

Abstract
The homomeric α7 nicotinic receptor (nAChR) is one of the most abundant nAChRs in the central nervous system where it contributes to cognition, attention, and working memory. α7 nAChR is also present in lymphocytes, dendritic cells (DCs), and macrophages and it is emerging as an important drug target for intervention in inflammation and sepsis. Natural killer (NK) cells display cytotoxic activity against susceptible target cells and modulate innate and adaptive immune responses through their interaction with DCs. We here show that human NK cells also express α7 nAChR. α7 nAChR mRNA is detected by RT-PCR and cell surface expression of α7 nAChR is detected by confocal microscopy and flow cytometry using α-bungarotoxin, a specific antagonist. Both mRNA and protein levels increase during NK stimulation with cytokines (IL-12, IL-18, and IL-15). Exposure of cytokine-stimulated NK cells to PNU-282987, a specific α7 nAChR agonist, increases intracellular calcium concentration ([Ca2+]i) mainly released from intracellular stores, indicating that α7 nAChR is functional. Moreover, its activation by PNU-282987 plus a specific positive allosteric modulator greatly enhances the Ca2+ responses in NK cells. Stimulation of NK cells with cytokines and PNU-282987 decreases NF-κB levels and nuclear mobilization, down-regulates NKG2D receptors, and decreases NKG2D-dependent cell-mediated cytotoxicity and IFN-γ production. Also, such NK cells are less efficient to trigger DC maturation. Thus, our results demonstrate the anti-inflammatory role of α7 nAChR in NK cells and suggest that modulation of its activity in these cells may constitute a novel target for regulation of the immune response.
Keywords: calcium intracellular release, cellular immune response, Cys-loop receptor, natural killer cells (NK cells), nicotinic acetylcholine receptors (nAChR)
Introduction
Neurotransmitter-gated ion channels of the Cys-loop receptor family are synaptic receptors that convert a chemical signal into an electrical one by rapidly opening a channel that allows the flux of ions through the membrane (1). They are pentameric proteins composed of an extracellular domain that carries the agonist binding sites and a transmembrane domain that forms the ion-conducting pore (2). The nAChRs are cationic channels of the Cys-loop receptor family activated by the endogenous neurotransmitter acetylcholine (ACh).5 They are involved in a wide variety of physiological processes, including learning, memory, sensory processing, and neuromuscular transmission (1). These receptors can be homomeric, such as the α7 nAChR, or heteromeric, such as the muscle and the neuronal α4β2 nAChRs (3). The activation of the different nAChRs may produce different effects because each nAChR subtype differs in ligand-binding affinities, biophysical properties, and downstream effectors (4–6). α7 nAChR is one of the most abundant nAChRs in the central nervous system, and contributes to cognitive functioning, sensory information processing, attention, working memory, and reward pathways (7). It has the highest Ca2+ permeability among Cys-loop receptors; its activation triggers a rise in intracellular Ca2+ levels and a series of Ca2+-dependent intracellular processes (6).
Several lines of evidence obtained in the last years, including those from our laboratory (8, 9), have demonstrated that the cholinergic system is not only confined to the nervous system, and that immune cells also express nAChRs, enzymes, and proteins involved in synthesis, storage, transport, and degradation/hydrolysis of ACh (10, 11). Although the functional relevance of this extra-neuronal cholinergic system remains ill-defined, it might be relevant as a mechanism of local modulation of the immune response through autocrine and paracrine circuits.
Previous studies from our laboratory have shown that α7 nAChR in human lymphocytes modulates cortisol-induced apoptosis and lymphocyte activation (8, 9). In addition, it has been demonstrated that activation of α7 nAChR mediates anti-inflammatory effects (12–17). Therefore, targeting of α7 nAChR has emerged as a promising pharmacological strategy for the treatment of inflammation in a variety of human diseases, such as obesity, atherosclerosis, diabetes, asthma, cystic fibrosis, Alzheimer disease, Parkinson disease, sepsis, ulcerative colitis, psoriasis, pancreatitis, and arthritis (12–17). Despite the relevance of α7 nAChR as a novel immunomodulatory target, its presence in human NK cells has not been yet determined.
NK cells constitute 5–15% of lymphoid cells in human peripheral blood and are critical during immunity against intracellular infectious agents and tumors (18, 19). NK cells have also immunoregulatory properties due to the secretion of multiple cytokines such as IFN-γ, TNF-α, GM-CSF, and chemokines that contribute to modulate the innate immune response and to shift the adaptive immune response to a Th1-biased profile through interactions with DCs and macrophages (18, 20–23). Accumulating evidence indicates that NK cells may also play an important role during autoimmune disorders (19, 24, 25). The activity of NK cells is controlled by a dynamic balance of signals elicited upon engagement of activating and inhibitory receptors by discrete ligands expressed on target cells (23, 26). Very limited information suggests that cholinergic receptors may modulate NK cell activity (27, 28). It has been demonstrated that mouse NK cells express β2 nAChR subunit and that this subunit is involved in nicotine-induced attenuation of mouse NK cell functions (29).
Therefore, the aim of this work was to investigate the expression of α7 nAChR in human NK cells and the regulation of NK cell effector functions by α7 nAChR ligands. We report here the first experimental evidence demonstrating the expression of α7 nAChR on human NK cells and the biological consequences of its activation.
Results
Human NK Cells Express α7 nAChR
First, we isolated NK cells from healthy donors and assessed the expression of α7 nAChR mRNA by RT-PCR. As shown in Fig. 1A, freshly isolated human NK cells express α7 nAChR mRNA. By analogy with other immune cells, we hypothesized that the expression of nAChRs in NK cells changes upon cell activation (8, 30–32). As human NK cells become activated by cytokines, particularly IL-12, IL-18, and IL-15 (23, 33, 34), and considering that these cytokines increase during inflammation, we stimulated NK cells with these cytokines and performed a kinetic analysis of α7 nAChR mRNA expression by quantitative PCR (qPCR). The results show that α7 nAChR mRNA peaks at 48 h after cell stimulation and returns to background levels after 72 h (data not shown). The peak corresponds to a ∼3.1-fold increase in α7 nAChR mRNA with respect to that of fresh cells (Fig. 1B). Therefore, we chose 48 h stimulation with cytokines for all our experiments.
FIGURE 1.
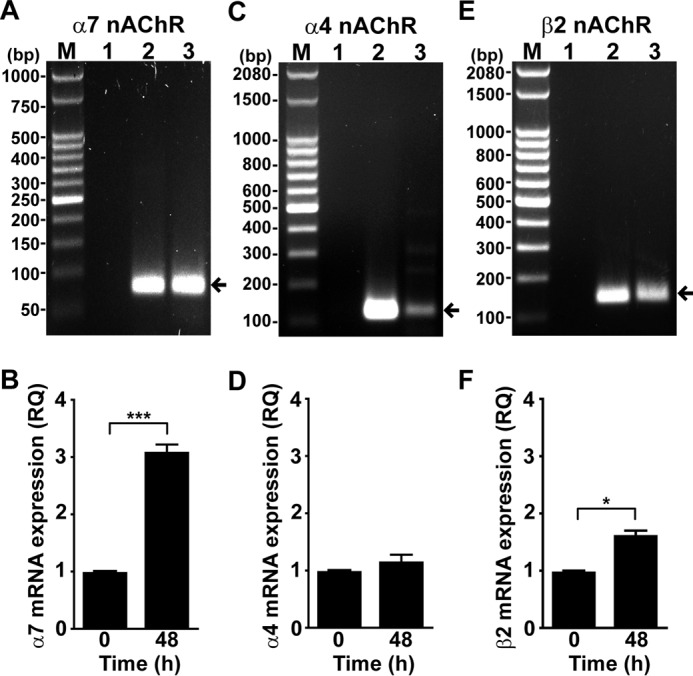
Human NK cells express mRNA for the α7, α4, and β2 nAChR subunits. Expression of α7, α4, and β2 nAChR subunits mRNA was assessed in human NK cells by RT-PCR. A, C, and E, transcripts of α7, α4, and β2 nAChR subunits were detected by electrophoresis in agarose gel as bands of 75, 121, and 136 bp, respectively (arrows); M, DNA ladder (50 bp (A) and 100 bp (C and E)); 1, PCR negative control; 2, PCR positive control (A, human α7 cDNA; B, human α4 cDNA; C, human β2 cDNA); 3, human NK cells. B, D, and F, relative quantification (RQ) of mRNA expression of α7, α4, and β2 nAChR subunit mRNA in human NK cells incubated with and without IL-12, IL-18, and IL-15 for 48 h. qPCR was carried out as described under “Experimental Procedures.” The results are the mean ± S.E., and are cumulative of at least three independent experiments performed with NK cells from different donors (n = 3–5). *, p < 0.05; ***, p < 0.001; paired t test.
We also detected in freshly isolated NK cells mRNA corresponding to α4 (Fig. 1C) and β2 (Fig. 1E) nAChR subunits. By qPCR we determined that after cytokine stimulation of NK cells, the mRNA level of α4 nAChR does not change significantly (Fig. 1D), whereas that of β2 slightly increases with respect to fresh cells (Fig. 1F). As different nAChR types are present in human NK cells, we used a selective α7 nAChR agonist (PNU-282987) and a specific antagonist (α-BTX) for further functional experiments.
Cell surface expression of α7 nAChR was analyzed using Alexa Fluor 488-labeled α-BTX, a selective α7 nAChR antagonist, in NK cells cultured in the absence or presence of IL-12, IL-18, and IL-15 for 48 h and assessed by fluorescence microscopy (Fig. 2A) and flow cytometry (FC) (Fig. 2B). In line with the qPCR results, an increased number of fluorescent cells and higher fluorescence per cell are observed on stimulated NK cells compared with resting cells. The pretreatment with α-BTX markedly reduces the binding of Alexa Fluor 488-labeled α-BTX to the cells (Fig. 2). These results indicate that NK cells express surface α7 nAChR that is up-regulated during NK stimulation with cytokines.
FIGURE 2.

Human NK cells express cell surface α7 nAChR. Human NK cells were incubated with and without IL-12/IL-18/IL-15 for 48 h and stained with Alexa Fluor 488-labeled α-BTX. Then the cells were analyzed by fluorescence microscopy (A) and FC (B). A, representative fluorescence microscopy images of resting NK cells (upper panel) or NK cells stimulated with cytokines (lower panel) and incubated with labeled α-BTX. Left images correspond to human NK cells preincubated with unlabeled α-BTX before the addition of Alexa Fluor 488-labeled α-BTX as a specificity control. Insets, phase-contrast micrographs of the same cells. B, FC analysis of resting or cytokine-stimulated NK cells stained with labeled α-BTX. Gray histograms, unstained cells; blue dashed line, resting NK cells preincubated with unlabeled α-BTX; red dashed line, stimulated NK cells preincubated with unlabeled α-BTX; blue line, resting NK cells; red line, stimulated NK cells. Data shown in A and B are representative of three independent experiments performed with NK cells from different donors (n = 5). **, p < 0.01; ***, p < 0.001; paired t test. Bar graphs show results as mean ± S.E of the average fluorescence intensity of cell surface-labeled α-BTX.
Activation of α7 nAChR Triggers Ca2+ Mobilization in Human NK Cells
To investigate whether α7 nAChR in NK cells is functional and its activation triggers the typical changes in [Ca2+]i, we conducted confocal microscopy analysis of NK cells cultured in the presence of cytokines during 48 h and loaded with Fluo-3/AM. PNU-282987 leads to a slow and continuous increase of [Ca2+]i (Fig. 3, B, E, and F). Although the magnitude of the responses vary among individual cells, all cells show a clear change in [Ca2+]i after agonist addition (Fig. 3, A and B). The Ca2+ responses are not detected in resting cells, probably due to the lower expression level of α7 nAChR as also described in lymphocytes (8, 31).
FIGURE 3.

Activation of α7 nAChR in human NK cells triggers Ca2+ mobilization. Human NK cells were stimulated with IL-12/IL-18/IL-15 for 48 h, loaded with Fluo-3/AM, and analyzed by laser scanning microscopy as described under “Experimental Procedures.” The images were obtained immediately before (0 s) and after 80 s (80 s) of exposure to the drugs, which corresponds to the time at which the maximum Ca2+ increase was typically observed. The color scale represents the relative fluorescence intensity of Fluo-3/AM-loaded cells, with black representing the lowest and white the highest [Ca2+]i. The cells were incubated in the absence (−) (A and E) or presence of: 30 μm PNU-282987 (P) (B and F), 30 μm PNU-282987 and 3 μm PNU-120596 (P + P120596) (C and G), 30 μm PNU-282987 after 15 min preincubation with 1 μm α-BTX (α-BTX + P) (D and H). Relative fluorescence values were calculated as F/F0 to represent the relative [Ca2+]i levels. Each curve represents the variations of intracellular Ca2+ in one cell, and the responses of three representative cells are shown for each condition. The arrow indicates the time of drug application. I, bars represent mean ± S.E. of the relative fluorescence intensity of each cell (measured at the maximum value) from each condition. The data are representative of three independent experiments performed with NK cells from different donors (n = 5). At least 100 cells/condition were analyzed. ***, p < 0.001; one-way ANOVA test with Bonferroni post hoc test.
To further confirm that the increase in [Ca2+]i is mediated by α7 nAChR, we co-applied PNU-282987 with PNU-120596, which is a specific α7 nAChR positive allosteric modulator (PAM) (35). As expected, the presence of the PAM leads to an enhanced increase of [Ca2+]i (Fig. 3, C and G), which is revealed by a slight increase in maximum fluorescence intensity (Fig. 3I) and an increase in the rate of change in Ca2+ signal (Fig. 3, F and G). The area under the curve is 20% bigger in the presence of the agonist and PAM with respect to the agonist alone. Moreover, the change in [Ca2+]i induced by PNU-282987 is abolished if cells are preincubated 15 min with 1 μm α-BTX, again confirming that the effect is specifically mediated by α7 nAChR activation (Fig. 3, D, H, and I).
To further elucidate the origin of the [Ca2+]i increased by PNU-282987, we stimulated human NK cells with this agonist in Ca2+-free buffer containing 5 mm EGTA, an extracellular Ca2+ chelator. The PNU282987-elicited increase in [Ca2+]i is not affected by the absence of extracellular Ca2+ (Fig. 4, A, C, and E). In contrast, the increase in [Ca2+]i is inhibited by preincubation of cells with 5 μm BAPTA-AM, a cell-permeable intracellular Ca2+ chelator (Fig. 4, B and D). These results demonstrate that α7 nAChR in NK cells is functional and its activation releases Ca2+ from intracellular stores, which, in turn, may modulate cell function.
FIGURE 4.

α7 nAChR activation induces Ca2+ release from intracellular Ca2+ stores. Human NK cells were stimulated with IL-12/IL-18/IL-15 for 48 h, loaded with Fluo-3/AM, and analyzed by laser scanning microscopy as described under “Experimental Procedures.” The images were obtained immediately before (0 s) and after 80 s (80 s) of exposure to the drugs. The color scale represents the relative fluorescence intensity of Fluo-3/AM-loaded cells, with black representing the lowest and white the highest [Ca2+]i. PNU-282987 (30 μm) was added to cells preincubated with 5 mm EGTA-containing Ca2+-free buffer for 5 min (A and C) or with 5 μm BAPTA-AM for 15 min (B and D). Relative fluorescence values were calculated as F/F0 to represent the relative [Ca2+]i levels. Each curve represents the variations of intracellular Ca2+ in one cell, and the responses of three representative cells are shown for each condition. The arrow indicates the time of drug application. E, bars represent mean ± S.E. of the relative fluorescence intensity of each cell (measured at the maximum value) from each condition. Dashed line represents the mean value relative fluorescence intensity in cells exposed to 30 μm PNU-282987 (control). The data are representative of three independent experiments performed with NK cells from different donors (n = 3). At least 100 cells/condition were analyzed. **, p < 0.01; paired t test.
Activation of α7 nAChR in Human NK Cells Regulates NKG2D Expression
We next examined whether activation of α7 nAChR affects the expression of some major NK cell activating receptors such as NKG2D, NKp46, and DNAM-1. To achieve maximal effects, we first incubated NK cells with IL-12, IL-18, and IL-15 for 48 h to promote maximal α7 nAChR expression. Thereafter, we treated the cells for 18 h in the presence of the cytokines with the agonist and/or antagonist, and assessed their effects on the expression of the NK cell receptors by FC. We observed that PNU-282987 induces down-regulation of NKG2D, and is abolished by preincubation of cells with α-BTX (Fig. 5A). In contrast, the expression levels of the other NK cell receptors, NKp46 and DNAM-1, are not affected by the presence of PNU-282987 (Fig. 5, B and C). Of note, PNU-282987 does not affect NK cell viability (11.3 ± 2.2% of cell death in control versus 12.9 ± 2.1% in PNU-282987-treated cells). These experiments indicate that activation of α7 nAChR down-regulates selectively the expression of NKG2D, which is one of the major activating receptors involved in NK cell function.
FIGURE 5.

Activation of α7 nAChR down-regulates NKG2D expression in human NK cells. Human NK cells were stimulated for 48 h with IL-12/IL-18/IL-15, and further cultured for 18 h with cytokines in the absence (−) or presence of PNU-282987 (P), α-BTX and PNU-282987 (α-BTX + P) or α-BTX alone (α-BTX). Thereafter, cell surface expression of NKG2D, NKp46, and DNAM-1 was assessed by FC. The upper panels show expression of NKG2D (A), NKp46 (B) and DNAM-1 (C) in NK cells in the absence or presence of α7 nAChR ligands. Bar graphs show results as mean ± S.E., and are cumulative of three independent experiments performed with NK cells from different donors (n = 5). **, p < 0.01; one-way ANOVA test with Bonferroni post hoc test. The lower panels show representative histograms. Gray histogram, IC mAb; black line, NK cells stimulated with cytokines; dashed line, NK cells stimulated with cytokines and cultured in the presence of PNU-282987; gray line, NK cells stimulated with cytokines and cultured in the presence of α-BTX + PNU-282987.
α7 nAChR Activation Affects NK Cell-mediated Cytotoxicity, IFN-γ Production, and Impacts on Maturation of Human DCs
To determine the physiological consequences of activation of α7 nAChR on NK cells, we assessed their NKG2D-dependent cytotoxic activity and IFN-γ production. In both studies, NK cells were stimulated with cytokines for 48 h and further cultured for 18 h with cytokines in the absence or presence of α7 nAChR agonist or antagonist. To assess NKG2D-dependent cytotoxicity, we used a mouse urothelial carcinoma cell (MB49) transduced to stably express MICA on the cell surface (one of the known ligands for NKG2D (36, 37)) or its corresponding negative control transduced with empty vector as target cells (Fig. 6A). We observed that NK cells display a higher cytotoxic activity against MB49-MICA cells than against MB49-pMSCV (negative control) cells, confirming the role of the NKG2D-MICA receptor-ligand pair in this response (Fig. 6B). However, NK cells exposed previously to the specific α7 nAChR agonist display a significantly reduced cytotoxic activity against MB-49-MICA cells, close to the background cytotoxicity observed with MB49-pMSCV cells (Fig. 6B). We conclude that PNU-282987 blunts NKG2D-dependent cytotoxicity of NK cells, which is in line with the observed down-regulation of NKG2D induced by the α7 nAChR agonist. Similarly, the percentage of IFN-γ-producing NK cells is significantly lower if NK cells are stimulated with cytokines and exposed to PNU-282987 (Fig. 6C). In both functional assays, the effect of PNU-282987 is prevented by α-BTX (Fig. 6, B and C), indicating that α7 nAChR negatively regulates NKG2D-dependent cytotoxicity and IFN-γ production of NK cells.
FIGURE 6.

Activation of α7 nAChR decreases NK cell-mediated cytotoxicity and IFN-γ production. A, expression of MICA in MB49 cells. MB49 were transduced with a retroviral vector encoding MICA*008 (left panel) or with empty vector (right panel). Gray histogram, IC mAb; black line, MICA-specific mAb. Numbers within histograms represent the MFI for MICA in each condition. B and C, human NK cells were stimulated for 48 h with IL-12/IL-18/IL-15, and further cultured for 18 h with cytokines in the absence (−) or presence of PNU-282987 (P), α-BTX and PNU-282987 (α-BTX + P) or α-BTX alone (α-BTX). Thereafter, cytotoxic activity and IFN-γ production by NK were assessed. B, NK cell-mediated cytotoxicity was assessed after co-culture for 5 h with MB49 cells transduced to stably express MICA on the cell surface (MB49-MICA, black bars) or its corresponding negative control (MB49-pMSCV, gray bars) as explained under “Experimental Procedures.” Data are shown as mean ± S.E., and are cumulative of at least three independent experiments performed with NK cells from different donors (n = 6). Representative histograms of the assays with MB49-MICA cells are shown on the right. Numbers within histograms, percentage of positive cells for Zombie Green in each condition. *, p < 0.05; **, p < 0.01; two-way ANOVA test with Sidak post hoc test. C, production of IFN-γ by NK cells was assessed by FC as explained under “Experimental Procedures.” Data are shown as mean ± S.E., and are cumulative of three independent experiments performed with NK cells from different donors (n = 5). Representative dot plots of NK cells (CD56+) stimulated with IL-12/IL-18/IL-15 cultured in the absence (−) or presence of PNU-282987 or α-BTX + PNU-282987 are shown on the right. **, p < 0.01; one-way ANOVA test with Bonferroni post hoc test.
To gain further insight into the biological consequences of α7 nAChR activation on human NK cells and its repercussion on their cross-talk with DCs, human NK cells were stimulated for 48 h with IL-12/IL-18/IL-15, and further cultured for 18 h with cytokines in the absence or presence of PNU-282987. Thereafter, these NK cells were co-cultured with immature DCs (iDCs) for 4 h and we evaluated the expression of MHC-II, CD86 (a costimulatory molecule), and CD83 (DC maturation marker) on DCs (Fig. 7). We observed that pre-treatment of cytokine-stimulated NK cells with PNU-282987 results in significantly lower expression of MHC-II and CD83 in DCs, and less percentage of CD86high DCs (Fig. 7) compared with cytokine-stimulated NK cells not exposed to the α7 nAChR agonist. These data indicate that activation of α7 nAChR on NK cells attenuates DC activation/maturation.
FIGURE 7.

Activation of α7 nAChR decreases NK cell ability to promote DC activation/maturation. Human NK cells were stimulated for 48 h with IL-12/IL-18/IL-15, and further cultured for 18 h with cytokines in the absence (−) or presence of PNU-282987 (P). Thereafter, NK cells were co-cultured with iDCs for 4 h as described under “Experimental Procedures” and DCs were used to assess cell surface expression of MHC-II (A), CD83 (B), and CD86 (C) (n = 15, 16, and 8 for MHC-II, CD83, and CD86, respectively). Data are shown as mean ± S.E., and are cumulative of at least three independent experiments performed with NK cells from different donors. Dots linked with a line represent paired data from the same donor of NK cells. *, p < 0.05; **, p < 0.01; paired one-tailed t test.
α7 nAChR Stimulation Reduces Expression and Nuclear Mobilization of NF-κB
Stimulation of NK cells with cytokines triggers intracellular pathways, some of which converge in NF-κB activation. Besides, activation of α7 nAChR inhibits NF-κB activation in macrophages (38). Therefore, we explored whether activation of this transcription factor is affected by α7 nAChR activation in human NK cells stimulated with IL-12, IL-18, and IL-15 (Fig. 8). We observed that NK cells stimulated with cytokines display an intense nuclear staining and that PNU-282987 produces a reduction in the nuclear mobilization of NF-κB p65 (Fig. 8, A, B, and D). This effect is abolished if cells are preincubated 15 min with α-BTX (Fig. 8C). In addition, the expression of NF-κB p65 is lower in cytokine-stimulated NK cells exposed to PNU-282987 than in those not exposed to the agonist as assessed by both fluorescence microscopy (Fig. 8D) and FC (Fig. 8, E and F).
FIGURE 8.

Activation of α7 nAChR inhibits nuclear mobilization and decreases the expression of NF-κB in human NK cells. Human NK cells were stimulated for 48 h with IL-12, IL-18, and IL-15, and further cultured for 18 h with cytokines in the absence (−) or presence of PNU-282987 (P), PNU-282987+α-BTX (α-BTX + P). Then, NK cells were processed for NF-κB p65 detection and analyzed by confocal microscopy (A-D) and FC (E and F). Upper panels, representative fluorescence microscopy images of NK cells in the absence (A) or presence of PNU-282987 (P) (B) or α-BTX + PNU-282987 (C). D, fluorescence was quantified as detailed under “Experimental Procedures” and graphs show mean fluorescence intensity (AU) of NK cells in cytoplasm (gray bars) and nucleus (black bars). The results are the mean ± S.E., and are cumulative of three independent experiments performed with NK cells from different donors (n = 5). At least 100 cells/condition were analyzed. E, representative histograms of the expression of NF-κB p65 determined by FC. Gray histogram, IC mAb; black line, NK cells stimulated with cytokines; dashed line, NK cells stimulated with cytokines and cultured in the presence of PNU-282987; gray line, NK cells stimulated with cytokines and cultured in the presence of α-BTX + PNU-282987. F, expression of NF-κB p65 assessed by FC. Data are shown as mean ± S.E., and are cumulative of three independent experiments performed with NK cells from different donors (n = 3). *, p < 0.05; ***, p < 0.001; one-way ANOVA test with Bonferroni post hoc test.
Discussion
The present study is, to our knowledge, the first to demonstrate the presence of functional α7 nAChR on human NK cells, which is up-regulated after cytokine stimulation. We show that α7 nAChR activation in cytokine-stimulated NK cells triggers Ca2+ mobilization from intracellular stores, decreases IFN-γ production probably by reducing NF-κB levels and its nuclear translocation, decreases cell-cytotoxic activity due to down-regulation of NKG2D receptor, and attenuates the activation/maturation of DCs.
Several lines of evidence, including those from our laboratory (8, 9), have demonstrated that human immune cells, particularly lymphocytes (T and B cells), DCs and macrophages express nAChRs (10, 11, 13). Thus, the current results extend the expression of α7, α4, and β2 nAChRs to NK cells.
Interestingly, NK cells from mouse do not express α7 nAChR mRNA (29). Such a difference in α7 nAChR expression between these two species has also been shown in lymphocytes (8, 30, 32, 39, 40), and may underlie a different neuroimmunomodulatory potential of lymphoid cells in them. In mouse, nicotine exposure impairs the ability of NK cells to kill target cells and release cytokines through β2-containing nAChRs (29). Because it has been proposed that β2 and α7 subunits contribute differently to the nicotine-dependent modulation of immune cell functions (41, 42), it would be required to further explore the functional role of α4β2 in human NK cells to understand the effects of nonspecific nAChRs ligands, such as nicotine. Our findings indicate that human NK cells can adjust to environmental changes because α7 nAChR is up-regulated by the presence of IL-12, IL-18, and IL-15, probably to control excessive IFN-γ secretion and cytotoxicity that could induce damage in the surrounding healthy cells. This phenomenon is not unique to NK cells as it has been shown that stimulation of T cells alters the expression of nAChR subunits (8, 30–32).
We show that specific activation of α7 nAChR mediates Ca2+ signaling in NK cells. PNU-282987 causes a sustained increase in [Ca2+]i in human NK cells, which is larger in the presence of a PAM. Similar types of responses were reported in other human non-neuronal cells, such as mesenchymal stem cells (43), endothelial cells (44), peripheral blood lymphocytes and leukemic cell lines (31, 45), and platelets (46). The relatively slow response may be due to the fact that the release of Ca2+ from intracellular stores is the underlying mechanism associated to α7 nAChR activation in these cells. This is in line with previous reports showing that activation of α7 nAChR leads to Ca2+ mobilization from intracellular stores through a Ca2+-induced Ca2+ release mechanism, probably through inositol 1,4,5-trisphosphate and ryanodine receptors (6, 47, 48). Similar to our results, in lymphocyte T cells mobilization of Ca2+ through the α7 nAChR channel is not necessarily required for the α7-induced release of Ca2+ from the internal stores (45).
NK cells are crucial components of the immune response against tumors and virus-infected cells (49), and they play a key role in the regulation of the adaptive immune response (19, 25). The stimulation of NK cells can occur through various cytokines, such as IL-12, IL-18, IL-2, IL-15, and type I IFNs (23, 33), and through the engagement of activating receptors (23, 26), triggering their effector functions. We found that activation of α7 nAChR in cytokine-stimulated NK cells decreases the expression of NKG2D, which constitutes the underlying mechanism for the observed decrease of the NKG2D-dependent cytotoxicity against MICA-expressing target cells. Activation of α7 nAChR also decreases the production of IFN-γ in response to cytokines. To elucidate the mechanism underlying this effect, we measured NF-κB because in macrophages it has been associated with the reduction in pro-inflammatory cytokine production, such as TNF-α (16, 38, 50). We found that in human stimulated NK cells, α7 nAChR activation leads to reduced levels and reduced nuclear translocation of NF-κB p65, thus identifying one of the mechanisms underlying the anti-inflammatory effect of α7 nAChR in these cells.
T-cell priming in vivo requires DCs maturation, which is in turn induced upon recognition of pathogen-derived molecular patterns as well as inflammatory cytokines (51–53). NK cells have been shown to contribute to DC maturation and in this manner, contribute to shift the adaptive immune response toward a Th1-biased profile (20–22). As our results show that activation of α7 nAChR in NK cells impairs DCs maturation, we can speculate that this may impact on the ability of DCs cells to induce optimal activation and differentiation of T cells into Th1 cells during the course of the adaptive immune response against different pathogens or tumor cells (51–53).
Due to the high complexity and the multiple possible pathways involved, even in neuronal cells Ca2+ signaling triggered by α7 nAChR has not been completely resolved (6). The mechanisms that link the early increase in cytosolic Ca2+ with the final anti-inflammatory effects have not yet been completely elucidated (14, 54–56). α7 nAChR has been shown to modulate several signaling pathways, such as JAK2/STAT3/NF-κB, PLC/inositol 1,4,5-trisphosphate, and PI3K/Akt/Nrf-2 pathways, which in immune cells result in potent anti-inflammatory effects through inflammatory cytokine production inhibition and overexpression of hemooxigenase-1 (54, 57–59). Also, a direct coupling of α7 nAChR to G-protein has been recently described (60, 61). Thus, our study opens doors to decipher in human NK cells the highly complex signaling pathways triggered by α7 nAChR and their association to different cell functions.
Our results extend the previous findings that indicate that signaling through α7 nAChR in immune cells mediates anti-inflammatory effects (12–17). This “cholinergic anti-inflammatory pathway” and its role in immunity and inflammation is associated to various human pathologies, including obesity, atherosclerosis, diabetes, asthma, cystic fibrosis, Alzheimer disease, Parkinson disease, sepsis, ulcerative colitis, psoriasis, pancreatitis and arthritis sepsis, diabetes, neurodegenerative diseases, osteoarthritis, and inflammatory bowel disease (12–17).
Blood concentrations of ACh and choline are probably low to efficiently activate α7 nAChRs of immune cells (62–65). However, immune cells, including T and B cells, macrophages, and DCs have been shown to be capable of synthesizing and releasing ACh, a process that is up-regulated during inflammation (66). Also, lymph nodes have an additional source of ACh from autonomic nerve terminals (67). Local levels of choline, another full agonist of α7 nAChR, can be elevated under conditions associated with ischemia, stroke, and substantial plasma membrane damage (65, 68). Therefore, it is likely that ACh and choline primarily act on the immune system by wiring transmission between close interactions between leukocytes or leukocyte and neurons (see reviews Refs. 65 and 69). Besides, immune cells also express SLURP-1 (secreted mammalian Ly-6/urokinase plasminogen activator receptor-related protein 1), a positive allosteric modulator of α7 nAChR (70, 71). Moreover, SLURP-1 has also been detected in human blood and plasma (63, 72). Accordingly, α7 nAChR is therefore emerging as an important drug target for the modulation of inflammation in different physiopathological contexts (12–17). Thus, based on our results, NK cells may constitute an additional player of the cholinergic anti-inflammatory pathway.
Potentiation of α7 nAChR can be achieved by the use of agonists or PAMs, and both types of ligands emerge as potential therapeutic drugs (16, 73, 74). In particular, PAMs are of interest because they enhance α7 nAChR responses maintaining the temporal spatial characteristics of the endogenous activation, and they show higher specificity than agonists. We here show that PNU-120596 potentiates α7 nAChR responses in NK cells. Thus, α7 nAChR in NK cells could constitute another important drug target for immune modulation.
The anti-inflammatory effects of α7 nAChR signaling in NK cells observed in this study may constitute a physiological strategy to prevent or limit excessive tissue damage during the immune response against tumors and virus-infected cells. The present study opens the possibility to specifically modulate α7 nAChR activity as a novel pathway to regulate human NK cells.
Experimental Procedures
Materials
Recombinant human IL-12 and IL-15 were from PeproTech; recombinant human IL-18 was from MBL International. The following monoclonal antibodies (mAbs) against human molecules were used: FITC- and PE-labeled anti-CD3 (UCHT-1, Southern Biotech); PE/Cy5- and PE/Cy7-labeled anti-CD56 (N901, Beckman Coulter); PE-labeled anti-IFN-γ (4S.B3, Biolegend); PE-labeled anti-NKG2D mAb (clone 1D11, Biolegend); PE-labeled anti-NKp46 (clone 9E2; Biolegend); FITC-labeled anti-CD226 (DNAM-1, clone DX11, BD Pharmingen); FITC anti-CD14 (clone HCD14, Biolegend); FITC anti-HLA-DR (clone L243, Biolegend); PE anti-CD86 (clone IT2.2, Biolegend); FITC anti-CD83 (clone HB15e, Biolegend); Alexa 488-labeled anti-MICA (clone 159227, R&D Systems); isotype-matched negative control mAb (IC mAb, eBioscience). α-BTX and Alexa Fluor 488-labeled α-BTX were from Molecular Probes; PNU-120596 and PNU-282987 were from Santa Cruz Biotechnology; EGTA was from Sigma; and BAPTA/AM was from Calbiochem (San Diego, CA). pMSCV and pMSCV/MICA*008 retroviral DNA (negative control or encoding MICA*008, respectively) were kindly provided by Dr. Alessandra Zingoni and Dr. Angela Santoni from the Laboratory of Molecular Immunology and Immunopathology, Department of Molecular Medicine, Sapienza University of Rome, Italy.
Cell Lines
MB49-pMSCV or MB49-MICA were generated as follows. For retrovirus production, the retrovirus packaging cell line PT67 was transfected with viral DNA (pMSCV and pMSCV/MICA*008) and the packaging vectors (pCMVgag-pol and pMD2.G) using Lipofectamine Plus (Invitrogen). After 48 h, virus-containing supernatants were harvested, filtered, and used for infection as follows: 1 ml of viral supernatant containing PolyGram (8 mg/ml) was used to infect 5 × 105 MB49 cells for 2 h. MICA+ cells (assessed by FC) were sorted in a FACSAria IIplus flow cytometer (BD Bioscience).
Isolation and Culture of NK Cells
NK cells were isolated from blood of healthy volunteers provided by the Hemotherapy Service of the Hospital Interzonal Dr. José Penna, Bahía Blanca, Province of Buenos Aires, Argentina, and the Hemotherapy Unit of the Hospital Carlos Durand or the Service of Transfusion Medicine of the Hospital Churruca-Visca, both of Buenos Aires, Argentina. NK cells were isolated using the RosetteSep NK cell enrichment mixture (StemCell) and Ficoll-PaqueTM Plus (Amersham Biosciences) centrifugation. Purity of isolated cells was always above 90%, as assessed by flow cytometry (as CD3−CD56+ for cells). NK cells were cultured in RPMI 1640 (Sigma) supplemented with 10% inactivated fetal bovine serum (NatoCor, Córdoba, Argentina), sodium pyruvate, glutamine, and gentamicin (Sigma) for 48 h in the absence (control cells) and presence of 10 ng/ml of IL-12, 10 ng/ml of IL-18, and 1 ng/ml of IL-15 (cytokine-stimulated cells). The α7 nAChR-specific ligands, 10 μm PNU-282987 or 0.5 μm α-BTX, were added to the cultures as indicated in each figure. Viability of NK cells was assessed by flow cytometry using Zombie Green (Biolegend).
RT-PCR Analysis
Total RNA was extracted with TRIzol reagent (Invitrogen Life Technologies) following the manufacturer's instructions. RNA was converted into cDNA using the Molony murine leukemia virus reverse transcriptase (Promega) and random primers (Promega). End point PCR was run for 40 cycles in a Mini Cycler (MJ Research). Real time qPCR was performed using a Rotor-Gene 6000 (Corbett Research). Amplification included 2 min at 94 °C followed by 40 cycles of a three-step loop: 20 s at 94 °C, 40 s at 56 °C, and 40 s at 72 °C. Results were expressed as the fold-increase of gene expression of cytokine-treated cells versus control (non-treated) cells. Results of gene expression were normalized against the 18S rRNA gene, the expression of which was not changed in stimulated cells. The 18S rRNA primer sequences used were: 5′-TCGAGGCCCTGTAATTGGAA-3′ (sense) and 5′-CCCTCCAATGGATCCTCGTT-3′ (antisense); the α7 nAChR subunit primer sequences used were: 5′-CCAATGACTCGCAACCACTC-3′ (sense) and 5′-GGTTCTTCTCATCCACGTCC-3′ (antisense); the α4 nAChR subunit primer sequences used were: 5′-GGATGAGAAGAACCAGATGATG-3′ (sense) and 5′-CTCGGAGGGGATGCGGAT-3′ (antisense); and the β2 nAChR subunit primer sequences used were: 5′-CGGATACAGAGGAGCGGC-3′ (sense) and 5′-TGCACACTGATGAGCTGG-3′ (antisense). To avoid contamination of genomic DNA, the sense and antisense primers annealed to sequences in different exons, respectively.
Flow Cytometry
Cells were stained with fluorochrome-labeled mAbs, analyzed in a FACSCalibur or a FACSAria flow cytometers (BD Biosciences), and data were processed with the FlowJo software (Tree Star Inc., Ashland, OR). Expression of cell surface receptors on NK cells was analyzed by FC as previously described (75) and results were expressed as mean fluorescence intensity (MFI).
IFN-γ Production by NK Cells
NK cells from healthy donors were stimulated for 48 h at 37 °C with 10 ng/ml of IL-12, 10 ng/ml of IL-18, and 1 ng/ml of IL-15, were incubated for 18 h in the absence or presence of α7 nAChR-specific drugs. During the last 14 and 4 h of culture, GolgiPlug (BD) and GolgiStop (BD) were added, respectively. Cells were harvested and stained with anti-CD56 mAb, permeabilized with Cytofix/Cytoperm (BD Biosciences), and stained with the anti-IFN-γ mAb to assess IFNγ-producing cells by FC.
NK Cell-mediated Cytotoxicity
NK cells from healthy donors were stimulated for 48 h at 37 °C with 10 ng/ml of IL-12, 10 ng/ml of IL-18, and 1 ng/ml of IL-15, were incubated for 18 h in the absence or presence of α7 nAChR-specific drugs. Then, NK cells were washed and co-cultured with eFluor Dye 670-labeled MB49-pMSCV or MB49-MICA cells at a 1:1 ratio. After 5 h, cells were labeled with Zombie Green and analyzed by FC. Percentage of cytotoxicity was calculated as 100× percentage of eFluor Dye 670+Zombie Greenhigh cells/percentage of eFluor Dye 670+ cells (76).
Isolation and Culture of DCs
Monocytes were isolated from the blood of healthy volunteers provided by the Hemotherapy Unit of the Hospital Carlos Durand or the Service of Transfusion Medicine of the Hospital Churruca-Visca, both of Buenos Aires, Argentina, by immunomagnetic selection of CD14+ cells using MACS (Miltenyi Biotech, Bergisch Gladbach, Germany). Purity of isolated cells was always above 90%, as assessed by FC as CD14+ cells. Monocytes were cultured for 6 days with GM-CSF and IL-4 to obtain iDCs. These iDCs were cultured with NK cells (previously stimulated with cytokines in the absence or presence of α7 nAChR-specific ligands as described in the previous section) at a 1:1 ratio for 4 h in RPMI 1640 supplemented with 10% inactivated fetal bovine serum, sodium pyruvate, glutamine, and gentamicin. Thereafter, DCs were harvested and used for analysis of cell surface markers by FC.
α-BTX Binding Assays
Cultured NK cells were harvested, washed, and incubated in the absence (total binding) or presence of 1 μm α-BTX (nonspecific binding) for 30 min. Then, cells were incubated with Alexa Fluor 488-labeled α-BTX (1 μg/ml) for 1 h. Thereafter, fluorescence was assessed by FC and fluorescence microscopy. For fluorescence microscopy, cells were mounted and analyzed by fluorescence microscopy using a Nikon Eclipse E-600 microscope. Images were obtained with a SBIG Astronomical Instrument (Santa Barbara, CA) provided with a CCDOPS software package (version 5.02) to drive a model ST-7 digital charge-coupled device camera. Appropriate dichroic and emission filters were used and images were analyzed using the ImageJ software (National Institutes of Health, Bethesda, MD).
Measurement of Changes in [Ca2+]i
NK cells from healthy donors were stimulated for 48 h at 37 °C with 10 ng/ml of IL-12, 10 ng/ml of IL-18, and 1 ng/ml of IL-15 and transferred to coverglass bottomed culture dishes coated with poly-l-lysine for 2 h (77) and incubated with 3 μm of the Ca2+-sensitive fluorescent indicator Fluo-3/AM (Molecular Probes). [Ca2+]i was assessed using a confocal laser scanning microscope (Leica DMIRE2). Fluo-3/AM-loaded cells were excited at 488 nm, and the fluorescence emission above 530 nm was acquired using a ×20 objective. Images of fluorescence were obtained at a rate of one frame every 4.2 s during 152.8 s. PNU-282987 (30 μm) and PNU-120596 (3 μm) were added directly to the samples. Basal fluorescence (F0) was calculated prior to the addition of α7 nAChR ligands. Data were normalized to F0 (F/F0 ratio) to control for variations in basal fluorescence. The F/F0 ratio was used to express the relative change in intracellular Ca2+ over time.
Detection of NF-κB
NK cells were stimulated for 48 h at 37 °C with 10 ng/ml of IL-12, 10 ng/ml of IL-18, and 1 ng/ml of IL-15 and transferred to coverglass bottomed culture dishes coated with poly-l-lysine for 2 h. Then, NK were incubated for 2 h in the absence or presence of α7-nAChR specific ligands, fixed with 2% of PFA, permeabilized with 0.1% saponin for 10 min, and stained with anti-p65 mAb (sc-109, Santa Cruz Biotechnology, Santa Cruz, CA) and FITC-labeled goat anti-rabbit IgG (Jackson ImmunoResearch Laboratories). Thereafter, NF-κB was assessed by FC and fluorescence microscopy. For fluorescence microscopy, cells were mounted with Dabco, and observed in a confocal laser scanning microscope (Leica DMIRE2) using a ×60 objective. Fluorescence images were analyzed with the software ImageJ. For each cell, regions of interest were drawn around the nucleus and cytoplasm. The average fluorescence intensity for each regions of interest was measured from 8-bit images in at least 100 cells for each experimental condition.
Statistical Analysis
Data were plotted as mean ± S.E. A one-way ANOVA test with Bonferroni post hoc test was used when three or more experimental groups were compared; a two-way ANOVA with repeated measures matched by both factors and Sidak post hoc test was used for the cytotoxicity experiment and a paired t test was used when two experimental groups were compared.
Author Contributions
S. R. Z. participated in the design of the study, performed and analyzed the experiments shown in Figs. 1–6 and 8, participated in the writing of the manuscript, and prepared the figures. A. Z. performed and analyzed the experiments shown in Figs. 1, 6, and 7. N. I. T. performed and analyzed the experiments shown in Figs. 6 and 7. N. W. Z. and C. B. participated in the design of the study, analysis, and writing of the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank the staff of Hemotherapy Service, Hospital Interzonal Dr. José Penna, Bahía Blanca, Buenos Aires, Argentina, for their valuable assistance. We thank the people of the Blood Bank of the “Carlos Durand” Hospital and of the “Complejo Médico Churruca-Visca” for providing the buffy coats. We thank Fundación René Barón and Fundación Williams for additional support, Prof. Dr. Gabriela Salvador for providing anti-p65 NF-κB mAb, Dr. Verónica Gonzalez-Pardo for providing BAPTA-AM, and Alessandra Zingoni and Angela Santoni from the Laboratory of Molecular Immunology and Immunopathology, Department of Molecular Medicine, Sapienza University of Rome, Italy, for providing the retroviral vectors for the transduction experiments.
This work was supported by grants from the Fundación Alberto J. Roemmers de Argentina and Agencia Nacional de Promoción Científica y Tecnológica de Argentina (ANPCYT) (to S. R. Z.), ANPCYT and Universidad de Buenos Aires (to N. W. Z.), and ANPCYT, Universidad Nacional del Sur-Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), and Universidad Nacional del Sur (to C. B.). The authors declare that they have no conflicts of interest with the contents of this article.
- ACh
- acetylcholine
- nAChR
- nicotinic receptor
- NK
- natural killer cell
- α-BTX
- α-bungarotoxin
- PAM
- positive allosteric modulator
- FC
- flow cytometry
- IC mAb
- isotype-matched negative control mAb
- MFI
- mean fluorescence intensity
- DC
- dendritic cells
- qPCR
- quantitative PCR
- BAPTA
- 1,2-bis(2-aminophenoxyl)ethane-N,N,N′,N′-tetraacetic acid
- ANOVA
- analysis of variance
- PE
- phycoerythrin
- Cy
- cyanine.
References
- 1. Bouzat C. (2012) New insights into the structural bases of activation of Cys-loop receptors. J. Physiol. Paris 106, 23–33 [DOI] [PubMed] [Google Scholar]
- 2. Unwin N. (2005) Refined structure of the nicotinic acetylcholine receptor at 4-Å resolution. J. Mol. Biol. 346, 967–989 [DOI] [PubMed] [Google Scholar]
- 3. Albuquerque E. X., Pereira E. F., Alkondon M., and Rogers S. W. (2009) Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol. Rev. 89, 73–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chernyavsky A. I., Arredondo J., Marubio L. M., and Grando S. A. (2004) Differential regulation of keratinocyte chemokinesis and chemotaxis through distinct nicotinic receptor subtypes. J. Cell Sci. 117, 5665–5679 [DOI] [PubMed] [Google Scholar]
- 5. Fasoli F., and Gotti C. (2015) Structure of neuronal nicotinic receptors. Curr. Top. Behav. Neurosci. 23, 1–17 [DOI] [PubMed] [Google Scholar]
- 6. Shen J. X., and Yakel J. L. (2009) Nicotinic acetylcholine receptor-mediated calcium signaling in the nervous system. Acta Pharmacol. Sin. 30, 673–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Andersen N., Corradi J., Sine S. M., and Bouzat C. (2013) Stoichiometry for activation of neuronal α7 nicotinic receptors. Proc. Natl. Acad. Sci. U.S.A. 110, 20819–20824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. De Rosa M. J., Dionisio L., Agriello E., Bouzat C., and Esandi Mdel C. (2009) α7 nicotinic acetylcholine receptor modulates lymphocyte activation. Life Sci. 85, 444–449 [DOI] [PubMed] [Google Scholar]
- 9. De Rosa M. J., Esandi Mdel C., Garelli A., Rayes D., and Bouzat C. (2005) Relationship between α7 nAChR and apoptosis in human lymphocytes. J. Neuroimmunol. 160, 154–161 [DOI] [PubMed] [Google Scholar]
- 10. Kawashima K., and Fujii T. (2004) Expression of non-neuronal acetylcholine in lymphocytes and its contribution to the regulation of immune function. Front. Biosci. 9, 2063–2085 [DOI] [PubMed] [Google Scholar]
- 11. Kawashima K., Fujii T., Moriwaki Y., and Misawa H. (2012) Critical roles of acetylcholine and the muscarinic and nicotinic acetylcholine receptors in the regulation of immune function. Life Sci. 91, 1027–1032 [DOI] [PubMed] [Google Scholar]
- 12. Bencherif M., Lippiello P. M., Lucas R., and Marrero M. B. (2011) α7 nicotinic receptors as novel therapeutic targets for inflammation-based diseases. Cell Mol. Life Sci. 68, 931–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. de Jonge W. J., and Ulloa L. (2007) The α7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br. J. Pharmacol. 151, 915–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Filippini P., Cesario A., Fini M., Locatelli F., and Rutella S. (2012) The Yin and Yang of non-neuronal α7-nicotinic receptors in inflammation and autoimmunity. Curr. Drug Targets 13, 644–655 [DOI] [PubMed] [Google Scholar]
- 15. Nizri E., and Brenner T. (2013) Modulation of inflammatory pathways by the immune cholinergic system. Amino Acids 45, 73–85 [DOI] [PubMed] [Google Scholar]
- 16. Pohanka M. (2012) α7 nicotinic acetylcholine receptor is a target in pharmacology and toxicology. Int. J. Mol. Sci. 13, 2219–2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tracey K. J. (2009) Reflex control of immunity. Nat. Rev. Immunol. 9, 418–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Campbell K. S., and Hasegawa J. (2013) Natural killer cell biology: an update and future directions. J. Allergy Clin. Immunol. 132, 536–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Poggi A., and Zocchi M. R. (2014) NK cell autoreactivity and autoimmune diseases. Front. Immunol. 5, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ferlazzo G., and Morandi B. (2014) Cross-talks between natural killer cells and distinct subsets of dendritic cells. Front. Immunol. 5, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Barreira da Silva R., and Münz C. (2011) Natural killer cell activation by dendritic cells: balancing inhibitory and activating signals. Cell Mol. Life Sci. 68, 3505–3518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ferlazzo G., and Münz C. (2009) Dendritic cell interactions with NK cells from different tissues. J. Clin. Immunol. 29, 265–273 [DOI] [PubMed] [Google Scholar]
- 23. Zwirner N. W., and Domaica C. I. (2010) Cytokine regulation of natural killer cell effector functions. Biofactors 36, 274–288 [DOI] [PubMed] [Google Scholar]
- 24. Fogel L. A., Yokoyama W. M., and French A. R. (2013) Natural killer cells in human autoimmune disorders. Arthritis Res. Ther. 15, 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tian Z., Gershwin M. E., and Zhang C. (2012) Regulatory NK cells in autoimmune disease. J. Autoimmun. 39, 206–215 [DOI] [PubMed] [Google Scholar]
- 26. Shifrin N., Raulet D. H., and Ardolino M. (2014) NK cell self tolerance, responsiveness and missing self recognition. Semin. Immunol. 26, 138–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hsueh C. M., Chen S. F., Lin R. J., and Chao H. J. (2002) Cholinergic and serotonergic activities are required in triggering conditioned NK cell response. J. Neuroimmunol. 123, 102–111 [DOI] [PubMed] [Google Scholar]
- 28. Qiu Y. H., Peng Y. P., Jiang J. L., and Wang J. J. (2004) Effect of acetylcholine on in vitro IL-2 production and NK cell cytotoxicity of rats. Lymphology 37, 31–38 [PubMed] [Google Scholar]
- 29. Hao J., Shi F. D., Abdelwahab M., Shi S. X., Simard A., Whiteaker P., Lukas R., and Zhou Q. (2013) Nicotinic receptor β2 determines NK cell-dependent metastasis in a murine model of metastatic lung cancer. PLoS ONE 8, e57495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chernyavsky A. I., Arredondo J., Galitovskiy V., Qian J., and Grando S. A. (2009) Structure and function of the nicotinic arm of acetylcholine regulatory axis in human leukemic T cells. Int. J. Immunopathol. Pharmacol. 22, 461–472 [DOI] [PubMed] [Google Scholar]
- 31. Landais E., Liautaud-Roger F., and Antonicelli F. (2010) Lymphocytes prime activation is required for nicotine-induced calcium waves. Front. Biosci. 2, 928–939 [DOI] [PubMed] [Google Scholar]
- 32. Qian J., Galitovskiy V., Chernyavsky A. I., Marchenko S., and Grando S. A. (2011) Plasticity of the murine spleen T-cell cholinergic receptors and their role in in vitro differentiation of naive CD4 T cells toward the Th1, Th2 and Th17 lineages. Genes Immun. 12, 222–230 [DOI] [PubMed] [Google Scholar]
- 33. Chijioke O., and Münz C. (2013) Dendritic cell derived cytokines in human natural killer cell differentiation and activation. Front. Immunol. 4, 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Romee R., Leong J. W., and Fehniger T. A. (2014) Utilizing cytokines to function-enable human NK cells for the immunotherapy of cancer. Scientifica 2014, 205796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. daCosta C. J., Free C. R., Corradi J., Bouzat C., and Sine S. M. (2011) Single-channel and structural foundations of neuronal α7 acetylcholine receptor potentiation. J. Neurosci. 31, 13870–13879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lam R. A., Chwee J. Y., Le Bert N., Sauer M., Pogge von Strandmann E., and Gasser S. (2013) Regulation of self-ligands for activating natural killer cell receptors. Ann. Med. 45, 384–394 [DOI] [PubMed] [Google Scholar]
- 37. Mistry A. R., and O'Callaghan C. A. (2007) Regulation of ligands for the activating receptor NKG2D. Immunology 121, 439–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang H., Liao H., Ochani M., Justiniani M., Lin X., Yang L., Al-Abed Y., Wang H., Metz C., Miller E. J., Tracey K. J., and Ulloa L. (2004) Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat. Med. 10, 1216–1221 [DOI] [PubMed] [Google Scholar]
- 39. Kawashima K., and Fujii T. (2003) The lymphocytic cholinergic system and its contribution to the regulation of immune activity. Life Sci. 74, 675–696 [DOI] [PubMed] [Google Scholar]
- 40. Sun Z., Smyth K., Garcia K., Mattson E., Li L., and Xiao Z. (2013) Nicotine inhibits memory CTL programming. PLoS ONE 8, e68183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hao J., Simard A. R., Turner G. H., Wu J., Whiteaker P., Lukas R. J., and Shi F. D. (2011) Attenuation of CNS inflammatory responses by nicotine involves α7 and non-α7 nicotinic receptors. Exp. Neurol. 227, 110–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Simard A. R., Gan Y., St-Pierre S., Kousari A., Patel V., Whiteaker P., Morley B. J., Lukas R. J., and Shi F. D. (2013) Differential modulation of EAE by α9*- and β2*-nicotinic acetylcholine receptors. Immunol. Cell Biol. 91, 195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hoogduijn M. J., Cheng A., and Genever P. G. (2009) Functional nicotinic and muscarinic receptors on mesenchymal stem cells. Stem Cells Dev. 18, 103–112 [DOI] [PubMed] [Google Scholar]
- 44. Wu J. C., Chruscinski A., De Jesus Perez V. A., Singh H., Pitsiouni M., Rabinovitch M., Utz P. J., and Cooke J. P. (2009) Cholinergic modulation of angiogenesis: role of the 7 nicotinic acetylcholine receptor. J. Cell Biochem. 108, 433–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Razani-Boroujerdi S., Boyd R. T., Dávila-García M. I., Nandi J. S., Mishra N. C., Singh S. P., Pena-Philippides J. C., Langley R., and Sopori M. L. (2007) T cells express α7-nicotinic acetylcholine receptor subunits that require a functional TCR and leukocyte-specific protein tyrosine kinase for nicotine-induced Ca2+ response. J. Immunol. 179, 2889–2898 [DOI] [PubMed] [Google Scholar]
- 46. Schedel A., Thornton S., Schloss P., Klüter H., and Bugert P. (2011) Human platelets express functional α7-nicotinic acetylcholine receptors. Arterioscler. Thromb. Vasc. Biol. 31, 928–934 [DOI] [PubMed] [Google Scholar]
- 47. Dajas-Bailador F., and Wonnacott S. (2004) Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol. Sci. 25, 317–324 [DOI] [PubMed] [Google Scholar]
- 48. Sharma G., and Vijayaraghavan S. (2002) Nicotinic receptor signaling in nonexcitable cells. J. Neurobiol. 53, 524–534 [DOI] [PubMed] [Google Scholar]
- 49. Mandal A., and Viswanathan C. (2015) Natural killer cells: in health and disease. Hematol. Oncol. Stem Cell Ther. 8, 47–55 [DOI] [PubMed] [Google Scholar]
- 50. Pavlov V. A., and Tracey K. J. (2005) The cholinergic anti-inflammatory pathway. Brain. Behav. Immun. 19, 493–499 [DOI] [PubMed] [Google Scholar]
- 51. Banchereau J., Briere F., Caux C., Davoust J., Lebecque S., Liu Y. J., Pulendran B., and Palucka K. (2000) Immunobiology of dendritic cells. Annu. Rev. Immunol. 18, 767–811 [DOI] [PubMed] [Google Scholar]
- 52. Banchereau J., and Steinman R. M. (1998) Dendritic cells and the control of immunity. Nature 392, 245–252 [DOI] [PubMed] [Google Scholar]
- 53. Cella M., Sallusto F., and Lanzavecchia A. (1997) Origin, maturation and antigen presenting function of dendritic cells. Curr. Opin. Immunol. 9, 10–16 [DOI] [PubMed] [Google Scholar]
- 54. Egea J., Buendia I., Parada E., Navarro E., León R., and Lopez M. G. (2015) Anti-inflammatory role of microglial α7 nAChRs and its role in neuroprotection. Biochem. Pharmacol. 97, 463–472 [DOI] [PubMed] [Google Scholar]
- 55. Kalkman H. O., and Feuerbach D. (2016) Modulatory effects of α7 nAChRs on the immune system and its relevance for CNS disorders. Cell Mol. Life Sci. 73, 2511–2530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hurst R., Rollema H., and Bertrand D. (2013) Nicotinic acetylcholine receptors: from basic science to therapeutics. Pharmacol. Ther. 137, 22–54 [DOI] [PubMed] [Google Scholar]
- 57. Corradi J., and Bouzat C. (2016) Understanding the bases of function and modulation of α7 nicotinic receptors: implications for drug discovery. Mol. Pharmacol. 10.1124/mol.116.104240 [DOI] [PubMed] [Google Scholar]
- 58. Suzuki T., Hide I., Matsubara A., Hama C., Harada K., Miyano K., Andrä M., Matsubayashi H., Sakai N., Kohsaka S., Inoue K., and Nakata Y. (2006) Microglial α7 nicotinic acetylcholine receptors drive a phospholipase C/IP3 pathway and modulate the cell activation toward a neuroprotective role. J. Neurosci. Res. 83, 1461–1470 [DOI] [PubMed] [Google Scholar]
- 59. Zdanowski R., Krzyzowska M., Ujazdowska D., Lewicka A., and Lewicki S. (2015) Role of α7 nicotinic receptor in the immune system and intracellular signaling pathways. Cent. Eur. J. Immunol. 40, 373–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. King J. R., and Kabbani N. (2016) α7 nicotinic receptor coupling to heterotrimeric G proteins modulates RhoA activation, cytoskeletal motility, and structural growth. J. Neurochem. 10.1111/jnc.13660 [DOI] [PubMed] [Google Scholar]
- 61. King J. R., Nordman J. C., Bridges S. P., Lin M. K., and Kabbani N. (2015) Identification and characterization of a G protein-binding cluster in α7 nicotinic acetylcholine receptors. J. Biol. Chem. 290, 20060–20070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Adamczyk M., Brashear R. J., and Mattingly P. G. (2006) Choline concentration in normal blood donor and cardiac troponin-positive plasma samples. Clin. Chem. 52, 2123–2124 [DOI] [PubMed] [Google Scholar]
- 63. Kawashima K., Fujii T., Moriwaki Y., Misawa H., and Horiguchi K. (2015) Non-neuronal cholinergic system in regulation of immune function with a focus on α7 nAChRs. Int. Immunopharmacol. 29, 127–134 [DOI] [PubMed] [Google Scholar]
- 64. Lueders C. D., Müller O., Storm C., and Möckel M. (2007) Evaluation of a chemiluminescent assay for analysis of choline in human plasma and whole blood. Lab. Med. 38, 726–728 [Google Scholar]
- 65. Pacheco R., Riquelme E., and Kalergis A. M. (2010) Emerging evidence for the role of neurotransmitters in the modulation of T cell responses to cognate ligands. Cent. Nerv. Syst. Agents Med. Chem. 10, 65–83 [DOI] [PubMed] [Google Scholar]
- 66. Weinstein L. I., Revuelta A., and Pando R. H. (2015) Catecholamines and acetylcholine are key regulators of the interaction between microbes and the immune system. Ann. N.Y. Acad. Sci. 1351, 39–51 [DOI] [PubMed] [Google Scholar]
- 67. Mignini F., Streccioni V., and Amenta F. (2003) Autonomic innervation of immune organs and neuroimmune modulation. Auton. Autacoid Pharmacol. 23, 1–25 [DOI] [PubMed] [Google Scholar]
- 68. Uteshev V. V. (2012) α7 nicotinic ACh receptors as a ligand-gated source of Ca2+ ions: the search for a Ca2+ optimum. Adv. Exp. Med. Biol. 740, 603–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Uteshev V. V. (2014) The therapeutic promise of positive allosteric modulation of nicotinic receptors. Eur. J. Pharmacol. 727, 181–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Arredondo J., Chernyavsky A. I., Webber R. J., and Grando S. A. (2005) Biological effects of SLURP-1 on human keratinocytes. J. Invest. Dermatol. 125, 1236–1241 [DOI] [PubMed] [Google Scholar]
- 71. Chimienti F., Hogg R. C., Plantard L., Lehmann C., Brakch N., Fischer J., Huber M., Bertrand D., and Hohl D. (2003) Identification of SLURP-1 as an epidermal neuromodulator explains the clinical phenotype of Mal de Meleda. Hum. Mol. Genet. 12, 3017–3024 [DOI] [PubMed] [Google Scholar]
- 72. Kawashima K., Fujii T., Moriwaki Y., Misawa H., and Horiguchi K. (2012) Reconciling neuronally and nonneuronally derived acetylcholine in the regulation of immune function. Ann. N.Y. Acad. Sci. 1261, 7–17 [DOI] [PubMed] [Google Scholar]
- 73. Chatzidaki A., and Millar N. S. (2015) Allosteric modulation of nicotinic acetylcholine receptors. Biochem. Pharmacol. 97, 408–417 [DOI] [PubMed] [Google Scholar]
- 74. Williams D. K., Wang J., and Papke R. L. (2011) Positive allosteric modulators as an approach to nicotinic acetylcholine receptor-targeted therapeutics: advantages and limitations. Biochem. Pharmacol. 82, 915–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Domaica C. I., Fuertes M. B., Uriarte I., Girart M. V., Sardañons J., Comas D. I., Di Giovanni D., Gaillard M. I., Bezrodnik L., and Zwirner N. W. (2012) Human natural killer cell maturation defect supports in vivo CD56(bright) to CD56(dim) lineage development. PLoS ONE 7, e51677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ziblat A., Domaica C. I., Spallanzani R. G., Iraolagoitia X. L., Rossi L. E., Avila D. E., Torres N. I., Fuertes M. B., and Zwirner N. W. (2015) IL-27 stimulates human NK-cell effector functions and primes NK cells for IL-18 responsiveness. Eur. J. Immunol. 45, 192–202 [DOI] [PubMed] [Google Scholar]
- 77. Rossi L. E., Avila D. E., Spallanzani R. G., Ziblat A., Fuertes M. B., Lapyckyj L., Croci D. O., Rabinovich G. A., Domaica C. I., and Zwirner N. W. (2012) Histone deacetylase inhibitors impair NK cell viability and effector functions through inhibition of activation and receptor expression. J. Leukoc. Biol. 91, 321–331 [DOI] [PubMed] [Google Scholar]