12. Traverso J. M., Donnay I., and Lequarre A. S. (2005) Effects of polyadenylation inhibition on meiosis progression in relation to the polyadenylation status of cyclins A2 and B1 during in vitro maturation of bovine oocytes. Mol. Reprod. Dev. 71, 107–114 [DOI] [PubMed] [Google Scholar]
- 13. Zhang D. X., Cui X. S., and Kim N. H. (2010) Molecular characterization and polyadenylation-regulated expression of cyclin B1 and Cdc2 in porcine oocytes and early parthenotes. Mol. Reprod. Dev. 77, 38–50 [DOI] [PubMed] [Google Scholar]
- 14. Chen J., Torcia S., Xie F., Lin C. J., Cakmak H., Franciosi F., Horner K., Onodera C., Song J. S., Cedars M. I., Ramalho-Santos M., and Conti M. (2013) Somatic cells regulate maternal mRNA translation and developmental competence of mouse oocytes. Nature Cell Biol. 15, 1415–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wühr M., Freeman R. M. Jr., Presler M., Horb M. E., Peshkin L., Gygi S. P., and Kirschner M. W. (2014) Deep proteomics of the Xenopus laevis egg using an mRNA-derived reference database. Curr. Biol. 24, 1467–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Virant-Klun I., and Krijgsveld J. (2014) Proteomes of animal oocytes: What can we learn for human oocytes in the in vitro fertilization programme? BioMed Res. Intl. 2014, 856907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang S., Kou Z., Jing Z., Zhang Y., Guo X., Dong M., Wilmut I., and Gao S. (2010) Proteome of mouse oocytes at different developmental stages. Proc. Natl. Acad. Sci. U.S.A. 107, 17639–17644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pfeiffer M. J., Siatkowski M., Paudel Y., Balbach S. T., Baeumer N., Crosetto N., Drexler H. C., Fuellen G., and Boiani M. (2011) Proteomic analysis of mouse oocytes reveals 28 candidate factors of the “reprogrammome.” J. Proteome Res. 10, 2140–2153 [DOI] [PubMed] [Google Scholar]
- 19. Albe K. R., Butler M. H., and Wright B. E. (1990) Cellular concentrations of enzymes and their substrates. J. Theoret. Biol. 143, 163–195 [DOI] [PubMed] [Google Scholar]
- 20. Grealy M., Diskin M. G., and Sreenan J. M. (1996) Protein content of cattle oocytes and embryos from the two-cell to the elongated blastocyst stage at day 16. J. Reprod. Fertility 107, 229–233 [DOI] [PubMed] [Google Scholar]
- 21. Pujol M., López-Béjar M., and Paramio M. T. (2004) Developmental competence of heifer oocytes selected using the brilliant cresyl blue (BCB) test. Theriogenology 61, 735–744 [DOI] [PubMed] [Google Scholar]
- 22. Lin-Moshier Y., and Marchant J. S. (2013) The Xenopus oocyte: A single-cell model for studying Ca2+ signaling. Cold Spring Harbor Protocols 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McGivern J. V., Swaney D. L., Coon J. J., and Sheets M. D. (2009) Toward defining the phosphoproteome of Xenopus laevis embryos. Dev. Dynamics 238, 1433–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wühr M., Güttler T., Peshkin L., McAlister G. C., Sonnett M., Ishihara K., Groen A. C., Presler M., Erickson B. K., Mitchison T. J., Kirschner M. W., and Gygi S. P. (2015) The nuclear proteome of a vertebrate. Curr. Biol. 25, 2663–2671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Smits A. H., Lindeboom R. G., Perino M., van Heeringen S. J., Veenstra G. J., and Vermeulen M. (2014) Global absolute quantification reveals tight regulation of protein expression in single Xenopus eggs. Nucleic Acids Res. 42, 9880–9891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sun L., Bertke M. M., Champion M. M., Zhu G., Huber P. W., and Dovichi N. J. (2014) Quantitative proteomics of Xenopus laevis embryos: Expression kinetics of nearly 4000 proteins during early development. Scientific Rep. 4, 4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hughes C. S., Foehr S., Garfield D. A., Furlong E. E., Steinmetz L. M., and Krijgsveld J. (2014) Ultrasensitive proteome analysis using paramagnetic bead technology. Mol. Syst. Biol. 10, 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 29. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., and Mann M. (2011) Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 30. Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., and Selbach M. (2011) Global quantification of mammalian gene expression control. Nature 473, 337–342 [DOI] [PubMed] [Google Scholar]
- 31. Cox J., Hein M. Y., Luber C. A., Paron I., Nagaraj N., and Mann M. (2014) Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 13, 2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Petersen T. N., Brunak S., von Heijne G., and Nielsen H. (2011) SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786 [DOI] [PubMed] [Google Scholar]
- 33. Huang da W., Sherman B. T., and Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protocols 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 34. Kanehisa M., and Goto S. (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Geiger T., Wehner A., Schaab C., Cox J., and Mann M. (2012) Comparative proteomic analysis of eleven common cell lines reveals ubiquitous but varying expression of most proteins. Mol. Cell. Proteomics 11, M111 014050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dunning K. R., and Robker R. L. (2012) Promoting lipid utilization with l-carnitine to improve oocyte quality. Animal Reprod. Sci. 134, 69–75 [DOI] [PubMed] [Google Scholar]
- 37. Dunning K. R., Russell D. L., and Robker R. L. (2014) Lipids and oocyte developmental competence: The role of fatty acids and beta-oxidation. Reproduction 148, R15–R27 [DOI] [PubMed] [Google Scholar]
- 38. Phanstiel D. H., Brumbaugh J., Wenger C. D., Tian S., Probasco M. D., Bailey D. J., Swaney D. L., Tervo M. A., Bolin J. M., Ruotti V., Stewart R., Thomson J. A., and Coon J. J. (2011) Proteomic and phosphoproteomic comparison of human ES and iPS cells. Nat. Methods 8, 821–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Amaral A., Castillo J., Ramalho-Santos J., and Oliva R. (2014) The combined human sperm proteome: Cellular pathways and implications for basic and clinical science. Human Reprod. Update 20, 40–62 [DOI] [PubMed] [Google Scholar]
- 40. Jensen P. L., Beck H. C., Petersen J., Hreinsson J., Wånggren K., Laursen S. B., Sorensen P. D., Christensen S. T., and Andersen C. Y. (2013) Proteomic analysis of human blastocoel fluid and blastocyst cells. Stem Cell Dev. 22, 1126–1135 [DOI] [PubMed] [Google Scholar]
- 41. Wu X., Viveiros M. M., Eppig J. J., Bai Y., Fitzpatrick S. L., and Matzuk M. M. (2003) Zygote arrest 1 (Zar1) is a novel maternal-effect gene critical for the oocyte-to-embryo transition. Nat. Genetics 33, 187–191 [DOI] [PubMed] [Google Scholar]
- 42. Bakhtari A., and Ross P. J. (2014) DPPA3 prevents cytosine hydroxymethylation of the maternal pronucleus and is required for normal development in bovine embryos. Epigenetics 9, 1271–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ratnam S., Mertineit C., Ding F., Howell C. Y., Clarke H. J., Bestor T. H., Chaillet J. R., and Trasler J. M. (2002) Dynamics of DNMT1 methyltransferase expression and intracellular localization during oogenesis and preimplantation development. Dev. Biol. 245, 304–314 [DOI] [PubMed] [Google Scholar]
- 44. Bebbere D., Ariu F., Bogliolo L., Masala L., Murrone O., Fattorini M., Falchi L., and Ledda S. (2014) Expression of maternally derived KHDC3, NLRP5, OOEP and TLE6 is associated with oocyte developmental competence in the ovine species. BMC Dev. Biol. 14, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang X., Park K. E., Koser S., Liu S., Magnani L., and Cabot R. A. (2012) KPNA7, an oocyte- and embryo-specific karyopherin alpha subtype, is required for porcine embryo development. Reprod. Fertility Dev. 24, 382–391 [DOI] [PubMed] [Google Scholar]
- 46. Hu J., Wang F., Yuan Y., Zhu X., Wang Y., Zhang Y., Kou Z., Wang S., and Gao S. (2010) Novel importin-alpha family member Kpna7 is required for normal fertility and fecundity in the mouse. J. Biol. Chem. 285, 33113–33122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ezkurdia I., Vázquez J., Valencia A., and Tress M. (2014) Analyzing the first drafts of the human proteome. J. Proteome Res. 13, 3854–3855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Savitski M. M., M W. I., Hahne H., Kuster B., and Bantscheff M. (2015) A scalable approach for protein false discovery rate estimation in large proteomic data sets. Mol. Cell Proteomics 14, 2394–2404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Roovers E. F., Rosenkranz D., Mahdipour M., Han C. T., He N., Chuva de Sousa Lopes S. M., van der Westerlaken L. A., Zischler H., Butter F., Roelen B. A., and Ketting R. F. (2015) Piwi proteins and piRNAs in mammalian oocytes and early embryos. Cell Reports 10, 2069–2082 [DOI] [PubMed] [Google Scholar]
- 50. Fallahian M., Sebire N. J., Savage P. M., Seckl M. J., and Fisher R. A. (2013) Mutations in NLRP7 and KHDC3L confer a complete hydatidiform mole phenotype on digynic triploid conceptions. Human Mutation 34, 301–308 [DOI] [PubMed] [Google Scholar]
- 51. Parry D. A., Logan C. V., Hayward B. E., Shires M., Landolsi H., Diggle C., Carr I., Rittore C., Touitou I., Philibert L., Fisher R. A., Fallahian M., Huntriss J. D., Picton H. M., Malik S., Taylor G. R., Johnson C. A., Bonthron D. T., and Sheridan E. G. (2011) Mutations causing familial biparental hydatidiform mole implicate c6orf221 as a possible regulator of genomic imprinting in the human oocyte. Am. J. Human Genetics 89, 451–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Reddy R., Akoury E., Phuong Nguyen N. M., Abdul-Rahman O. A., Dery C., Gupta N., Daley W. P., Ao A., Landolsi H., Ann Fisher R., Touitou I., and Slim R. (2013) Report of four new patients with protein-truncating mutations in C6orf221/KHDC3L and colocalization with NLRP7. Eur. J. Human Genetics 21, 957–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chhikara N., Saraswat M., Tomar A. K., Dey S., Singh S., and Yadav S. (2012) Human epididymis protein-4 (HE-4): A novel cross-class protease inhibitor. PloS One 7, e47672. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 54. Chudecka-Glaz A. M., Cymbaluk-Ploska A. A., Menkiszak J. L., Sompolska-Rzechula A. M., Toloczko-Grabarek A. I., and Rzepka-Górska I. A. (2014) Serum HE4, CA125, YKL-40, bcl-2, cathepsin-L and prediction optimal debulking surgery, response to chemotherapy in ovarian cancer. J. Ovarian Res. 7, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhang J., Zhang W., Zou D., Chen G., Wan T., Li N., and Cao X. (2003) Cloning and functional characterization of GNPI2, a novel human homolog of glucosamine-6-phosphate isomerase/oscillin. J. Cell. Biochem. 88, 932–940 [DOI] [PubMed] [Google Scholar]
- 56. Cohen L. S., and Studzinski G. P. (1967) Correlation between cell enlargement and nucleic acid and protein content of HeLa cells in unbalanced growth produced by inhibitors of DNA synthesis. J. Cell. Physiol. 69, 331–339 [DOI] [PubMed] [Google Scholar]
- 57. Zhao L., Kroenke C. D., Song J., Piwnica-Worms D., Ackerman J. J., and Neil J. J. (2008) Intracellular water-specific MR of microbead-adherent cells: The HeLa cell intracellular water exchange lifetime. NMR in Biomed. 21, 159–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Saito K., and Siomi M. C. (2010) Small RNA-mediated quiescence of transposable elements in animals. Dev. Cell 19, 687–697 [DOI] [PubMed] [Google Scholar]
- 59. Lombard-Banek C., Moody S. A., and Nemes P. (2016) Single-cell mass spectrometry for discovery proteomics: Quantifying translational cell heterogeneity in the 16-cell frog (Xenopus) embryo. Angew. Chem. 55, 2454–2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.