

Abstract
Protein kinase Cϵ (PKCϵ) promotes synaptic maturation and synaptogenesis via activation of synaptic growth factors such as BDNF, NGF, and IGF. However, many of the detailed mechanisms by which PKCϵ induces synaptogenesis are not fully understood. Accumulation of PSD-95 to the postsynaptic density (PSD) is known to lead to synaptic maturation and strengthening of excitatory synapses. Here we investigated the relationship between PKCϵ and PSD-95. We show that the PKCϵ activators dicyclopropanated linoleic acid methyl ester and bryostatin 1 induce phosphorylation of PSD-95 at the serine 295 residue, increase the levels of PSD-95, and enhance its membrane localization. Elimination of the serine 295 residue in PSD-95 abolished PKCϵ-induced membrane accumulation. Knockdown of either PKCϵ or JNK1 prevented PKCϵ activator-mediated membrane accumulation of PSD-95. PKCϵ directly phosphorylated PSD-95 and JNK1 in vitro. Inhibiting PKCϵ, JNK, or calcium/calmodulin-dependent kinase II activity prevented the effects of PKCϵ activators on PSD-95 phosphorylation. Increase in membrane accumulation of PKCϵ and phosphorylated PSD-95 (p-PSD-95S295) coincided with an increased number of synapses and increased amplitudes of excitatory post-synaptic potentials (EPSPs) in adult rat hippocampal slices. Knockdown of PKCϵ also reduced the synthesis of PSD-95 and the presynaptic protein synaptophysin by 30 and 44%, respectively. Prolonged activation of PKCϵ increased synapse number by 2-fold, increased presynaptic vesicle density, and greatly increased PSD-95 clustering. These results indicate that PKCϵ promotes synaptogenesis by activating PSD-95 phosphorylation directly through JNK1 and calcium/calmodulin-dependent kinase II and also by inducing expression of PSD-95 and synaptophysin.
Keywords: phosphorylation, protein kinase CϵPKC), protein translocation, scaffold protein, synapse, PSD-95
Introduction
Protein kinase Cϵ (PKCϵ) is one of the novel PKC isotypes and is characterized as a calcium-independent and phorbol ester/diacylglycerol-sensitive serine/threonine kinase. Among the novel PKCs, PKCϵ is the most abundant species in the central nervous system, mediating various neuronal functions (1, 2). In neuroblastoma cells overexpression of PKCϵ, but not PKCα, -βII, or -δ leads to neurite outgrowth through interaction of actin filaments and the C1 domain of PKCϵ (3–5). The actin binding site of PKCϵ is also implicated in exocytosis of neurotransmitters (6). PKCϵ is essential for many types of learning and memory (7, 8) and neuroprotection (9–13). Neuronal contact with astrocytes also promotes global synaptogenesis through PKCϵ signaling (14). PKCϵ activation has been shown to promote the maturation of dendritic synapses during associative learning (9). PKCϵ activation also protects against neurodegeneration (10, 15). Phosphorylation of long-tailed AMPA receptors GluA4 and GluA1 by PKC promotes their surface expression (16, 17). PKC activation induces protein synthesis required for long term memory (12, 18). PKCϵ activation is also required for HuD-mediated mRNA stabilization of neurotrophic factors (19) and apoE-mediated epigenetic regulation of BDNF (20). PKC activation induces translocation of calcium/calmodulin-dependent kinase II (CaMKII)2 to synapses (21) where it participates in PSD-95-induced synaptic strengthening (22). PKC also promotes NMDA receptor trafficking by indirectly triggering CaMKII autophosphorylation and subsequent increased association with NMDA receptors (23).
Thus, a number of studies have suggested that PKC activators such as bryostatin and dicyclopropanated linoleic acid methyl ester (DCPLA-ME) may be useful therapeutic candidates for the treatment of Alzheimer disease and other causes of synaptic loss such as ischemia, stroke, and fragile X syndrome (5, 6, 14, 24). Some of these benefits have been attributed to induction of neurotrophic factors such as BDNF or the activation of anti-Aβ repair pathways and anti-apoptotic activity (10, 13, 20, 25). However, the biochemical mechanisms by which PKCϵ induces synaptogenesis and mediates neuroprotection are still not fully understood.
At excitatory synapses, the postsynaptic density is characterized by an electron-dense thick matrix that contains key molecules involved in the regulation of glutamate receptor targeting and trafficking (26). PSD-95 is an abundant scaffold protein in excitatory synapses, where it functions to cluster proteins such as glutamate receptors on the postsynaptic membrane and couples them to downstream signaling molecules, thereby inducing the surface expression and synaptic insertion of glutamate receptors (27–29). In addition to its role in synaptic function, PSD-95 has also been proposed to affect synapse maturation and stabilization (30–32) and, thus, synapse number. Phosphorylation of the serine 295 residue of PSD-95 enhances the synaptic accumulation of PSD-95 and its ability to recruit surface α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors and potentiate excitatory postsynaptic currents (33).
In the present study we examined the role of PKCϵ signaling and PKCϵ activators in PSD-95 regulation and induction of synaptogenesis in cultured neurons and CA1 hippocampal slices. We report that PKCϵ activation induces membrane translocation and phosphorylation of PSD-95 at the serine 295 residue, coinciding with an increased number of synapses. Our data suggest that an important mechanism by which PKCϵ induces synaptogenesis is by increasing the phosphorylation of PSD-95 at the postsynaptic site and by regulating the expression of synaptophysin at the presynaptic site.
Results
PKC Activation Prevents Degradation of Primary Human Neurons
PKCϵ is present in high concentration in central neuronal tissues and has been implicated in broad spectrum neuronal functions. To determine the effect of PKCϵ activation on survival and maintenance, primary human neurons were treated for 40 days with two different PKC activators (bryostatin 1 and DCPLA-ME, which are relatively specific for PKCϵ) (13, 34–36). Culture media and activators were changed every 3 days. Cells were imaged from three independent wells every 5 days, and neurite-positive cells were counted from 508-μm2 field images. Cells treated with either DCPLA-ME (100 nm) or bryostatin 1 (0.27 nm) showed an improved survival with increased neuritic branching (Fig. 1A). Untreated cells showed degeneration and 50% cell loss by 36 days, whereas the treated cells remained healthy for at least 40 days (Fig. 1B). The number of viable neurite-positive cells was also significantly higher at 40 days (F(2,6) = 705.4; ANOVA, p < 0.0001) in the activator-treated cells than untreated cells (bryostatin 1, 369.7 ± 12.2; DCPLA-ME, 334.7 ± 1.8; untreated 109.7 ± 6.4).
FIGURE 1.

PKCϵ activation prevents degeneration of human primary neurons. Primary human neurons were treated with either DCPLA-ME (100 nm) or bryostatin 1 (0.27 nm) for 40 days. Fresh drug was added every third day with 50% media change. A, image of 40-day-old untreated (Control) and bryostatin 1- and DCPLA-ME-treated neurons. B, number of neurite-positive cells counted from three 20× fields (508 μm2) over time. DCPLA-ME and bryostatin 1 treatment stabilized cellular viability for at least 40 days. Viability of untreated cells declined after 20 days. C–E, PKCϵ, PSD-95 and synaptophysin mRNA levels in 40-day-old neurons compared with 1-day neurons. F, immunoblot analysis of PKCϵ, p-PSD-95S295, PSD-95, and synaptophysin in 40-day-old neurons compared with 1-day neurons. M, mass markers. G–J, immunostaining of p-PSD-95S295, PSD-95, and synaptophysin calculated from immunoblots. Staining is significantly higher in DCPLA-ME- and bryostatin 1-treated cells. Data are represented as the mean ± S.E. of three independent experiments (Student's t test. *, p < 0.05; **, p < 0.005).
Prolonged PKCϵ Activation Prevents Loss of Synaptic Proteins
We quantified the mRNA expression of PKCϵ, PSD-95, and synaptophysin at 40 days in untreated and PKCϵ activator-treated neurons. At 40 days the mRNA levels of PKCϵ (F(3,8) = 18.3; p = 0.0006) and PSD-95 (F(3,8) = 44.6; p < 0.0001) were significantly higher in the PKCϵ activator-treated cells compared with 40 day control cells (Fig. 1, C and D). Synaptophysin mRNA showed no significant change in between treated and untreated groups (Fig. 1E). We also quantified the protein expression of phosphorylated PSD-95 (p-PSD-95S295), PSD-95, and synaptophysin at 40 days in untreated and PKCϵ activator-treated neurons by immunoblot (Fig. 1F). Expression levels of PKCϵ (F(3,8) = 16.60; p < 0.001), p-PSD-95S295 (F(3,8) = 66.83; p < 0.0001), PSD-95 (F(3,8) = 21.22; p < 0.001) and synaptophysin were significantly higher in the 40-day PKCϵ activator-treated cells compared with 40-day control cells (Fig. 1, G–J). Moreover, protein expression levels of PKCϵ, PSD-95, and synaptophysin showed a marked decrease in 40-day untreated cells compared with 1-day cells, even after correction for total protein, whereas PKCϵ activation prevented the time-dependent loss. This indicates an essential role of PKCϵ in maintenance of synapses and preserving normal levels of both PSD-95 and synaptophysin.
Bryostatin 1 and DCPLA-ME Specifically Activate PKCϵ
We then investigated whether this phenomenon is specific to PKCϵ or whether other PKC isozymes are involved. PKC translocation to the plasma membrane generally has been considered the hallmark of activation and frequently has been used as a surrogate measure of PKC isoform activation in cells (37). Expression levels of PKCα, PKCϵ, and PKCδ in the soluble (cytosol) and particulate (membrane) were measured by immunoblot at 1, 4, and 24 h after either bryostatin 1 (0.27 nm) or DCPLA-ME (100 nm) treatment (Fig. 2, A and C). Both DCPLA-ME and bryostatin 1 increased membrane translocation of PKCϵ but not PKCα or PKCδ (Fig. 2, B and D), confirming that both the compounds activate PKCϵ but not PKCα or PKCδ.
FIGURE 2.
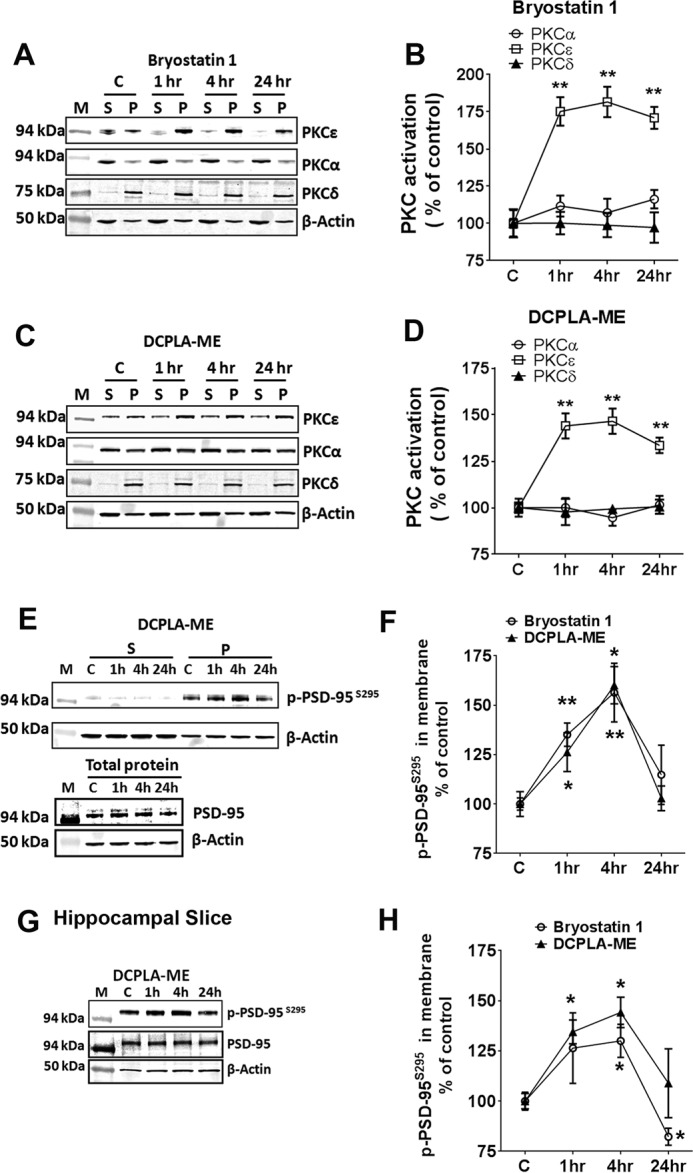
PKCϵ activation induces membrane accumulation of p-PSD-95S295. Primary human neurons were treated with ethanol (C), bryostatin 1 (0.27 nm), or DCPLA-ME (100 nm) for 1. 4, and 24 h. Neurons were separated into soluble (S) and membrane (P) fractions and immunoblotted against PKCα, PKCϵ, and PKCδ. A–C, and p-PSD-95S295, PSD-95 and β-actin. E, M represents molecular weight markers. PKC activation is reported as the percentage of total protein in the membrane. Bryostatin 1-treated neurons showed PKCϵ activation at 1 h (175.2 ± 9.5%; p = 0.002), 4 h (181.6 ± 10.2%; p = 0.0016), and 24 h (170.9 ± 7.4%; p = 0.001) compared with the untreated neurons (100.0 ± 3.1%; F(3,8) = 22.5; ANOVA, p < 0.0005) (B), and DCPLA-ME treated neurons showed a significant increase in PKCϵ activation at 1 h (144.0 ± 6.8%;p = 0.004), 4 h (146.5 ± 6.8%; p = 0.003), and 24 h (133.6 ± 4.2%; p = 0.003) compared with the untreated neurons (F(3,8) = 15.7; ANOVA, p = 0.001). D, induced PKCϵ activation at 1, 4, and 24 h. PKCϵ activation significantly increased the p-PSD-95S295 expression in the membranes at 1 h and 4 h. F, adult rat hippocampal organotypic slices were treated with ethanol (C), bryostatin 1 (0.27 nm), or DCPLA-ME (100 nm) for 1, 4, and 24 h. Protein lysates were immunoblotted against p-PSD-95S295, PSD-95, and β-actin. G, PKCϵ activation significantly increased the p-PSD-95S295 expression in the membranes at 1 and 4 h. H, data are represented as the mean ± S.E. of three independent experiments (Student's t test. *, p < 0.05; **, p < 0.005).
PKCϵ Activation Induces Membrane Translocation of Phosphorylated PSD-95 (Serine 295)
Phosphorylation of PSD-95 on serine 295 is known to promote localization of PSD-95 in the postsynaptic density (PSD), strengthening the excitatory synapse (33). To determine whether time-dependent PKCϵ activation has an effect on localization and expression of p-PSD-95S295, we measured the expression of p-PSD-95S295 in the soluble and particulate fractions of the primary human neurons at 1, 4, and 24 h post PKC activator treatment (Fig. 2, E and F). PKCϵ activation increased the level of p-PSD-95S295 in the particulate fraction of both bryostatin 1 (F(3,8) = 4.9; ANOVA, p = 0.03) and DCPLA-ME-treated cells (F(3,8) = 11.7; ANOVA, p = 0.003) (Fig. 2F). The total PSD-95 expression in whole cell lysate from primary human neurons was unchanged among different groups (Fig. 2E). At 4 h p-PSD-95S295 levels were significantly higher in bryostatin 1 (156.4 ± 14.9%; p = 0.01)- and DCPLA-ME (160.1 ± 9.5%; p = 0.003)-treated neurons compared with untreated neurons. In adult rat hippocampal slices PKCϵ activation increased p-PSD-95S295 expression at 1 and 4 h (bryostatin 1, F(3,8) = 4.95; ANOVA p = 0.031; DCPLA-ME: F(3,8) = 4.34; ANOVA p = 0.043) (Fig. 2, G and H). Negligible amounts of p-PSD-95S295 were detected in the soluble fraction. These results show that the increase in membrane localization of p-PSD-95S295 corresponded with the kinetics of PKCϵ activation at 1 and 4 h.
PKCϵ-mediated Phosphorylation of PSD-95 at Serine 295 Is Essential for Its Membrane Association
Purified recombinant human PSD-95 (r-PSD-95) protein was readily phosphorylated by activated recombinant PKCϵ (r-PKCϵ) in vitro, and the PKC inhibitor bisindolylmaleimide I (Go 6850) (BisI, 100 nm) blocked the reaction (Fig. 3A). Both bryostatin 1 and DCPLA-ME increased the amount of p-PSD-95S295 in vitro compared with unactivated PKC alone (bryostatin 1 + r-PKCϵ + r-PSD-95, 200.3 ± 5.06%, p = 0.004; DCPLA-ME + r-PKCϵ + r-PSD-95, 194.6 ± 12.95%, p = 0.032; r-PKCϵ + r-PSD-95 control, 146.9 ± 7.06%) (Fig. 3B). The PKC inhibitor BisI blocked the phosphorylation. These results show that PKCϵ can phosphorylate PSD-95 at serine 295 in vitro.
FIGURE 3.

PKCϵ-mediated phosphorylation of PSD-95 at serine 295 is essential for its membrane accumulation. A, immunoblot representing p-PSD-95S295 and PSD-95 expression after incubation of the different combinations of recombinant PKCϵ, PSD-95, PKCϵ activators, and PKCϵ inhibitors (inh) mentioned above at 37 °C for 10 min in vitro. B, bryostatin 1 (Bry, 0.27 nm) and DCPLA-ME (100 nm) induced the phosphorylation of PSD-95 at serine 295 position. C, expression of p-PSD-95S295 and PSD-95 and β-actin in HEK-293 cells transfected with empty vector, wild type human PSD-95, and mutant PSD-95S295K. D, expression of PSD-95, p-PSD-95S295, PKCϵ, and β-actin in the soluble (S) and particulate (P) fraction of wild-PSD-95- and PSD-95S295K-transfected HEK-293 cells treated with bryostatin 1 and DCPLA-ME for 4 h in the presence or absence of EAVSLKPT (5 μm). S, soluble fractions; P, membrane fractions. Percentage of total protein in the membrane; PKCϵ (E), PSD-95 (F) and p-PSD-95S295 (G). Data are represented as the mean ± S.E. of three independent experiments (Student's t test. *, p < 0.05; **, p < 0.005; ***, p < 0.005).
Next we tested if the serine 295 residue in PSD-95 is essential for its membrane translocation. We created two separate clones, one containing the wild type human PSD-95 and the other containing mutant-PSD-95S295K in which the serine residue at 295 (AGT) was changed to lysine (AAA). Both the clones were transfected and expressed in HEK-293 cells, and their expression was measured by immunoblot. Both transfected cell lines showed PSD-95 immunoreactivity against a PSD-95 antibody raised against the N-terminal region of PSD-95. The anti-p-PSD-95S295 antibody showed positive bands only with the wild type PSD-95-transfected cell lysate. Untransfected HEK-293 cells showed no PSD-95 expression (Fig. 3C). The wild PSD-95 and PSD-95S295K-expressing HEK-293 cells were then treated with bryostatin 1 and DCPLA-ME for 4 h in presence or absence of PKCϵ translocation inhibitor (EAVSLKPT; 5 μm) and fractionated into cytosol and membrane fractions. Only small amounts of p-PSD-95 were found in soluble fractions (Fig. 3D). As expected, bryostatin 1 and DCPLA-ME significantly increased membrane translocation of PKCϵ in both PSD-95- and PSD-95S295K-expressing cells (in wild type cells, bryostatin 1 = +54.4 ± 4.9%, p = 0.0004; DCPLA-ME = +19.1 ± 4.2%, p = 0.01; F(4,10) = 28.8, ANOVA, p < 0.0001) (Fig. 3E). PKCϵ activators also increased translocation of wild type PSD-95 (bryostatin 1, +29.9 ± 2.3%, p = 0.0002; DCPLA-ME, +20.5 ± 2.5%, p = 0.001; F(4,10) = 35, ANOVA, p < 0.0001) but not mutated PSD-95S295K, which lacks the PKCϵ phosphorylation site (F(2,6) = 0.75, ANOVA p = 0.51) (Fig. 3F).
To further verify that phosphorylation at serine 295 is essential for membrane association, we immunoblotted the membrane fraction against anti-p-PSD-95S295 antibody. The level of p-PSD-95S295 in the membrane was increased in the PKCϵ activator-treated cells (bryostatin 1, 151.7 ± 6.1%, p = 0.002; DCPLA-ME, 161.1 ± 10.4%, p = 0.004 (100 ± 2.8%); F(4,10) = 27.4, ANOVA, p < 0.0001) (Fig. 3G). Membrane p-PSD-95S295 translocation was blocked by the PKC inhibitor. Together, these results indicate that PKCϵ activation phosphorylates the serine 295 residue of PSD-95, and this phosphorylation is necessary for membrane accumulation of PSD-95.
PKCϵ-mediated Membrane Localization of p-PSD-95S295 Involves JNKI and CaMKII
Previously it has been reported that accumulation of PSD-95 in the PSD is increased by synaptic activity and by a Rac1-JNK1 signaling pathway (33). PKCϵ is involved in JNK activation in macrophages (38, 39), and CaMKII inhibitors inhibit PKC-mediated signaling in hippocampal neurons (40). Thus we investigated the involvement of PKCϵ, JNK, and CaMKII in PSD-95S295 translocation in primary human neurons. Cells were pretreated for 30 min with BisI (Go 6850) (100 nm, PKC inhibitor), SP600125 (20 μm, JNK inhibitor), or KN-93 (10 μm, CaMKII inhibitor) and then treated with PKCϵ activators for 4 h. Cells were fractioned into cytosolic and membrane fractions, and the membrane fractions were analyzed for the expression of p-PSD-95S295. The inhibitors alone reduced the expression of membrane-bound p-PSD-95S295 (F(4,10) = 23.04; ANOVA, p < 0.0001) (Fig. 4, A and C). DCPLA-ME treatment increased membrane localization of p-PSD-95S295 (147.3 ± 2.8%; F(5, 12) = 39.2; ANOVA, p < 0.0001). PSD-95 phosphorylation was prevented by blocking PKC activation using bisindolylmaleimide I (Fig. 4, B and D), confirming the involvement of PKCϵ in localization of p-PSD-95S295 in the membrane. The JNK inhibitor SP600125 and the CaMKII inhibitor K-93 also prevented PKCϵ-mediated phosphorylation and translocation of PSD-95 (Fig. 4, B and D).
FIGURE 4.

PKCϵ-mediated membrane localization of p-PSD-95S295 involves JNK1 and CaMKII. Immunoblot showing p-PSD-95S295 expression in the membrane of neurons treated with vehicle, bisindolylmaleimide I (100 nm, PKC inhibitor), SP600125 (20 μm, JNK inhibitor), and K-93 (10 μm, CaMKII inhibitor) for 4 h (A). B, p-PSD-95S295 expression in the membrane of neurons treated with DCPLA-ME in presence or absence of different inhibitors. C, the inhibitors alone reduced the expression of membrane-bound p-PSD-95S295. D, PKC, JNK, and CaMKII inhibitors prevented the effect of PKCϵ activation on p-PSD-95. Activated PKCϵ increased phosphorylation of PSD-95 (E), JNK1 (F), and CaMKII (G). H, immunoblot showing the down-regulation of PKCϵ and JNK in PKCϵ and JNK siRNA-treated neurons, respectively. I. Protein expression of p-PSD-95S295 and β-actin in the membrane of control, PKCϵ KD, and JNK KD neurons in the presence or absence of DCPLA-ME. J, either in PKCϵ or JNK knockdown human neurons, DCPLA-ME failed to induce the membrane accumulation of p-PSD-95S295. K, diagram representing PKCϵ-mediated membrane translocation of p-PSD-95S295 and involvement of JNK1 and CaMKII in the pathway. Data are represented as the mean ± S.E. of three independent experiments (Student's t test. *, p < 0.05; **, p < 0.005; ***, p < 0.0005). N.S., not significant; M, mass markers.
PKCϵ phosphorylated PSD-95 in vitro, incorporating 1.46 ± 0.05 mol of [32P]ATP/mol of PSD-95. Western blotting with p-PSD-95S295-specific antibody confirmed that this included the Ser-295 site (Fig. 3, A and B). PKC and JNK inhibitors fully inhibited the PKCϵ-mediated PSD-95 phosphorylation, whereas a CaMKII inhibitor partially prevented PSD-95 phosphorylation (Fig. 4E). PKCϵ also phosphorylated JNK1 in vitro, incorporating 1.02 ± 0.04 mol of [32P]ATP per mol of JNK1; BisI prevented JNK1 phosphorylation (Fig. 4F). PKC is also reported to phosphorylate CaMKII in vitro (41); we also found an increase in phosphorylation of CaMKII by PKCϵ (Fig. 4G). Because both JNK and CaMKII inhibitors prevented PSD-95 phosphorylation by PKCϵ (Fig. 4E), we considered the possibility that the JNK inhibitor might not be specific. Therefore, we performed a siRNA knockdown of PKCϵ and JNK in human neurons. PKCϵ or JNK knockdown caused a 50% reduction in their respective protein expression (Fig. 4H). DCPLA-ME failed to induce the membrane accumulation of p-PSD-95S295 in PKCϵ and JNK knockdown human neurons (F(5,12) = 24.6; ANOVA, p < 0.0001(Fig. 4, I and J). These results confirm that PKCϵ is required for membrane translocation of p-PSD-95S295 and that JNK and CaMKII are intermediates in the pathway (Fig. 4K).
PKCϵ Activation Induces Synaptogenesis in Adult Hippocampal Slices
Next we investigated if PKCϵ-mediated phosphorylation of PSD-95 at serine 295 leads to synaptogenesis. Because 4-h PKCϵ activator treatment produced the highest p-PSD-95S295 level, we quantified the number of synapses from within 100 μm2 of 30–35 CA1 regions from untreated, bryostatin 1, and DCPLA-ME-treated slices using electron microscopy (3 different slices in each group) (Fig. 5A). Both bryostatin 1 and DCPLA-ME increased the number of synapses at 4 h compared with only vehicle-treated control (8.97 ± 0.63, p = 0.002, n = 35; 6.97 ± 0.50, p = 0.04, n = 30; 5.77 ± 0.50; n = 35 CA1 areas, respectively) (Fig. 5B). Presynaptic vesicle density was measured in a series of three-dimensional stacked images from 6–10 presynaptic boutons from three different hippocampal slices. Bryostatin 1 treatment increased presynaptic vesicle density at 4 h (93.23 ± 4.1, p < 0.001, n = 30 presynaptic boutons) in comparison to control (71.33 ± 4.45, n = 22 presynaptic boutons).
FIGURE 5.

PKCϵ activation induces synaptogenesis. Adult rat hippocampal organotypic slices were treated with ethanol (C), bryostatin 1 (0.27 nm), or DCPLA-ME (100 nm) for 1 and 4 h. A, electron micrographs of the stratum radiatum in the hippocampal CA1 area (100 μm2 CA1 area at ×5000) treated with bryostatin 1 and DCPLA-ME for 4 h. 30–35 CA1 areas each from three different hippocampal slices were analyzed. Dendritic spines showing synapse are highlighted in yellow. B, PKCϵ activation increased the synapse number at 4 h (F(2,96) = 9.05; ANOVA p < 0.0005) in bryostatin 1 (8.97 ± 0.63, p < 0.005, n = 34 CA1 area) and DCPLA-ME (6.97 ± 0.5; p < 0.05, n = 30 CA1 area)-treated slices compared with control (5.77 ± 0.50). Typical traces of EPSPs evoked at a stimulus intensity of 200 μA from bryostatin 1-treated hippocampal slices after 1 h (C) and 4 h (F). The input-output response, reflecting basal synaptic transmission, increased after treatment with bryostatin1 (0.27 nm) after 1 h (D) and 4 h (G). Areas under the curves (AUC) were calculated to compare the basal levels of synaptic transmission. Bryostatin 1 increased the EPSP slope significantly at 1 h and 4 h (E and H). Data are represented as the mean ± S.E. of three independent experiments (Student's t test. *, p < 0.05; **, p < 0.005; ***, p < 0.0005).
Next we investigated the effect of bryostatin 1 on basal synaptic transmission of hippocampal CA1 pyramidal neurons to determine whether the new synapses are functional. Field potential recordings were measured from rat hippocampal slices. An input-output curve was calculated with stimulus intensity versus the slope of excitatory synaptic potentials (EPSPs) elicited in response to increasing intensity of stimulation to the Schaffer collateral. The mean EPSP slope increased with stronger intensity of stimulus. Slices preincubated with bryostatin 1 for 1 h exhibited greater EPSP slope without any change in fiber volley amplitude. This was abolished with 30-min pretreatment with the PKC inhibitor bisindolylmaleimide I (Go 6850) (BisI, 100 nm) (Fig. 5, C and D). Bryostatin 1 increased the area under the curve, which represents the overall basal synaptic transmission, and a PKC inhibitor prevented the increase (bryostatin1, 0.71 ± 0.08, p = 0.03; Bis1 + bryostatin 1, 0.49 ± 0.07; untreated (ethanol only), 0.51 ± 0.06) (Fig. 5E). Treatment of slices for 4 h with bryostatin (12 slices, 3 rats) dramatically increased the EPSP slope compared with the ethanol-treated slices (6 slices, 3 rats) (Fig. 5, F and G). The smaller response in the 4-h untreated slices compared with 1-h untreated slices may be attributed to the vehicle (ethanol) added to the slices. EPSPs in hippocampal slices are reduced by a smaller percentage after ethanol treatment (42). Thus, the prolonged treatment of slices with ethanol for 4 h may have slightly reduced the EPSP slope in these groups. Bryostatin increased the area under the curve by nearly 2-fold (p < 0.0001, Fig. 5H). These results suggest that bryostatin 1 treatment facilitates basal synaptic transmission in the Schaffer collateral commissural pathway of rat hippocampus and that the increase in EPSP slope is independent of the fiber volley.
Our results indicate that increased phosphorylation of PSD-95 by PKCϵ leads to an increase in synapse number with increased synaptic activity. Together these data demonstrate that the new synapses are functional.
PKCϵ Knockdown Reduces the Expression of PSD-95 and Synaptophysin
PKCϵ is known to perform important functions both in presynaptic (14) and postsynaptic sites. To investigate whether PKCϵ is essential for the expression of synaptic proteins, we measured the effect of PKCϵ knockdown (PKCϵ KD) and PKCϵ overexpression (PKCϵ OE) on the expression of postsynaptic PSD-95 and presynaptic synaptophysin. Knockdown of PKCϵ was achieved by transfecting the neurons with a mixture of siRNA containing a pool of three to five siRNAs. PKCϵ siRNA effectively reduced PKCϵ expression both at the mRNA and protein levels by 2- and 3.4-fold (Fig. 6, A and I) after 72 h. Scrambled siRNA did not cause any change in PKCϵ expression (Fig. 6, F and G). PKCϵ overexpression in the neurons was obtained by transfecting pCMV6-ENTRY vector containing human PKCϵ cDNA. Transfected neurons showed an ∼7.4 fold increase in PKCϵ mRNA level (Fig. 6A) and a 3.6-fold increase in PKCϵ protein level (Fig. 6, H and I). Overexpressing PKCϵ by 7-fold increased the level of synaptophysin mRNA by 59.3 ± 1.3% and also increased the level of PSD-95 by 71.6 ± 3.8%. Knockdown of PKCϵ had opposite effects (Fig. 6, B and C). PKCϵ overexpression or knockdown did not alter SNAP-25 and syntaxin-1 mRNA levels (Fig. 6, D and E). Loss of PKCϵ expression reduced the protein levels of PSD-95 by 30% (0.71 ± 0.07; p = 0.043 (Fig. 6K) and synaptophysin by 44% (0.56 ± 0.08; p = 0.021) (Fig. 6J) compared with controls transfected with scrambled siRNA. PKCϵ OE produced a 50% increase in synaptophysin (1.51 ± 0.1 versus 1.0 ± 0.1 in control; p = 0.015) (Fig. 6J) and a 30% increase in PSD-95 expression (1.31 ± 0.08 versus 1.0 ± 0.07 in control; p = 0.024) compared with vector-only transfected cells (Fig. 6K).
FIGURE 6.

PKCϵ is essential for the expression of PSD-95 and synaptophysin. Primary human neurons were transfected with empty vector/scrambled siRNA (C), PKCϵ siRNA (PKCϵ KD), or a PKCϵ overexpression vector (PKCϵ OE) following the method described under “Experimental Procedures.” Cells were analyzed 72 h after treatment. A, mRNA transcript levels of PKCϵ. B, PSD-95. C, synaptophysin. D, SNAP-25. E, syntaxin-1 in PKCϵ KD and PKCϵ OE neurons. PKCϵ KD suppressed, while PKCϵ OE induced PSD-95 and synaptophysin mRNA transcript. F, agarose gel image showing no effect of scrambled siRNA on PKCϵ mRNA. G, immunoblot showing protein expression of PKCϵ in untreated, PKCϵ siRNA, and scrambled siRNA-treated human neurons. H, immunoblot staining of PKCϵ, PSD-95, synaptophysin, and actin in control, PKCϵ KD, and PKCϵ OE neurons. I–K, graphical representation of protein expression levels of PKCϵ, PSD-95, synaptophysin in control, PKCϵ KD, and PKCϵ OE neurons. Data are represented as the mean ± S.E. of three independent experiments (Student's t test. *, p < 0.05; **, p < 0.05; ***, p < 0.0005).
Knockdown of PKCϵ Reduces Synaptogenesis
To further establish the role of PKCϵ in synaptogenesis and its underlying role in expression of PSD-95 and synaptophysin we used confocal microscopy to measure the effect of PKCϵ knockdown on the localization of PSD-95 and colocalization of PSD-95 and synaptophysin. Punctate colocalization (clusters of proximal pre- and post-synaptic markers on neurites) of PSD-95 and synaptophysin is widely accepted as an indicator of synapses (43, 44). Primary human neurons were treated with bryostatin 1or DCPLA-ME alone or after PKCϵ KD for 10 days. PSD-95 clusters and colocalized PSD-95 and synaptophysin (as recognized by staining grains along a 40-μm length of neurite, n = 10) were counted in 4 independent experiments (Fig. 7A). In normal cells, PKCϵ activation by bryostatin 1 and DCPLA-ME significantly induced PSD-95 clustering in the neurites compared with untreated controls (p < 0.05) (Fig. 7B). The number of synapses was also significantly higher in cells treated with bryostatin1 and DCPLA-ME than in untreated neurons at 10 days (Fig. 7C). The increase in the number of synapses was independent of the neuron density. We found no change in neuron density (measured by NeuN staining) after 10 days of PKCϵ activator treatment (supplemental Fig. 1). In PKCϵ KD cells, immunofluorescence staining of human neurons showed a loss of synaptic networks, and bryostatin 1 and DCPLA-ME had no effect. PKCϵ KD prevented the effect of PKCϵ activators, and more importantly, reduced the basal level of PSD-95 clusters and synapses by 50%. (Fig. 7, A–C).
FIGURE 7.

Loss of PKCϵ prevents synaptogenesis. A, confocal images of untreated and DCPLA-ME (100 nm)-, bryostatin 1 (0.27 nm)-, PKCϵ siRNA + DCPLA-ME (100 nm)-, and PKCϵ siRNA + bryostatin 1 (0.27 nm)-treated primary human neurons (10 days). Each condition is represented by five panels. Four square panels represent nucleus (blue), PSD-95 (green), synaptophysin (red), and merged image. The rectangular panel represents a magnified image of a 40-μm neurite. B, number of PSD 95 signal grains was measured along 40-μm neurite length (10 individual neurites from 4 independent slides). Bryostatin 1 and DCPLA-ME significantly increased the PSD-95 clusters per 40 μm neurite (F(2,9) = 4.5; ANOVA p < 0.05). C, synapses were quantified by the number of colocalized PSD-95 and synaptophysin signals. PKCϵ activation increased synapse number (F(2,9) = 6.1; ANOVA p < 0.05), and PKCϵ KO prevented the synaptogenic effect of PKC activators. D, immunoblot analysis of PKCϵ, p-PSD-95S295, PSD-95, and synaptophysin. PKCϵ knockdown (KD) reduced PKCϵ expression by 50% after 10 days in human neurons. E–G, at 10 days PKCϵ activation increased the expression of PSD-95 and synaptophysin significantly, but in PKCϵ KD cells their expressions were lower even after treatment with activators. Data are represented as the mean ± S.E. of at least three independent experiments (Student's t test. *, p < 0.05; **, p < 0.005; ***, p < 0.0005).
We also quantified the expression levels of PKCϵ, p-PSD-95S295, PSD-95, and synaptophysin by immunoblot after 10 days of PKCϵ-siRNA transfection (Fig. 7D). PKCϵ KD cells expressed significantly lower amounts of PSD-95 (F(5,12) = 19.24, ANOVA p < 0.0001) (Fig. 7, E and F) and synaptophysin (F(5,12) = 12.79, ANOVA p = 0.0002) (Fig. 7G). Bryostatin 1 and DCPLA-ME failed to induce PSD-95 and synaptophysin expression in PKCϵ KD neurons. Bryostatin 1, but not DCPLA-ME, produced a 40% decrease in PKCϵ protein staining (Fig. 7D). No loss in PKCϵ mRNA was found in bryostatin 1-treated neurons (data not shown). Down-regulation of PKC after activation by bryostatin 1 is a well documented phenomenon (45, 46).
We further confirmed the effect of long term PKCϵ activation on synaptogenesis using rat hippocampal brain slices. Slices were treated with bryostatin 1 and DCPLA-ME for 10 days. The serum-free culture medium was changed every 3 days with fresh additions of activators. Synapse number in each case was quantified using electron microscopy (Fig. 8A). Bryostatin 1 (7.97 ± 0.68, p = 0.013, n = 29 CA1 areas) and DCPLA-ME (8.71 ± 0.78, p = 0.001, n = 24 CA1 areas) treatment increased the number of synapses in hippocampal slices compared with vehicle-only treated slices (4.5 ± 0.45; n = 24 CA1 area) (Fig. 8B). Presynaptic vesicle density was also significantly higher in the bryostatin 1 (59.6 ± 6.4, p < 0.05, n = 19 presynaptic boutons)- and DCPLA-ME (60.4 ± 5.1, p = 0.04, n = 19 presynaptic boutons)-treated slices than vehicle-treated controls (48.4 ± 4.3, n = 20 presynaptic boutons) (Fig. 8, C and D). Together, these findings confirm that PKCϵ is essential for bryostatin 1- and DCPLA-ME-mediated increase in PSD-95 and synaptophysin expression leading to increased synaptogenesis at 10 days.
FIGURE 8.

PKCϵ activation induces synaptogenesis in hippocampal slices. A, electron microscopy of the hippocampal CA1 area from adult organotypic brain slices treated with vehicle (Control), DCPLA-ME (100 nm), and bryostatin 1 (0.27 nm) for 10 days. Dendritic spines showing synapse are highlighted in yellow. B, PKCϵ activation by bryostatin 1 and DCPLA-ME increased synapse number in adult organotypic brain slices (F(2,6) = 11.9; ANOVA p < 0.01). C, electron micrograph showing increased presynaptic vesicle density in PKC activator treated slices. The gray level of presynaptic vesicle stack from six to seven presynaptic boutons was measured from three different hippocampal slices. D represents dendritic spine, the red arrow marks synapse, and yellow marks presynaptic vesicles. D, bryostatin 1 and DCPLA-ME significantly induced the presynaptic vesicle density at 10 days. Data are represented as the mean ± S.E. of at least three independent experiments (Student's t test. *, p < 0.05; **, p < 0.005).
Discussion
The outgrowth of neurites and formation of synapses depends on interactions among a number of regulatory proteins. These interactions are required for synaptic structure rearrangement, spinogenesis, and synaptogenesis. PKCϵ is one of the key regulators of synaptogenesis (3, 24), and PKCϵ activators promote the maturation of dendritic spines (9, 47). PSD-95 is a scaffold protein that also plays an important role in formation of excitatory synapses (48, 49).
Here we showed that PKCϵ activation induces translocation and phosphorylation of PSD-95 at the serine 295 residue leading to PSD-95 accumulation at the postsynaptic density. Our findings showed that PKCϵ activation not only increased the survival of neurons but also preserved the neuronal structure. Untreated cells showed gradual degeneration over 25 days, suggesting that PKCϵ activation is beneficial for both maturation and survival of neurons, confirming a previous report by Hama et al. (14). We have shown that short term acute changes in PKCϵ activity induce structural and biochemical changes in post-synaptic density scaffolding protein PSD-95 as well as increased synaptic activity. Synaptic activity is important for neuronal survival. Synaptic activity induces expression of survival genes and suppresses pro-death genes (50). Therefore, the increased survival of neurons treated with PKCϵ activators may be due to the increased connectivity induced in the early stages; however, other factors such as elevated neurotrophins may also play a role. PKCϵ induces BDNF (10, 19), and elevated expression and release of BDNF is associated with elevated synaptic activity, which contributes to neuroprotection (51, 52).
PKCϵ activation and membrane translocation occur both presynaptically (14, 53) and postsynaptically (8) where it phosphorylates important substrate proteins required for synaptic facilitation and synaptogenesis. We found that p-PSD-95S295 accumulation increased in the membrane of PKCϵ-activated neurons and followed the same time course as PKCϵ activation at 1 h and 4 h. The serine 295 residue was essential for the PKCϵ-mediated membrane accumulation of PSD-95. In vitro, PKCϵ phosphorylated both PSD-95 and JNK1. The JNK1 inhibitor also prevented PKCϵ activation-mediated increase in p-PSD-95S295, confirming previous findings showing that serine 295 phosphorylation of PSD-95 is regulated by Rac1-JNK1 and PP1/PP2A signaling (33, 54). PKCϵ is involved in JNK activation; PKD, a downstream effector of PKC, also regulates JNK (38, 39, 55). Knockdown of either PKCϵ or JNK inhibited the PKCϵ activator-mediated p-PSD-95S295 accumulation in the membrane, thus confirming that PKCϵ and JNK act collectively in regulating PSD-95. Although it has been reported that synaptic localization of PSD-95 is regulated by JNK signaling and not by CaMKII (33, 56), our data demonstrate a role of both JNKI and CaMKII. This is possible as PKC activation induces a simultaneous translocation of CaMKII to synapses (21), and CaMKII activation is needed for PSD-95-induced synaptic strengthening (22). CaMKII is a downstream target of PKCϵ in many pathways, including the events responsible for the induction of neuroplastic changes associated with hyperalgesic priming (57). In this study we found that both JNK1 and CaMKII inhibitors prevented the PKCϵ-mediated membrane association of p-PSD-95S295. These results suggest that JNK1 and CaMKII are downstream to PKCϵ in events responsible for phosphorylation and membrane accumulation of PSD-95.
We also demonstrated that PKCϵ activation increases the levels of PSD-95 and the number of synapses. In adult hippocampal slices, bryostatin 1 increased basal synaptic activity. Our results indicate an important link between PKCϵ activation and the membrane localization of PSD-95, specifically enriching the membrane with the p-PSD-95S295 form, which is known to strengthen the excitatory synapses (33). PSD-95 also regulates membrane insertion of AMPA receptor and dendritic spine morphology during synaptic plasticity (22, 30–32).
Overexpression of PSD-95 converts silent synapses to functional synapses (58), whereas synaptophysin may be required for increased presynaptic vesicle density, thereby facilitating neurotransmitter release (59). We found that overexpressing PKCϵ in primary human neurons induces the mRNA and protein levels of PSD-95 and synaptophysin, whereas knockdown of PKCϵ reduces PSD-95 and synaptophysin mRNA and protein levels. Our results indicate that PKCϵ regulates the gene expression of PSD-95 and synaptophysin. PKCϵ may play a critical role in synapse maintenance by regulating the synthesis of PSD-95 and synaptophysin (18). PKCϵ is known to drive the mitogenic response and DNA synthesis (60) via the Raf-MEK-ERK cascade and regulates transcription of essential genes through JNK/AP1, NF-κB, and JAK/STAT cascades (61, 62). PSD-95 is a critical transcriptional target of NF-κB, which is known to induce excitatory synapse formation and regulate dendritic spine formation and morphology in murine hippocampal neurons (63). Synaptophysin mRNA expression is induced by the BDNF-cFos pathway (64). NF-κB and synaptophysin have a common regulator in BDNF (65). PKCϵ up-regulates BDNF expression (19–20, 66).
In conclusion, PKCϵ has two specific roles in synaptogenesis; at the postsynaptic site it regulates PSD-95, either directly or through JNKI and CaMKII, and at the presynaptic site it induces the expression of synaptophysin. Repeated treatment with PKCϵ activators induces synthesis of PKCϵ, PSD-95, and synaptophysin, resulting in an increased number of synapses. PKCϵ knockdown inhibits the synthesis of PSD-95 and synaptophysin leading to a reduced number of synapses. Besides the PKC-JNK1/CaMKII- PSD-95 pathway, PKCϵ can also induce synaptogenesis through the HuD-BDNF pathway. PKCϵ stabilizes HuD, which increases the stability and rate of translocation of target mRNAs. HuD increases as a result of PKCϵ activation after learning (67) and stabilizes the mRNA for BDNF, nerve growth factor (NGF), and neurotrophin-3 (NT-3) (19). PKCϵ activation induces the synthesis of BDNF (10, 20, 47), and BDNF induces transport of PSD-95 to the dendrites (68), which is required for maintenance of mature spines (69). Deficits of PKCϵ function could also contribute to the synapse loss in Alzheimer disease (15), whereas the therapeutic elimination of such deficits may offer a strategy for the treatment of synaptic loss in Alzheimer disease and other synaptic disorders.
Experimental Procedures
Materials
Bryostatin 1 was purchased from Biomol International (Farmingdale, NY). DCPLA-ME was synthesized in our laboratory following the method described earlier (34, 70) and shown to be specific for PKCϵ. Primary antibodies (rabbit polyclonal anti-PKCϵ (sc-214), rabbit polyclonal anti-PKCα (sc-208), rabbit polyclonal anti-PKCδ (sc-213), mouse monoclonal anti-synaptophysin (sc-17750), and mouse monoclonal anti-β-actin (sc-47778)) were obtained from Santa Cruz Biotechnology, Inc., (Santa Cruz, CA). Rabbit polyclonal anti-synaptophysin (TA300431) and rabbit polyclonal anti-phospho-PSD-95 (serine 295) (TA303850) were obtained from Origene (Rockville, MD), and rabbit polyclonal anti-PSD-95 (#3450) and rabbit polyclonal anti-JNK1/2 (#9258) were obtained from Cell Signaling Technology, Inc. (Danvers, MA). Chicken polyclonal anti-NeuN (ab134014) was obtained from Abcam (Cambridge, MA). All secondary antibodies were purchased from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA). The anti-chicken Cy5 conjugated antibody was purchased from Abcam. Bisindolylmaleimide I (Go 6850) and PKCϵ translocation inhibitor (EAVSLKPT) were obtained from Santa Cruz Biotechnology, and SP600125 and KN-93 were obtained from Cell Signaling Technology.
Cell Culture
Human primary neurons (hippocampal neurons, catalogue #1540, ScienCell Research Laboratories, Carlsbad, CA) were plated on poly-l-lysine-coated plates and were maintained in neuronal medium (ScienCell) supplemented with the neuronal growth supplement (NGS, ScienCell). For maintenance of neurons half of the media was changed every 3 days. Fresh activators were added with every media change. Human HEK-293 cells were obtained from ATCC, Manassas, VA. Cells were maintained in Eagle's minimum essential medium and 10% fetal bovine serum.
Organotypic Slice Culture
Organotypic hippocampal slices were prepared mainly according to the method described by Stoppini et al. (71) with slight modifications (72). Rats were sacrificed and immediately decapitated under sterile conditions. Brains were rapidly removed and placed into a chilled dissection medium composed of Hibernate A (BrainBits, Springfield, IL), 2% B27 supplement, 2 mm l-glutamine by GlutaMax and antibiotic-antimycotics (all from Invitrogen). The hippocampi were dissected out in fresh chilled dissection medium. Isolated hippocampi were washed in new chilled dissection medium and placed on a wet 3-mm paper on the Teflon stage of a manual tissue slice chopper (Vibratome Co., Saint Louis, MO) for coronal sectioning at 300 μm. Each slice with intact pyramidal and granular layers was transferred to one membrane insert (Millipore, Bedford, MA) in 12-well plates containing Neurobasal A, 20% horse serum, 2 mm l-glutamine, and antibiotics-antimycotics for 4 days. For long term maintenance slices were cultured in serum-free medium consisting of Neurobasal A with 2% B27, 2 mm l-glutamine, and antibiotic-antimycotics. Slices were incubated in a humidified 5% CO2 atmosphere at 37 °C. The entire medium was replaced with fresh medium at day 1. After that, half the medium was removed and replaced with fresh medium twice a week.
Cell Lysis and Western Blotting Analysis
Cells were harvested in homogenizing buffer containing 10 mm Tris-Cl (pH 7.4), 1 mm phenylmethylsulfonyl fluoride), 1 mm EGTA, 1 mm EDTA, 50 mm NaF, and 20 μm leupeptin and lysed by sonication. The homogenate was centrifuged at 100,000 × g for 15 min at 4 °C to obtain the cytosolic fraction (soluble) and membrane (particulate). The pellet was resuspended in the homogenizing buffer by sonication. For whole cell protein isolation from primary neurons the homogenizing buffer contained 1% Triton X-100. Protein concentration was measured using the Coomassie Plus (Bradford) Protein Assay kit (Pierce). After quantification, 20 μg of protein from each sample was subjected to SDS-PAGE analysis in a 4–20% gradient Tris-glycine polyacrylamide gel (Invitrogen). The separated protein was then transferred to a nitrocellulose membrane. The membrane was blocked with BSA and incubated with primary antibody overnight at 4 °C. All the primary antibodies were used at a 1:1000 dilution except rabbit polyclonal anti-p-PSD-95S295 (1:10000) and rabbit polyclonal anti-synaptophysin (1:10000). After incubation, it was washed 3× with Tris-buffered saline-Tween 20 and further incubated with alkaline phosphatase-conjugated secondary antibody at 1:10,000 dilution for 45 min. The membrane was finally washed 3× with Tris-buffered saline-Tween 20 and developed using the 1-step NBT-BCIP (nitro blue tetrazolium-5-bromo-4-chloro-3-indolyl phosphate) substrate (Pierce). The blot was imaged in an ImageQuant RT-ECL (GE Healthcare), and densitometric quantification was performed using IMAL software. For quantifying expression of a protein, the densitometric value for the protein of interest was normalized against β-actin (loading control).
Electrophysiology
Rats (1 month old) were euthanized, and hippocampus was isolated and sliced into 300-μm slices on a Leica VT1200S Vibratome. Slices were incubated in ACSF at room temperature for 1 h until recording (ACSF: 124 mm NaCl, 3 mm KCl, 1.2 mm MgSO4, 2.1 mm CaCl2, 1.4 mm NaH2PO4, 26 mm NaHCO2, and mm 20 dextrose, saturated with 95% O2 and 5% CO2, which maintains the pH at 7.4). Slices were treated with ethanol or bryostatin 1 for 1 h or 4 h. All recordings were made at room temperature. For synaptic stimulation and field EPSP recordings, pyramidal neurons in the CA1 field were identified with an Olympus BX50WI microscope. Field potential recordings were measured to determine synaptic function. A bipolar stimulating electrode (100-μm separation, FHC, Bowdoinham, ME) was placed in the hippocampal Schaffer collateral pathway to elicit EPSPs in CA1 stratum radiatum, EPSPs were recorded through patch pipettes (2–5 megaohms, 1.5 mm outer diameter, 0.86 mm inner diameter, P87 Brown-Flaming Puller, Sutter Instruments) filled with ACSF. All parameters including pulse duration, width, and frequency were computer-controlled. Constant-current pulse intensity was controlled by a stimulus isolation unit. Basal synaptic transmission, represented by input-output responses, was determined by the slopes of stabilized EPSP to different stimulus intensities. The strength of EPSPs was assessed by measuring the slopes (initial 20–80%) of the EPSPs rising phase.
PKC Assay
To measure PKC activity, 100 ng of recombinant PKCϵ (Sigma) was incubated for 15 min at 37 °C in the presence of 100 ng of JNK1 or 100 ng of PSD-95 or 100 ng of CaMKII, 4.89 mm CaCl2, 1.2 μg/μl phosphatidyl-l-serine, 0.18 μg/μl 1,2-dioctanoyl-sn-glycerol, 10 mm MgCl2, 20 mm HEPES (pH 7.4), 0.8 mm EDTA, 4 mm EGTA, 4% glycerol, 8 μg/ml aprotinin, 8 μg/ml leupeptin, 2 mm benzamidine, and 0.5 μCi of [γ-32P]ATP. [32P]Phosphoprotein formation was measured by adsorption onto phosphocellulose as described previously (70).
Knockdown and Overexpression
Human PSD-95 was cloned into pCDNA3.1 plasmid (GenScript, Piscataway, NJ). Mutant PSD-95 mutated at serine 295 residue was also cloned into pCDNA3.1 plasmid and was obtained from GenScript. PKCϵ knockdown was done using PKCϵ-siRNA constructs purchased from Santa Cruz Biotechnology. JNK knockdown was done using SAPK/JNK-siRNA from Cell Signaling Technology. Overexpression of PKCϵ was obtained by transfecting pCMV6-ENTRY vector containing human PKCϵ cDNA (Origene). Transfection was done using Lipofectamine 3000 (Invitrogen). Medium was changed after 6 h of Lipofectamine treatment. Protein expression was measured after 72 h of transfection.
Quantitative Real-time-PCR
Quantitative real-time-PCR was done following the method described earlier (13). Total RNA (500 ng) was reverse-transcribed using oligo(dT) and Superscript III (Invitrogen) at 50 °C for 1 h. The cDNA products were analyzed using a LightCycler 480 II (Roche Applied Science) PCR machine and LightCycler 480 SYBR Green 1 master mix following the manufacturer's protocol. Primers for PKCϵ (forward primer, AGCCTCGTTCACGGTTCTATGC; reverse primer, GCAGTGACCTTCTGCATCCAGA), PSD-95 (forward primer, TCCACTCTGACAGTGAGACCGA; reverse primer, CGTCACTGTCTCGTAGCTCAGA), synaptophysin (forward primer, TCGGCTTTGTGAAGGTGCTGCA; reverse primer, TCACTCTCGGTCTTGTTGGCAC), SNAP-25 (forward primer, CGTCGTATGCTGCAACTGGTTG; reverse primer, GGTTCATGCCTTCTTCGACACG), Syntaxin-1 (forward primer, TGGAGAACAGCATCCGTGAGCT; reverse primer, CCTCTCCACATAGTCTACCGCG); GAPDH (forward primer, GTCTCCTCTGACTTCAACAGCG; reverse primer, ACCACCCTGTTGCTGTAGCCAA) were purchased from Origene.
Electron Microscopy
Electron microscopy of slices were done following methods described earlier (9). Hippocampi were sectioned with a vibratome at 100 μm. Hippocampi were fixed in 1% OsO4. Electron micrographs (100 μm2 CA1 area at × 5000) were made of Epon-embedded hippocampal sections with a JEOL 200CX electron microscope. These sections were 90 nm thick and had been previously stained with uranyl acetate and lead citrate. During quantification, electron micrographs were digitally zoomed up to ×20,000 magnification. Spines were defined as structures that formed synapses with axon boutons and did not contain mitochondria. Presynaptic vesicle density was measured from within the presynaptic axonal boutons that were seen to form synapses with dendritic spines of diameter >600 nm. Increased numbers of presynaptic vesicles in axon boutons were measured as an increase in the frequency of axon boutons with presynaptic vesicles that occupied >50% of the cross-section space not occupied by other organelles.
Immunofluorescence and Confocal Microscopy
Cells were grown in four-chambered slides (Nunc) at low density. For immunofluorescence staining the cells were washed with PBS (pH 7.4) and fixed with 4% paraformaldehyde for 4 min. After fixation, cells were blocked and permeabilized with 5% horse serum and 0.3% Triton X-100 in 1× PBS for 30 min. Cells were washed 3× with 1 × PBS and incubated with primary antibodies (rabbit polyclonal anti-PSD-95, mouse monoclonal anti-synaptophysin, and chicken polyclonal anti-NeuN) for 1 h at 1:100 dilution. After the incubation slides were again washed 3× in 1 × PBS and incubated with the FITC anti-rabbit IgG, rhodamine anti-mouse IgG, and Cy5 anti-chicken IgY for 1 h at 1:400 dilution. Cells were further washed and mounted in Pro Long Gold antifade mounting solution (Invitrogen). Stained cells were viewed under the LSM 710 Meta confocal microscope (Zeiss) at 350-, 490-, 540-, and 650-nm excitation and 470-, 525-, 625-, and 667-nm emission for DAPI, FITC, rhodamine, and Cy5, respectively. Six individual fields at 40× or 63× oil lens magnification were analyzed for the mean fluorescence intensity in each channel. Punctate colocalization was done following the methods described earlier (43, 44).
Statistical Analysis
All experiments were performed at least three times. Data are represented as the mean ± S.E. All data were analyzed by one-way ANOVA and Newman-Keuls multiple comparison post test. Significantly different paired groups were further analyzed by two-tailed Student's t test using GraphPad Prism 6.1 software (La Jolla, CA). p values < 0.05 were considered statistically significant.
Author Contributions
A. S., T. J. N., and D. L. A. designed the study and wrote the paper. A. S. performed and analyzed all the biochemical and immunofluorescence experiments. J. H. performed and analyzed all the electron microscopy data. D. W. performed and analyzed the electrophysiology experiments.
Supplementary Material
The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Fig. 1.
- CaMKII
- calcium/calmodulin-dependent kinase II
- DCPLA-ME
- dicyclopropanated linoleic acid methyl ester
- ANOVA
- analysis of variance
- PSD
- postsynaptic density
- r-
- recombinant
- BisI
- bisindolylmaleimide I
- EPSP
- excitatory synaptic potential
- KD
- knockdown
- OE
- overexpression.
References
- 1. Akita Y. (2002) Protein kinase Cϵ (PKCϵ): its unique structure and function. J. Biochem. 132, 847–852 [DOI] [PubMed] [Google Scholar]
- 2. Chen Y., and Tian Q. (2011) The role of protein kinase Cϵ in neural signal transduction and neurogenic diseases. Front. Med. 5, 70–76 [DOI] [PubMed] [Google Scholar]
- 3. Zeidman R., Löfgren B., Pâhlman S., and Larsson C. (1999) PKCϵ, via its regulatory domain and independently of its catalytic domain, induces neurite-like processes in neuroblastoma cells. J. Cell Biol. 145, 713–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fagerström S., Påhlman S., Gestblom C., and Nånberg E. (1996) Protein kinase Cϵ is implicated in neurite outgrowth in differentiating human neuroblastoma cells. Cell Growth Differ. 7, 775–785 [PubMed] [Google Scholar]
- 5. Prekeris R., Hernandez R. M., Mayhew M. W., White M. K., and Terrian D. M. (1998) Molecular analysis of the interactions between protein kinase Cϵ and filamentous actin. J. Biol. Chem. 273, 26790–26798 [DOI] [PubMed] [Google Scholar]
- 6. Prekeris R., Mayhew M. W., Cooper J. B., and Terrian D. M. (1996) Identification and localization of an actin-binding motif that is unique to the ϵ isoform of protein kinase C and participates in the regulation of synaptic function. J. Cell Biol. 132, 77–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bank B., DeWeer A., Kuzirian A. M., Rasmussen H., and Alkon D. L. (1988) Classical conditioning induces long-term translocation of protein kinase C in rabbit hippocampal CA1 cells. Proc. Natl. Acad. Sci. U.S.A. 85, 1988–1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Olds J. L., Anderson M. L., McPhie D. L., Staten L. D., and Alkon D. L. (1989) Imaging of memory-specific changes in the distribution of protein kinase C in the hippocampus. Science 245, 866–869 [DOI] [PubMed] [Google Scholar]
- 9. Hongpaisan J., and Alkon D. L. (2007) A structural basis for enhancement of long-term associative memory in single dendritic spines regulated by PKC. Proc. Natl. Acad. Sci. U.S.A. 104, 19571–19576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hongpaisan J., Sun M. K., and Alkon D. L. (2011) PKCϵ activation prevents synaptic loss, Aβ elevation, and cognitive deficits in Alzheimer's disease transgenic mice. J. Neurosci. 31, 630–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alkon D. L., and Rasmussen H. (1988) A spatial-temporal model of cell activation. Science 239, 998–1005 [DOI] [PubMed] [Google Scholar]
- 12. Nelson T. J., Collin C., and Alkon D. L. (1990) Isolation of a G protein that is modified by learning and reduces potassium currents in Hermissenda. Science 247, 1479–1483 [DOI] [PubMed] [Google Scholar]
- 13. Sen A., Alkon D. L., and Nelson T. J. (2012) Apolipoprotein E3 (apoE3) but not apoE4 protects against synaptic loss through increased expression of protein kinase Cϵ. J. Biol. Chem. 287, 15947–15958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hama H., Hara C., Yamaguchi K., and Miyawaki A. (2004) PKC signaling mediates global enhancement of excitatory synaptogenesis in neurons triggered by local contact with astrocytes. Neuron 41, 405–415 [DOI] [PubMed] [Google Scholar]
- 15. Khan T. K., Sen A., Hongpaisan J., Lim C. S., Nelson T. J., and Alkon D. L. (2015) PKCϵ deficits in Alzheimer's disease brains and skin fibroblasts. J. Alzheimers Dis. 43, 491–509 [DOI] [PubMed] [Google Scholar]
- 16. Lin D. T., Makino Y., Sharma K., Hayashi T., Neve R., Takamiya K., and Huganir R. L. (2009) Regulation of AMPA receptor extrasynaptic insertion by 4.1N, phosphorylation and palmitoylation. Nat. Neurosci. 12, 879–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gomes A. R., Correia S. S., Esteban J. A., Duarte C. B., and Carvalho A. L. (2007) PKC anchoring to GluR4 AMPA receptor subunit modulates PKC-driven receptor phosphorylation and surface expression. Traffic 8, 259–269 [DOI] [PubMed] [Google Scholar]
- 18. Alkon D. L., Epstein H., Kuzirian A., Bennett M. C., and Nelson T. J. (2005) Protein synthesis required for long-term memory is induced by PKC activation on days before associative learning. Proc. Natl. Acad. Sci. U.S.A. 102, 16432–16437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lim C. S., and Alkon D. L. (2012) Protein kinase C stimulates HuD-mediated mRNA stability and protein expression of neurotrophic factors and enhances dendritic maturation of hippocampal neurons in culture. Hippocampus 22, 2303–2319 [DOI] [PubMed] [Google Scholar]
- 20. Sen A., Nelson T. J., and Alkon D. L. (2015) ApoE4 and Aβ oligomers reduce BDNF expression via HDAC nuclear translocation. J. Neurosci. 35, 7538–7551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fong D. K., Rao A., Crump F. T., and Craig A. M. (2002) Rapid synaptic remodeling by protein kinase C: reciprocal translocation of NMDA receptors and calcium/calmodulin-dependent kinase II. J. Neurosci. 22, 2153–2164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang P., and Lisman J. E. (2012) Activity-dependent regulation of synaptic strength by PSD-95 in CA1 neurons. J. Neurophysiol. 107, 1058–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yan J. Z., Xu Z., Ren S. Q., Hu B., Yao W., Wang S. H., Liu S. Y., and Lu W. (2011) Protein kinase C promotes N-methyl-d-aspartate (NMDA) receptor trafficking by indirectly triggering calcium/calmodulin-dependent protein kinase II (CaMKII) autophosphorylation. J. Biol. Chem. 286, 25187–25200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nelson T. J., and Alkon D. L. (2015) Molecular regulation of synaptogenesis during associative learning and memory. Brain Res. 1621, 239–251 [DOI] [PubMed] [Google Scholar]
- 25. Sun M. K., Nelson T. J., and Alkon D. L. (2015) Towards universal therapeutics for memory disorders. Trends Pharmacol. Sci. 36, 384–394 [DOI] [PubMed] [Google Scholar]
- 26. Li Z., and Sheng M. (2003) Some assembly required: the development of neuronal synapses. Nat. Rev. Mol. Cell Biol. 4, 833–841 [DOI] [PubMed] [Google Scholar]
- 27. Sheng M., and Hoogenraad C. C. (2007) The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu. Rev. Biochem. 76, 823–847 [DOI] [PubMed] [Google Scholar]
- 28. Funke L., Dakoji S., and Bredt D. S. (2005) Membrane-associated guanylate kinases regulate adhesion and plasticity at cell junctions. Annu. Rev. Biochem. 74, 219–245 [DOI] [PubMed] [Google Scholar]
- 29. Bats C., Groc L., and Choquet D. (2007) The interaction between Stargazin and PSD-95 regulates AMPA receptor surface trafficking. Neuron 53, 719–734 [DOI] [PubMed] [Google Scholar]
- 30. Ehrlich I., Klein M., Rumpel S., and Malinow R. (2007) PSD-95 is required for activity-driven synapse stabilization. Proc. Natl. Acad. Sci. U.S.A. 104, 4176–4181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. El-Husseini A. E., Schnell E., Chetkovich D. M., Nicoll R. A., and Bredt D. S. (2000) PSD-95 involvement in maturation of excitatory synapses. Science 290, 1364–1368 [PubMed] [Google Scholar]
- 32. De Roo M., Klauser P., Mendez P., Poglia L., and Muller D. (2008) Activity-dependent PSD formation and stabilization of newly formed spines in hippocampal slice cultures. Cereb. Cortex 18, 151–161 [DOI] [PubMed] [Google Scholar]
- 33. Kim M. J., Futai K., Jo J., Hayashi Y., Cho K., and Sheng M. (2007) Synaptic accumulation of PSD-95 and synaptic function regulated by phosphorylation of serine 295 of PSD-95. Neuron 56, 488–502 [DOI] [PubMed] [Google Scholar]
- 34. Kanno T., Yamamoto H., Yaguchi T., Hi R., Mukasa T., Fujikawa H., Nagata T., Yamamoto S., Tanaka A., and Nishizaki T. (2006) The linoleic acid derivative DCP-LA selectively activates PKCϵ, possibly binding to the phosphatidylserine binding site. J. Lipid Res. 47, 1146–1156 [DOI] [PubMed] [Google Scholar]
- 35. Sun M. K., Hongpaisan J., Nelson T. J., and Alkon D. L. (2008) Poststroke neuronal rescue and synaptogenesis mediated in vivo by protein kinase C in adult brains. Proc. Natl. Acad. Sci. U.S.A. 105, 13620–13625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nelson T. J., and Alkon D. L. (2009) Neuroprotective versus tumorigenic protein kinase C activators. Trends Biochem. Sci. 34, 136–145 [DOI] [PubMed] [Google Scholar]
- 37. Steinberg S. F. (2008) Structural basis of protein kinase C isoform function. Physiol. Rev. 88, 1341–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Comalada M., Xaus J., Valledor A. F., López-López C., Pennington D. J., and Celada A. (2003) PKCϵ is involved in JNK activation that mediates LPS-induced TNFα, which induces apoptosis in macrophages. Am. J. Physiol. Cell Physiol. 285, C1235–C1245 [DOI] [PubMed] [Google Scholar]
- 39. Lang W., Wang H., Ding L., and Xiao L. (2004) Cooperation between PKCα and PKCϵ in the regulation of JNK activation in human lung cancer cells. Cell. Signal. 16, 457–467 [DOI] [PubMed] [Google Scholar]
- 40. Brooks I. M., and Tavalin S. J. (2011) Ca2+/calmodulin-dependent protein kinase II inhibitors disrupt AKAP79-dependent PKC signaling to GluA1 AMPA receptors. J. Biol. Chem. 286, 6697–6706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Waxham M. N., and Aronowski J. (1993) Ca2+/calmodulin-dependent protein kinase II is phosphorylated by protein kinase C in vitro. Biochemistry 32, 2923–2930 [DOI] [PubMed] [Google Scholar]
- 42. Tokuda K., Izumi Y., and Zorumski C. F. (2013) Locally-generated acetaldehyde is involved in ethanol-mediated LTP inhibition in the hippocampus. Neurosci. Lett. 537, 40–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Barker A. J., Koch S. M., Reed J., Barres B. A., and Ullian E. M. (2008) Developmental control of synaptic receptivity. J. Neurosci. 28, 8150–8160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ippolito D. M., and Eroglu C. (2010) Quantifying synapses: an immunocytochemistry-based assay to quantify synapse number. J. Vis. Exp. 45, 2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kortmansky J., and Schwartz G. K. (2003) Bryostatin-1: a novel PKC inhibitor in clinical development. Cancer Invest. 21, 924–936 [DOI] [PubMed] [Google Scholar]
- 46. Nelson T. J., Sen A., Alkon D. L., and Sun M. K. (2014) Adduct formation in liquid chromatography-triple quadrupole mass spectrometric measurement of bryostatin 1. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 944, 55–62 [DOI] [PubMed] [Google Scholar]
- 47. Hongpaisan J., Xu C., Sen A., Nelson T. J., and Alkon D. L. (2013) PKC activation during training restores mushroom spine synapses and memory in the aged rat. Neurobiol. Dis. 55, 44–62 [DOI] [PubMed] [Google Scholar]
- 48. Cheng D., Hoogenraad C. C., Rush J., Ramm E., Schlager M. A., Duong D. M., Xu P., Wijayawardana S. R., Hanfelt J., Nakagawa T., Sheng M., and Peng J. (2006) Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol. Cell Proteomics 5, 1158–1170 [DOI] [PubMed] [Google Scholar]
- 49. Kim E., and Sheng M. (2004) PDZ domain proteins of synapses. Nat. Rev. Neurosci. 5, 771–781 [DOI] [PubMed] [Google Scholar]
- 50. Bell K. F., and Hardingham G. E. (2011) The influence of synaptic activity on neuronal health. Curr. Opin. Neurobiol. 21, 299–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lipsky R. H., and Marini A. M. (2007) Brain-derived neurotrophic factor in neuronal survival and behavior-related plasticity. Ann. N.Y. Acad. Sci. 1122, 130–143 [DOI] [PubMed] [Google Scholar]
- 52. Soriano F. X., Papadia S., Hofmann F., Hardingham N. R., Bading H., and Hardingham G. E. (2006) Preconditioning doses of NMDA promote neuroprotection by enhancing neuronal excitability. J. Neurosci. 26, 4509–4518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Akers R. F., Lovinger D. M., Colley P. A., Linden D. J., and Routtenberg A. (1986) Translocation of protein kinase C activity may mediate hippocampal long-term potentiation. Science 231, 587–589 [DOI] [PubMed] [Google Scholar]
- 54. Yang H., Courtney M. J., Martinsson P., and Manahan-Vaughan D. (2011) Hippocampal long-term depression is enhanced, depotentiation is inhibited and long-term potentiation is unaffected by the application of a selective c-Jun N-terminal kinase inhibitor to freely behaving rats. Eur. J. Neurosci. 33, 1647–1655 [DOI] [PubMed] [Google Scholar]
- 55. Hurd C., Waldron R. T., and Rozengurt E. (2002) Protein kinase D complexes with C-Jun N-terminal kinase via activation loop phosphorylation and phosphorylates the C-Jun N-terminus. Oncogene 21, 2154–2160 [DOI] [PubMed] [Google Scholar]
- 56. Farías G. G., Alfaro I. E., Cerpa W., Grabowski C. P., Godoy J. A., Bonansco C., and Inestrosa N. C. (2009) Wnt-5a/JNK signaling promotes the clustering of PSD-95 in hippocampal neurons. J. Biol. Chem. 284, 15857–15866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ferrari L. F., Bogen O., and Levine J. D. (2013) Role of nociceptor αCaMKII in transition from acute to chronic pain (hyperalgesic priming) in male and female rats. J. Neurosci. 33, 11002–11011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Stein V., House D. R., Bredt D. S., and Nicoll R. A. (2003) Postsynaptic density-95 mimics and occludes hippocampal long-term potentiation and enhances long-term depression. J. Neurosci. 23, 5503–5506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sun M. K., and Alkon D. L. (2009) Protein kinase C activators as synaptogenic and memory therapeutics. Arch. Pharm. (Weinheim) 342, 689–698 [DOI] [PubMed] [Google Scholar]
- 60. Li Y., Davis K. L., and Sytkowski A. J. (1996) Protein kinase Cϵ is necessary for erythropoietin's up-regulation of c-myc and for factor-dependent DNA synthesis: evidence for discrete signals for growth and differentiation. J. Biol. Chem. 271, 27025–27030 [PubMed] [Google Scholar]
- 61. Caino M. C., von Burstin V. A., Lopez-Haber C., and Kazanietz M. G. (2011) Differential regulation of gene expression by protein kinase C isozymes as determined by genome-wide expression analysis. J. Biol. Chem. 286, 11254–11264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mischak H., Goodnight J. A., Kolch W., Martiny-Baron G., Schaechtle C., Kazanietz M. G., Blumberg P. M., Pierce J. H., and Mushinski J. F. (1993) Overexpression of protein kinase Cδ and -ϵ in NIH 3T3 cells induces opposite effects on growth, morphology, anchorage dependence, and tumorigenicity. J. Biol. Chem. 268, 6090–6096 [PubMed] [Google Scholar]
- 63. Boersma M. C., Dresselhaus E. C., De Biase L. M., Mihalas A. B., Bergles D. E., and Meffert M. K. (2011) A requirement for nuclear factor-κB in developmental and plasticity-associated synaptogenesis. J. Neurosci. 31, 5414–5425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Coffey E. T., Akerman K. E., and Courtney M. J. (1997) Brain derived neurotrophic factor induces a rapid upregulation of synaptophysin and tau proteins via the neurotrophin receptor TrkB in rat cerebellar granule cells. Neurosci. Lett. 227, 177–180 [DOI] [PubMed] [Google Scholar]
- 65. Kajiya M., Shiba H., Fujita T., Takeda K., Uchida Y., Kawaguchi H., Kitagawa M., Takata T., and Kurihara H. (2009) Brain-derived neurotrophic factor protects cementoblasts from serum starvation-induced cell death. J. Cell Physiol. 221, 696–706 [DOI] [PubMed] [Google Scholar]
- 66. Sun M. K., Hongpaisan J., Lim C. S., and Alkon D. L. (2014) Bryostatin-1 restores hippocampal synapses and spatial learning and memory in adult fragile x mice. J. Pharmacol. Exp. Ther. 349, 393–401 [DOI] [PubMed] [Google Scholar]
- 67. Pascale A., Gusev P. A., Amadio M., Dottorini T., Govoni S., Alkon D. L., and Quattrone A. (2004) Increase of the RNA-binding protein HuD and posttranscriptional up-regulation of the GAP-43 gene during spatial memory. Proc. Natl. Acad. Sci. U.S.A. 101, 1217–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yoshii A., and Constantine-Paton M. (2007) BDNF induces transport of PSD-95 to dendrites through PI3K-AKT signaling after NMDA receptor activation. Nat. Neurosci. 10, 702–711 [DOI] [PubMed] [Google Scholar]
- 69. Kellner Y., Gödecke N., Dierkes T., Thieme N., Zagrebelsky M., and Korte M. (2014) The BDNF effects on dendritic spines of mature hippocampal neurons depend on neuronal activity. Front. Synaptic Neurosci. 6, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nelson T. J., Cui C., Luo Y., and Alkon D. L. (2009) Reduction of β-amyloid levels by novel protein kinase Cϵ activators. J. Biol. Chem. 284, 34514–34521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Stoppini L., Buchs P. A., and Muller D. (1991) A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods 37, 173–182 [DOI] [PubMed] [Google Scholar]
- 72. Kim H., Kim E., Park M., Lee E., and Namkoong K. (2013) Organotypic hippocampal slice culture from the adult mouse brain: a versatile tool for translational neuropsychopharmacology. Prog. Neuropsychopharmacol. Biol. Psychiatry 41, 36–43 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.