

Abstract
Our previous study suggested that ceramide synthase 6 (CerS6), an enzyme in sphingolipid biosynthesis, is regulated by p53: CerS6 was elevated in several cell lines in response to transient expression of p53 or in response to folate stress, which is known to activate p53. It was not clear, however, whether CerS6 gene is a direct transcriptional target of p53 or whether this was an indirect effect through additional regulatory factors. In the present study, we have shown that the CerS6 promoter is activated by p53 in luciferase assays, whereas transcriptionally inactive R175H p53 mutant failed to induce the luciferase expression from this promoter. In vitro immunoprecipitation assays and gel shift analyses have further demonstrated that purified p53 binds within the CerS6 promoter sequence spanning 91 bp upstream and 60 bp downstream of the transcription start site. The Promo 3.0.2 online tool for the prediction of transcription factor binding sites indicated the presence of numerous putative non-canonical p53 binding motifs in the CerS6 promoter. Luciferase assays and gel shift analysis have identified a single motif upstream of the transcription start as a key p53 response element. Treatment of cells with Nutlin-3 or low concentrations of actinomycin D resulted in a strong elevation of CerS6 mRNA and protein, thus demonstrating that CerS6 is a component of the non-genotoxic p53-dependent cellular stress response. This study has shown that by direct transcriptional activation of CerS6, p53 can regulate specific ceramide biosynthesis, which contributes to the pro-apoptotic cellular response.
Keywords: ceramide synthase, lipid signaling, p53, transcription factor, transcription regulation
Introduction
The tumor suppressor p53 is a transcription factor commonly activated upon certain types of cellular stress such as DNA damage, nutrient starvation, ribosomal stress, or oncogenic signaling (1–4). The list of p53-regulated targets includes several hundred genes (5), and the overall cellular response to p53 activation is an integral function determined by differential activation of target genes (6). Not all p53-responsive genes are direct transcriptional targets of p53, and indirect effects of the protein on transcription through other transcription factors or additional regulatory elements are common (7, 8). For example, one of the mechanisms multiplying the number of p53-responsive targets is the regulation of non-coding RNA transcription (9). A bioinformatics approach predicted that p53 is likely to regulate a handful of microRNAs, which in turn control expression of hundreds of genes (10), a phenomenon dramatically expanding the p53 network. The number of genes directly regulated by p53, however, could be much lower within a couple of hundred targets (11). Furthermore, although initially p53 was thought to act as the transcription activator, later it was demonstrated that it can also repress transcription through direct interaction with target genes as well as through indirect mechanisms (1, 3, 8). The direct transcriptional regulation by p53 itself is a complex process that could be affected by p53 modifications, the interaction with p53 co-regulators, and cooperation with other transcription factors (6).
Whether a given promoter is regulated by p53 depends on the amount of the protein, its modification state, the cofactors present at the promoter, or additional cooperating factors that enhance or repress the p53-induced transcription (12). These factors can also dictate which genes are targeted for transcriptional regulation (2, 7). However, the most important components defining the p53-directed transcriptional regulation are the p53 response elements on the DNA (13). The sequence-specific DNA binding activity of p53 is critical to its tumor suppressor activity in response to cellular and environmental stresses. Canonically, p53 binds as a tetramer to two decameric motifs, which have been defined by the consensus sequence RRRCWWGYYY with a separation of 0–13 bp, where R = purine, Y = pyrimidine, and W = A or T (14). Further studies have identified numerous non-canonical DNA binding motifs for p53 that can differ significantly in their affinity to the protein (2). For example, it has been demonstrated that a 10-base motif representing half of the canonical site can function as the p53 response element (15). Furthermore, most of the validated response elements have mismatches compared with the originally defined consensus motif, with 10% of such elements having novel sequences not clearly related to the consensus motif (2). Of note, functional activities of such non-canonical response elements can vary significantly, with lower activity sites requiring much higher levels of p53 protein for the transcriptional regulation (2).
Because p53 regulates transcription of so many genes involved in a variety of cellular processes, the protein has a very broad spectrum of its effect on cellular homeostasis. Pathways regulated by p53 include DNA repair, cell cycle arrest, apoptosis, senescence, autophagy, angiogenesis, and migration (1–4). More recently, the p53-dependent regulation of cellular metabolism has been also underscored (4). Thus, p53 regulates both glycolysis and oxidative phosphorylation shifting the energy production to the latter pathway (16, 17). Such regulation is achieved through the transcriptional repression of glucose transporters type 1, 3, and 4 and transcriptional activation of TIGAR, SCO2, GLS2, and Parkin (18–24). P53 also plays a key role in lipid metabolism activating fatty acid oxidation and inhibiting fatty acid synthesis (16), and it was further implicated in the regulation of sphingolipid metabolism (25). Sphingolipids play an important role in both the structural integrity of cellular membranes and as signaling molecules involved in the regulation of major cellular processes such as senescence, survival, and death (26, 27). Our previous study suggested that one of the enzymes in sphingolipid biosynthesis, ceramide synthase 6 (CerS6),4 is regulated by p53 (28). Thus, CerS6 was elevated in several cell lines in response to transient expression of p53 and also in response to folate stress, which is known to activate p53 (29, 30). For example, CerS6 was strongly elevated in response to the antifolate methotrexate in a p53-dependent manner (31). It was not clear, though, whether CerS6 gene is a direct transcriptional target of p53 or this was an indirect effect through additional regulatory factors. In the present study, we have investigated CerS6 promoter as a direct transcriptional target of p53.
Results
The CerS6 Promoter Is Regulated by p53
We have studied the effect of p53 on the CerS6 promoter regulation using a luciferase assay. The promoter has been previously cloned into pGL3 vector bearing firefly luciferase cDNA (32), and this construct was co-transfected with the p53-expressing vector in A549 cells. By itself, the CerS6 promoter displayed a weak transcriptional activity (Fig. 1A). However, we have observed a significant (>10-fold) increase in the luciferase activity in cells co-transfected for the p53 expression compared with cells co-transfected with the “empty” (negative control) plasmid (Fig. 1A). When cells were co-transfected with the luciferase-expressing reporter and the vector expressing the transcriptionally deficient R175H p53 mutant, no increase in the luciferase activity was recorded (Fig. 1A). To eliminate potential impact from endogenous p53 protein on CerS6 promoter, we have also performed luciferase assays in the p53-deficient A549 cell line. In this cell line p53 was silenced by the stable expression of the p53 shRNA, which completely prevented the protein expression (28, 33). Similar to p53-proficient A549 cells, strong activation of luciferase was seen in the p53-deficient cells upon co-expression with the p53-expressing vector but not the vector expressing mutant p53 (Fig. 1B). Of note, levels of expressed wild type and mutant p53 were similar in each cell line (Fig. 1, A and B, insets). Because the effect on the CerS6 promoter was comparable in both cell lines, in further studies we used regular A549 cells, which allow the evaluation of stress stimuli on the CerS6 expression.
FIGURE 1.
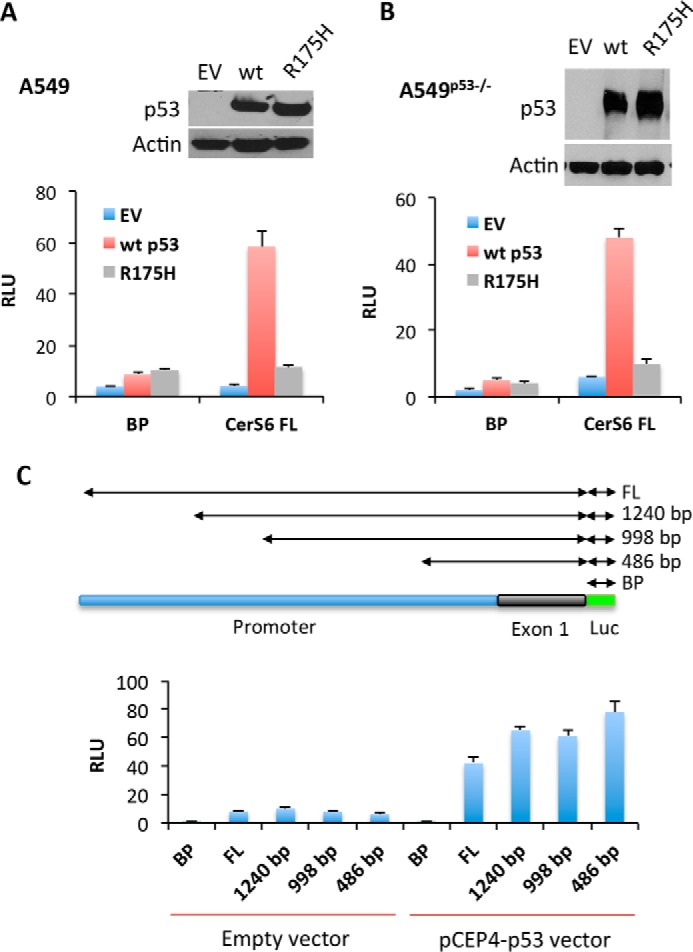
CerS6 promoter is activated by p53. A, activity of the CerS6 promoter in A549 cells with and without expressed p53, assessed as the relative luminescence units (RLU) in luciferase reporter assays. Co-transfections of CerS6 luciferase reporter with empty vector (EV), vector expressing wild type (wt) p53, or vector expressing the transcriptionally deficient R175H mutant were carried out, and luciferase activity was measured 48 h post-transfection. B, same as A, but experiments were performed in A549 p53 shRNA (p53−/−) cells. A reporter construct with the 1625-bp full-length (FL) promoter was used in experiments depicted in A and B; basic plasmid (BP, the pGL3 luciferase vector without the CerS6 promoter) served as a negative control. Insets show levels of p53 48 h post-transfection (Western blotting assays). C, activity of truncated CerS6 promoters measured in A549 cells by luciferase assays. Reporter constructs were co-transfected with either empty vector or p53-expressing vector. The diagram on the top schematically depicts the design of CerS6 promoter constructs used in these experiments. In each panel the means ± S.D. of a representative experiment performed in quadruplicate are shown. Experiments were repeated at least three times.
Previous study used four different length constructs of the CerS6 promoter to explore its regulation (32), and we tested these constructs for activation by p53. Luciferase assays have shown that all four promoter constructs resulted in a strong increase of luciferase activity when co-transfected with the p53 expression vector (Fig. 1C). Of note, the shortest construct was most active in the presence of p53 (Fig. 1C).
Identification of the p53 Binding Motif in the CerS6 Promoter
A previously published study of CerS6 promoter indicated its regulation by AP2, SP1/3, and CREB transcription factors but not by p53 (32). Our analysis of CerS6 promoter using several web-based tools for the prediction of transcription factor binding sites, including Patch 1, Match, Matrix Catch, Alibaba, and Transfac, did not produce hits corresponding to the p53 binding motifs. Thus, it appeared that this promoter does not have canonical p53-binding sites. Here we have applied Promo 3.0.2 online tool (34, 35) for additional analysis of the CerS6 promoter region, which included 1395 bp upstream of the transcription start and 284 bp of exon 1 (Fig. 2). Surprisingly, in contrast to other tools, Promo 3.0.2 has indicated multiple putative p53-binding sites in the CerS6 promoter (Fig. 2). Most of the hits (27 of the total of 32 sites) are located within of the proximal promoter (495 bp upstream of the transcription start) and 200 bp of exon 1. Of note, some of the predicted sites overlap within the same short contiguous sequence, suggesting that the real number of such sites is lower than the predicted number (Fig. 2).
FIGURE 2.

Analysis of the CerS6 promoter for the presence of p53-binding sites. Full-length CerS6 promoter sequence was analyzed using Promo 3.0.2 online tool. Putative p53 response elements are shown in red; predicted binding sites are indicated by double-arrow lines. The transcription start site is indicated by vertical arrow. The translation start in exon 1 (ATG) is shown in blue. The diagram on the top indicates positions of the putative p53 response elements (red squares). Triangles indicate number of binding sites within a contiguous sequence.
Defining the Minimal CerS6 Promoter Responsive to p53
To decipher the mechanism of the activation of CerS6 promoter by p53, we created a set of truncated promoter constructs (Fig. 3). The originally cloned CerS6 promoter included 1395 bp upstream of the transcription start site and a part of exon 1 (32). The portion of exon 1 in the promoter consists of the entire non-translatable (bp 1–200) and a part of translatable (bp 201–284) region. This promoter structure defined our strategy in the identification of the position of p53 response elements. By itself, the promoter has a weak transcriptional activity that is enhanced upon p53 expression by ∼6–10-fold (Fig. 3). The removal of the translatable portion of exon 1 has improved the response to p53 by ∼1.5-fold (Fig. 3). Whereas the precise mechanism of this effect is not clear, further promoter bashing was done with a construct lacking the translatable part of exon 1. Of note, the repressing effect of this translatable sequence on the promoter activity in the presence of p53 was observed for shorter promoter constructs as well; constructs III-V (Fig. 3) had ∼50% less activity if this portion of exon 1 was not removed (data not shown). Our experiments demonstrated that the p53 response elements are likely positioned in the CerS6 promoter region extended from −291 to 50 bp (Fig. 3); this construct revealed the strongest activation upon p53 expression, whereas further truncation of the promoter beyond this region impaired its activation by p53.
FIGURE 3.

Activation of truncated CerS6 promoter constructs by p53. The schematic on the left depicts promoter constructs used in this study (violet blocks correspond to the region upstream of the transcription start site; yellow blocks correspond to exon 1; the brown block in construct I indicates the translatable part of exon 1; numbers indicate position of corresponding base pairs). Luciferase activity (mean ± S.D.) of three independent experiments (each in quadruplicate) is shown. BP, basic plasmid (promoterless reporter). RLU, relative luminescence units.
Purified p53 Binds the Essential CerS6 Promoter
The activation of CerS6 promoter by p53 in the luciferase assay can proceed indirectly through regulatory elements activated by p53. To investigate whether p53 directly binds to the essential CerS6 promoter, we used the pulldown assay of the promoter fragment (from −204 to 60 relative to the transcription start site) on purified p53 protein. In these experiments the fragment corresponding to the above promoter region was excised from the plasmid using Sac1/HindIII endonucleases and purified from agarose gel after electrophoresis. The fragment was further incubated with full-length p53 protein and amplified by PCR with specific primers after the pulldown on p53-specific antibody and protein G-agarose (schematically depicted in Fig. 4A). Primers used for the amplification were fluorescently labeled, allowing the direct detection of PCR fragments (primer sequences are shown in Table 1). An intensive band corresponding to the CerS6 promoter was observed after the pulldown (Fig. 4B) that indicated the direct interaction between p53 and the promoter. The specificity of this interaction was demonstrated in control experiments, which used either ALDH1L1 protein (36) instead of p53 and the corresponding antibody or just IgG in the absence of p53; no amplified promoter sequences were observed under these conditions (Fig. 4B, lanes 2 and 3, correspondingly). These experiments also demonstrated that the interaction between p53 and the sequence corresponding to the CerS6 promoter is very strong; we were able to pull it down with p53 and its specific antibody even in the absence of cross-linking (Fig. 4B, lane 4).
FIGURE 4.

The p53 protein binds the essential CerS6 promoter. A, schematic depicting pulldown of CerS6 promoter fragment bound to p53 by immunoprecipitation with p53-specific antibody (Ab). The fragment corresponding to 264-bp CerS6 promoter was excised from pGL3/CerS6 vector, incubated with purified recombinant p53, cross-linked with formaldehyde, and pulled down using p53-specific antibody/protein G beads. After the pulldown, cross-linking was reversed by 5 m NaCl. The pulled down sequence was amplified by PCR using fluorescently labeled sequence-specific primers (shown in Table 1). B, agarose gel electrophoresis of the amplified CerS6 fragment after the pulldown (as depicted by panel A). Lane 1, pulldown of the CerS6 promoter using recombinant p53 and p53-specific antibody; lane 2, pulldown with ALDH1L1 protein and ALDH1L1-specific antibody; lane 3, pulldown with human IgG; lane 4, same as lane 1 but without cross-linking. The gel was stained with ethidium bromide, and the image was obtained using an Odyssey Fc instrument (LI-CORE). The green channel was assigned to image the ethidium bromide-stained double-stranded DNA; the red channel imaged the fluorescent primer. Double-stranded DNA containing the fluorescent label was seen in yellow due to the two-channel overlay.
TABLE 1.
Primers used for generation of CerS6 promoter constructs
Cy5 indicates fluorescent dye covalently attached to the 5′ end of corresponding oligonucleotides.
| Construct | Primers | Sequence |
|---|---|---|
| Reporters II-V | Sense | 5′-CAAAGCAAGATTCGAGATCTGC-3′ |
| Antisense | 5′-GCAGATCTCGAATCTTGCTTTG-3′ | |
| Reporter VI | Sense | 5′-CGGCGGCGGCATCGAGATCTGC-3′ |
| Antisense | 5′-GCAGATCTCGATGCCGCCGCCG-3′ | |
| Reporter VII | Sense | 5′-GCGTTAGCCAGTGGGCGAGTTG-3′ |
| Antisense | 5′-CAACTCGCCCACTGGCTAACGC-3′ | |
| Reporter VIII | Sense | 5′-CTTCGAGAGCAGCGGCGGCGGCAC-3′ |
| Antisense | 5′-GTGCCGCCGCCGCTGCTCTCGAAG-3′ | |
| Reporter IX | Sense | 5′-CTTTCCCCAGTCGAGATCTG-3′ |
| Antisense | 5′-CAGATCTCGACTGGGGAAAG-3′ | |
| Reporter X | Sense | 5′-CCCTGCGGGCGTTCGAGATCTGCG-3′ |
| Antisense | 5′-CGCAGATCTCGAACGCCCGCAGGG-3′ | |
| ΔRE1 | Sense | 5′-GACGAGCCAATCAGAGCTGCCATGTG-3′ |
| Antisense | 5′-CACATGGCAGCTCTGATTGGCTCGTC-3′ | |
| ΔRE2 | Sense | 5′-GAGCTGCCATGTGATTTCCCCAGCGCGG-3′ |
| Antisense | 5′-CCGCGCTGGGGAAATCACATGGCAGCTC-3′ | |
| ΔRE1/2 | Sense | 5′-GACGAGCCAATCATTTCCCCAGCGCG-3′ |
| Antisense | 5′-CGCGCTGGGGAAATGATTGGCTCGTC-3′ | |
| ΔRE3 | Sense | 5′-GAGGAGAGAACCGCCGCGAGCCTTCG-3′ |
| Antisense | 5′-CGAAGGCTCGCGGCGGTTCTCTCCTC-3′ | |
| 355 bp | Forward | Cy5-GTTAGCCAGCATTCTGAGCATGTG-3′ |
| 271 bp | Forward | Cy5-GCAAAGCGAAAGAGCGGTGAGAGC-3′ |
| 190 bp | Forward | Cy5-GACTGCGCCCTGCCCTGC-3′ |
| 151 bp | Forward | Cy5-GTGTGTGCCTCGCTGTTTGACGC-3 |
| 55 bp | Forward | Cy5-CAGAGGGAGGAGAGAACC |
| All | Reverse | Cy5-CTCGATGCCGCCGCCGCTGCTCTCG-3′ |
| 39 bp | Forward | Cy5-GAGCCTTCGAGAGCAGCG-3′ |
| Reverse | Cy5-CGCCGCCGCCTCCTCC |
The interaction between essential CerS6 promoter and the p53 protein was further confirmed by the gel-shift assays (Fig. 5). Six fragments corresponding to different promoter size and generated by the PCR amplification were tested in these experiments (Fig. 5A). Fluorescently labeled primers (Table 1) used in these amplifications allowed the direct imaging of PCR products. Gel-shift assays have shown a strong size-specific shift of the DNA in the presence of p53 for the four longest promoter constructs, 151 to 355 bp, whereas no such shift was observed for the two shortest constructs (Fig. 5B). A high molecular mass band was also seen for all pulled-down samples in the presence of p53, indicating that this band was a result of nonspecific aggregation (Fig. 5B). Additional nonspecific bands were observed for the shortest construct (Fig. 5B). Overall, these experiments have demonstrated that the essential CerS6 promoter interacts with p53 and indicated that the p53-binding site(s) is located within the 151-bp region extending from the position −91 to the position 60 relative to the transcription start site. Gel shift experiments performed in the presence of the p53-specific antibody resulted in the DNA supershift, further confirming the p53-promoter interaction (Fig. 5C). This interaction was disrupted in the presence of DNA duplex containing a canonical p53-binding site from the p21 promoter (Fig. 5C). The interaction of the 151-bp promoter with p53 as a function of the protein concentration was further examined in the gel shift assay, which indicated that the binding constant for the interaction is ∼20 nm (Fig. 5D).
FIGURE 5.

Analysis of the binding of p53 to CerS6 promoter using gel shift assay. A, schematic depicting DNA fragments used in these experiments. Fluorescent labeling of these fragments was achieved by the PCR amplification using primers with a covalently attached Cy5 fluorescent dye (Table 1). B, gel shift analysis of the interaction between CerS6 promoter fragments (from panel A) and purified p53 (50 nm). Arrows indicate respective fragments and DNA shifted upon the p53 binding. C, gel shift analysis of the interaction between the 151-bp promoter fragment (from panel A) and purified p53 (20–60 nm) in the presence or absence of p53-specific antibody (Ab). The last lane shows competition between the 151-bp fluorescent promoter fragment and non-fluorescent double-stranded DNA containing the canonical p53-binding motif from p21 promoter (shown in Table 2) for the p53 binding (60 nm). C, negative control, no p53. D, gel shift analysis of the interaction between the 151-bp promoter fragment (from panel A) and increasing concentrations of purified p53.
The p53 Response Elements in the CerS6 Promoter
Based on the results of the luciferase and gel-shift assays (above), we have further focused on a shorter part of the promoter, from the position −91 to the position 60. In this region three putative p53 binding motifs have been identified (Fig. 6A). To study whether the corresponding sequences are genuine p53 response elements (REs), we have created a set of promoter constructs that retained all three motifs or its different combinations (Fig. 6B) and tested them for the activation by p53 in luciferase assays. These assays have shown that putative p53 response element 2 is a key for the activation of CerS6 promoter by p53; the promoter construct retaining RE2 was as effective in luciferase induction in response to p53 as the construct with all three response elements, RE1–3 (Fig. 6C). Correspondingly, the deletion of this putative response element decreased the activation by ∼70% (Fig. 6C). Putative RE1 has shown some ability to activate the promoter, although to a lower extent than RE2 (Fig. 6C). In contrast, putative RE3 was not involved in the p53 response; its deletion did not affect the luciferase activation, and the construct retaining only RE3 was not induced by p53 (Fig. 6C). In agreement with the luciferase assay data, the gel-shift assay demonstrated the interaction between the short oligonucleotide bearing RE2 (Table 1) but not constructs bearing RE1 or RE3 (Fig. 6D). The interaction with RE2 was disrupted in the presence of DNA duplex containing a canonical p53-binding site from the p21 promoter (Fig. 6E).
FIGURE 6.

A single p53 response element upstream of the transcription start site regulates CerS6 promoter. A, position of three predicted putative p53 response elements in the minimal CerS6 promoter. The vertical arrow indicates the transcription start site; horizontal arrows correspond to duplexes (Table 2) used for the gel shift assay in panels D and E. B, schematic depicting CerS6 promoter constructs with different combination of the p53 response elements. C, activity of constructs (from panel B) with and without expressed p53, assessed as the relative luminescence units (RLU) in luciferase reporter assays. Co-transfections of luciferase reporters with empty vector (Vector), vector expressing wild type p53 or vector expressing the transcriptionally deficient R175H mutant were carried out, and luciferase activity was measured 48 h post-transfection. Luciferase activity (means ± S.D.) of a representative experiment performed in quadruplicate is shown. Experiments were repeated three times. FL, full length. D, gel shift analysis of the interaction between duplexes corresponding to RE1–3 (Table 2) and varying concentrations of purified p53 (0–60 nm). E, competition for the p53 binding between fluorescent RE2 duplex and non-fluorescent duplex containing the canonical p53-binding motif from p21 promoter (60 nm p53 was used; C, negative control, no p53 added).
CerS6 Transcription Is Induced upon p53 Activation
We further investigated whether the p53 activation in the cell induces the CerS6 promoter. One of the common ways to activate p53 is the treatment of cells with low concentrations of actinomycin D (37). Treatment of A549 cells with 500 ng/ml concentrations of the drug in our experiments resulted in elevation of p53 protein, which was accompanied by strong increase in CerS6 mRNA and protein (Fig. 7A). To assure that the elevation of CerS6 mRNA was associated with the p53 activation and was not a side effect of the drug, we also treated A549 cells with Nutlin-3, a specific activator of the p53 pathway (38). In agreement with the Nutlin-3 function, significant elevation of the p53 protein was seen in cells exposed to 10 μm concentrations of the drug (Fig. 7B). Similar to actinomycin D, Nutlin-3 has also evoked a strong increase in CerS6 mRNA, which was accompanied by the increase in the CerS6 protein levels (Fig. 7B).
FIGURE 7.
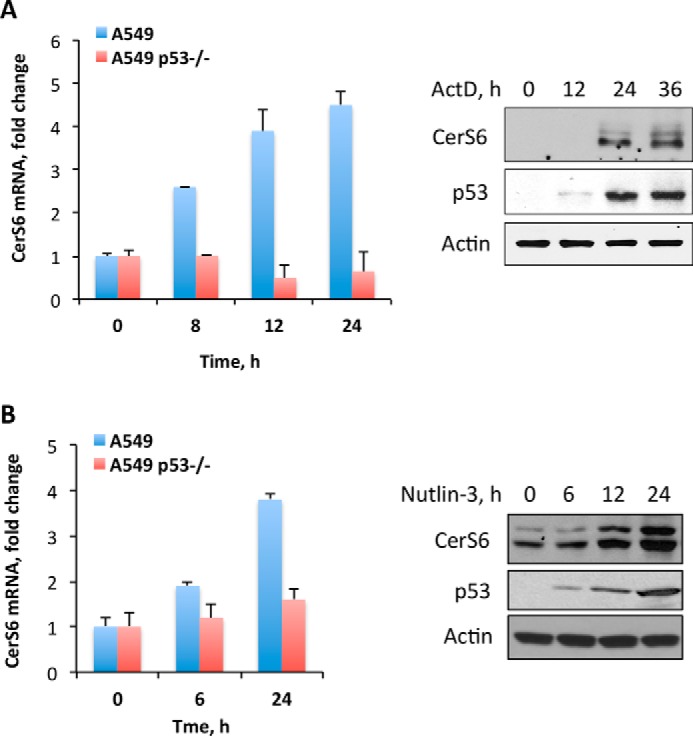
Drugs activating p53 elevate levels of CerS6 mRNA and protein. A, effect of actinomycin D (ActD, 500 ng/ml) on CerS6 mRNA (left panel) and protein (right panel) levels. B, effect of Nutlin-3 (10 μm) on CerS6 mRNA (left panel) and protein (right panel) levels. Error bars represent ±S.D., n = 3.
Discussion
Ceramides, a group of sphingolipids, play an important role as components of cellular membranes (39–42) and as signaling molecules (26, 43). In humans, there are six ceramide synthases, CerS1–6, which are involved in biosynthesis of ceramide molecules with different acyl chain length (44, 45). Expression and activity of these enzymes are regulated at several levels, including transcription (46). The information on transcriptional regulation of ceramide synthases, however, is scarce. The analysis of CerS1 and CerS6 promoters indicated their potential regulation by several common transcription factors, with Sp1 playing a key role in the activation of the CerS1 promoter (32). It has been recently shown that two other ceramide synthase genes, CerS4 and CerS5, are transcriptionally regulated by AP-1 in the estrogen receptor-dependent manner (47). Although alterations in biosynthesis and degradation of sphingolipids are associated with the p53 activation, genes encoding enzymes of the sphingolipid pathways, including ceramide synthases, are not known as transcriptional targets of p53. It has been reported, though, that p53 drives de novo ceramide synthesis by activating one of the ceramide synthases (48). The mechanism of such activation could be indirect, through the p53 effect on additional transcription regulators or repressors. For example, through the regulation of microRNAs p53 can affect the expression of a plethora of genes (10). In fact, such regulation has been shown for ceramide synthases: the alternative splice variant of CerS1 is inhibited by microRNA-574-5p (32). Furthermore, it has been recently reported that the elevation of CerS6 expression is associated with the reduction of microRNA-101 (49). Interestingly, this microRNA was predicted to have p53 response elements (10). The co-regulation of p53 and CerS6 and the role of microRNAs in this process, however, are not so clear. Thus, although CerS6 expression inversely correlates with miR-101, a positive correlation between the p53 accumulation and miR-101 has been recently reported (50). In this mechanism, miR-101 targets the proteasome maturation protein POMP, leading to impaired proteasome assembly and activity. So far there were no reports, which would specifically study the transcriptional regulation of CerS genes by p53, but a recent meta-analysis indicated the presence of p53 regulatory elements in these genes, although corresponding target scores were rather low (8).
We previously demonstrated that CerS6 is elevated in response to p53 activation or upon transient transfection of p53 (28). The mechanism of such regulation was not clear as the p53 binding motifs were not found in the CerS6 promoter region. Toward this end, indirect regulation of gene expression by p53, through the effect on other components of transcriptional machinery, is a common phenomenon (7, 8). In fact, much higher numbers of genes are indirect targets of p53 than its direct transcriptional targets (5). Interestingly, we have previously identified a putative p53-binding site in the CerS6 gene outside of the promoter region 3 kb downstream of the transcription start site (28) though mechanisms that would allow transcriptional regulation by utilizing such a remote site were not clear. An implication of CerS6 (LASS6) as a potential transcriptional target of p53 can be derived from the study, which used ChIP with the paired-end di-tag sequencing to create a global map of p53-binding sites in human genome (51). This high throughput screen, while not specifically identifying the CerS6 gene as one of the p53 targets, indicated a probability for the presence of the p53-binding sites in proximity to the gene.
Despite the fact that the canonical p53-binding sites were not present in CerS6 promoter, in our studies the promoter was strongly activated by p53 in luciferase reporter assays. This finding prompted us to further search for p53 binding motifs in the CerS6 promoter region. The Promo 3.0.2 online tool for the prediction of transcription factor (TF) binding sites in DNA sequences (34, 35) has identified multiple non-canonical p53-binding sites upstream of the transcription start site and in the non-translatable portion of exon 1. Although typically minimal promoter includes the proximal sequence upstream of the transcription site, in the CerS6 promoter the part of exon 1 was required for the full promoter activity. Of note, we have previously observed similar dependence for ALDH1L1 promoter (52). Our study demonstrated that of numerous non-canonical p53-binding sites in the CerS6 promoter; two upstream sites most closely located to the transcription start are important for the activation of the promoter by p53. This is in agreement with a general rule that the p53 response elements decrease their transactivation potential as they are distanced from the transcription start site (6, 11). Our data also indicate that one of these sites is a key response element of p53 whereas the other site might partially compensate for the absence of that key site.
Originally regarded as the regulator of the genotoxic stress response, in recent years p53 has been established as a key player in metabolism regulation (4, 7, 16, 17). The identification of novel p53 transcriptional targets involved in cellular metabolism provides a more integrative view of the effect of metabolic stress on homeostasis maintenance. Metabolic pathways regulated by p53 include glycolysis, oxidative phosphorylation, gluconeogenesis, serine biosynthesis, and fatty acid synthesis and degradation (4, 7, 16, 17). Although some circuits in the metabolic network are regulated by p53 indirectly, several key genes involved in metabolic pathways are direct transcriptional targets of p53, including those encoding for glucose transporters (23, 24), hexokinase 2 (53), mitochondrial glutaminase 2 (19, 20), carnitine palmitoyltransferase (54), pantothenate kinase-1 (55), phosphoglycerate dehydrogenase (56), and a component of the cystine/glutamate antiporter (57). Our study extends the p53 regulation to sphingolipid metabolism by demonstrating that CerS6 is a bona fide p53 target gene. CerS6 satisfies all four criteria for such genes (11): 1, it has p53 response elements in the promoter; 2, its promoter binds p53, which was demonstrated by several assays; 3, the promoter is activated by p53 in the luciferase assay; 4, stress stimuli activating the p53 response led to the elevation of CerS6. It is not clear at present whether p53 requires other co-regulators to activate the CerS6 promoter or whether p53 modifications (for example, phosphorylation at different residues) affect the interaction with the response elements in the promoter. Our in vitro experiments, however, indicated that non-modified p53 binds to the CerS6 promoter.
Interestingly, the transcriptional activation of CerS6 by p53 occurs in response to metabolic rather than genotoxic stress. Thus, our previous study demonstrated that CerS6 is elevated in a p53-dependent manner in response to folate stress enzyme ALDH1L1 (28); ALDH1L1, however, induces p53 through metabolic alterations in the absence of DNA damage (33, 58). In line with this finding, the treatment of cancer cells with antifolate methotrexate or folate withdrawal, two stimuli inducing alterations in folate, amino acid, and nucleotide metabolism, activated CerS6 in a p53-dependent fashion (28, 31). In the present study the transcriptional induction of CerS6 by p53 in the absence of genotoxic stress was further confirmed using two canonical activators of p53, actinomycin D and Nutlin-3, which do not induce DNA damage. Although actinomycin D at higher concentrations activates p53 through the induction of DNA damage (59), at low concentrations it mimics the effect of Nutlin-3 (37). In our experiments treatment of cells with Nutlin-3 or low concentrations of actinomycin D resulted in a strong elevation of both CerS6 mRNA and protein, thus demonstrating that CerS6 is a component of the non-genotoxic p53-dependent cellular stress response. It is still not fully understood how the metabolic functions of p53 contribute to its tumor suppression activity. Our present study has shown that by transcriptional activation of CerS6, the enzyme in sphingolipid metabolism, p53 can activate ceramide biosynthesis, which contributes to the pro-apoptotic cellular response.
Experimental Procedures
Cell Culture and Reagents
Actinomycin D and other chemicals were purchased from Sigma unless otherwise indicated. Nutlin-3 was obtained from Santa Cruz Biotechnology (Dallas, TX). Cell culture media and reagents were from Invitrogen Thermo Fisher (Waltham, MA). A549 cell line was from ATCC.
Generation of Truncated CerS6 Promoter Constructs
Truncations of the promoter were carried out by the PCR-based site-directed mutagenesis using Pfu Turbo polymerase (Agilent Technologies, Santa Clara, CA) as we previously described (52). PCR primers (Table 1) were from Eurofins (Huntsville, AL). All constructs were confirmed by sequencing.
Luciferase Reporter Assay
Reporter constructs were transfected in A549 cells using Neon nucleofector (Invitrogen) according to the manufacturer's manual. Luminescence was measured 48 h post-transfection using a Bright-Glo Luciferase assay system (Promega, Durham, NC). The pGL3-Basic plasmid (promoter-less) was used in each experiment to determine the basal levels of luciferase expression. Each construct was tested in three independent transfection experiments. A dual luciferase reporter assay system (Promega) was used to normalize experiments for transfection efficiency (the pRL-TK plasmid expressing Renilla Luciferase was co-transfected with each reporter vector at a ratio 1:10).
Expression and Purification of p53
The pCEP4-175 vector was a generous gift from Dr. Jennifer Pietenpol. Vector for the expression of wild type p53 was generated previously (33). Escherichia coli BL21(DE3) cells transformed with P53 expression construct were grown on an LB/ampicillin plate overnight. A single colony was inoculated into 5 ml of LB medium containing 50 μg/ml ampicillin and grown overnight at 37 °C. Then 500 ml of LB medium with ampicillin were inoculated with the overnight culture and incubated at 37 °C until A600 reached 0.7 followed by the addition of IPTG (1 mm final concentration, Fisher) and incubated additionally for 12 h at 25 °C. Cells were harvested by centrifugation (5000 × g, 10 min), resuspended in 50 ml of 50 mm NaH2PO4 buffer, pH 8.0, containing 300 mm NaCl and incubated for 30 min with 1 mg/ml lysozyme at 4 °C. The suspension was chilled on ice, sonicated, and spun down at 10,000 × g for 40 min). Supernatant was loaded on a PrepEase nickel-nitrilotriacetic acid high yield agarose (USB Corp.) column, and the His-tagged protein was purified per manufacturer's instructions. Protein preparations obtained after metal-affinity chromatography were further subjected to size-exclusion chromatography on Sephacryl S300 (GE Healthcare).
Promoter Pulldown Assays
The fragment corresponding to the promoter region −204 to 60 was excised from the pGL3/CerS6 vector using SacI and HindIII restriction endonucleases (both from New England BioLabs). The fragment was purified by electrophoresis in agarose gel (2%)/gel extraction kit (Qiagen) and incubated with pure p53 (20 ng). The p53-DNA complex was pulled down using p53-specific polyclonal antibody (Santa Cruz, 1:200 dilution) and a CHIP assay kit (EZChiP kit, Millipore). The procedure was done according to the manufacturer's manual. Purified DNA was amplified by PCR using fluorescently labeled primers (Table 1) and visualized using an Odyssey Fc imager (LI-COR Biosciences) after agarose gel electrophoresis. In the case of cross-linking, the p53-DNA complex was treated with 1% formaldehyde, and the reversal of cross-linking was done with 5 m NaCl.
Gel Shift Assay
Experiments were performed with synthetic oligonucleotides or PCR-amplified fragments (Tables 1 and 2). DNA and p53 were incubated in the reaction mixture containing 2 μl of 10× buffer (100 mm Tris, 500 mm KCl, 10 mm DTT, pH 7.5), 1 μl of poly(dI·dC) (1 μg/μl in 10 mm Tris-HCl, 1 mm EDTA, pH 7.5), 2 μl of 25 mm DTT, 1 μl of the corresponding DNA (20 pmol/μl) in 1× Tris-EDTA buffer, 2 μl of purified p53 protein (5 ng/μl in 20 mm Tris-HCl buffer, pH 7.5), and 12 μl of H2O. Reaction mixtures were incubated for 30 min at room temperature. Samples of DNA with or without pure p53 added were resolved using 4% native PAGE prepared in 50 mm Tris-HCl buffer, pH 7.5, 0.38 m glycine, and 2 mm EDTA). In titration experiments, increasing concentrations of p53 (2–60 nm) were used. The Kd was calculated according fluorescence intensity of shifted DNA bands. For supershift experiments, DNA and p53 were incubated as above in the presence of 1 or 2 μg of p53-specific polyclonal antibody (Santa Cruz), and the mixture was resolved using 3–8% gradient polyacrylamide gels. Synthetic primers were purchased from Eurofins. Longer promoter sequences were generated by PCR amplification using pGL3/CerS6 vector and primers shown in Table 1. The PCR mixture was resolved in 2% agarose gel, and the amplified fragment was purified from the gel using gel extraction kit (Qiagen).
TABLE 2.
Oligonucleotide duplexes corresponding to putative p53 response elements in the CerS6 promoter and the p53-binding motif in the p21 promoter (p21 RE)
Cy5 indicates fluorescent dye covalently attached to the 5′ end of corresponding oligonucleotides.
| Response element | Duplex |
|---|---|
| CerS6 RE1 | Cy5–5′-CGAAGACGAGCCAATCAGGGCGGGCGGCCCGAGCTGC-3′ |
| 3′-GCTTCTGCTCGGTTAGTCCCGCCCGCCGGGCTGGACG-5′ | |
| CerS6 RE2 | Cy5–5′-CATGTGACGGGCAAGGCGGCCCCTTTCCCCAGCGC-3′ |
| 3′-GTACACTGCCCGTTCCGCCGGGGAAAGGGGTCGCG-5′ | |
| CerS6 RE3 | Cy5–5′-GGCCAGAGGGAGGAGAGAACCGGGGCTCGCCGCGA-3′ |
| 3′-CCGGTCTCCCTCCTCTCTTGGCCCCGAGCGGCGCT-5′ | |
| p21 RE | 5′-CGAGGAACATGTCCCAACATGTTGCTCGAG-3′ |
| 3′-GCTCCTTGTACAGGGTTGTACAACGTGCTC-5′ |
Real Time PCR
Total RNA was purified using RNA Easy® Mini kit (Qiagen). Reverse transcriptase reaction was performed with an oligo(dT)18 primer using Advantage™ RT-for-PCR kit (Clontech). The resulting cDNA was used to measure CerS6 mRNA levels using MyiQ™ Single-Color Real-time PCR detection system (Bio-Rad) and the iQ5 optical system software (Bio-Rad). CerS6 RT-quantitative PCR primers were: 5′-GGG ATC TTA GCC TGG TTC TGG-3′ (forward), and 5′-GCC TCC GTG TTC TTC AG-3′ (reverse). β-Actin mRNA levels were used to normalize samples.
Western Blotting Assays
Cell lysates for Western blotting analysis were prepared in a buffer containing 50 mm Tris-HCl (pH 8.0), 150 mm NaCl, 2 mm EDTA, 1% Triton X-100, 0.1% SDS, 1 mm dithiothreitol, 1 mm PMSF, and protease inhibitor mixture (Sigma). Samples were subjected to SDS-PAGE followed by immunoblot with the corresponding antibodies. CerS6 polyclonal antibodies (1:1000) were purchased from Novus Biologicals (Littleton, CO). p53 was detected using monoclonal antibody (clone Pab421, 1:500). Actin was detected using a monoclonal antibody from Sigma (clone AC-15, 1:5000).
Author Contributions
B. F., K. A. J., and A. E. performed all the experiments, analyzed the data, and participated in the manuscript preparation. B. O. cloned the original CerS6 promoter and gave input on the overall promoter analysis. S. A. K. participated in the design of all experiments and data analysis, prepared the figures, and wrote the manuscript. N. I. K. conceived and coordinated the study, performed promoter analysis, participated in experimental design and data analysis, and wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant CA193782 (to N. I. K.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- CerS6
- ceramide synthase 6
- RE
- response element.
References
- 1. Laptenko O., and Prives C. (2006) Transcriptional regulation by p53: one protein, many possibilities. Cell Death Differ 13, 951–961 [DOI] [PubMed] [Google Scholar]
- 2. Menendez D., Inga A., and Resnick M. A. (2009) The expanding universe of p53 targets. Nat. Rev. Cancer 9, 724–737 [DOI] [PubMed] [Google Scholar]
- 3. Rinn J. L., and Huarte M. (2011) To repress or not to repress: this is the guardian's question. Trends Cell Biol. 21, 344–353 [DOI] [PubMed] [Google Scholar]
- 4. Kruiswijk F., Labuschagne C. F., and Vousden K. H. (2015) p53 in survival, death, and metabolic health: a lifeguard with a license to kill. Nat. Rev. Mol. Cell Biol. 16, 393–405 [DOI] [PubMed] [Google Scholar]
- 5. Neilsen P. M., Noll J. E., Suetani R. J., Schulz R. B., Al-Ejeh F., Evdokiou A., Lane D. P., and Callen D. F. (2011) Mutant p53 uses p63 as a molecular chaperone to alter gene expression and induce a pro-invasive secretome. Oncotarget 2, 1203–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beckerman R., and Prives C. (2010) Transcriptional regulation by p53. Cold Spring Harb. Perspect. Biol. 2, a000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goldstein I., and Rotter V. (2012) Regulation of lipid metabolism by p53: fighting two villains with one sword. Trends Endocrinol. Metab. 23, 567–575 [DOI] [PubMed] [Google Scholar]
- 8. Fischer M., Steiner L., and Engeland K. (2014) The transcription factor p53: not a repressor, solely an activator. Cell Cycle 13, 3037–3058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hermeking H. (2007) p53 enters the microRNA world. Cancer Cell 12, 414–418 [DOI] [PubMed] [Google Scholar]
- 10. Sinha A. U., Kaimal V., Chen J., and Jegga A. G. (2008) Dissecting microregulation of a master regulatory network. BMC Genomics 9, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Riley T., Sontag E., Chen P., and Levine A. (2008) Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 9, 402–412 [DOI] [PubMed] [Google Scholar]
- 12. Vousden K. H., and Prives C. (2009) Blinded by the light: the growing complexity of p53. Cell 137, 413–431 [DOI] [PubMed] [Google Scholar]
- 13. Menendez D., Inga A., and Resnick M. A. (2010) Potentiating the p53 network. Discov. Med. 10, 94–100 [PubMed] [Google Scholar]
- 14. el-Deiry W. S., Kern S. E., Pietenpol J. A., Kinzler K. W., and Vogelstein B. (1992) Definition of a consensus binding site for p53. Nat. Genet. 1, 45–49 [DOI] [PubMed] [Google Scholar]
- 15. Menendez D., Krysiak O., Inga A., Krysiak B., Resnick M. A., and Schönfelder G. (2006) A SNP in the flt-1 promoter integrates the VEGF system into the p53 transcriptional network. Proc. Natl. Acad. Sci. U.S.A. 103, 1406–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Berkers C. R., Maddocks O. D., Cheung E. C., Mor I., and Vousden K. H. (2013) Metabolic regulation by p53 family members. Cell Metab. 18, 617–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang S. J., and Gu W. (2014) To be, or not to be: functional dilemma of p53 metabolic regulation. Curr. Opin. Oncol. 26, 78–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Matoba S., Kang J. G., Patino W. D., Wragg A., Boehm M., Gavrilova O., Hurley P. J., Bunz F., and Hwang P. M. (2006) p53 regulates mitochondrial respiration. Science 312, 1650–1653 [DOI] [PubMed] [Google Scholar]
- 19. Suzuki S., Tanaka T., Poyurovsky M. V., Nagano H., Mayama T., Ohkubo S., Lokshin M., Hosokawa H., Nakayama T., Suzuki Y., Sugano S., Sato E., Nagao T., Yokote K., Tatsuno I., and Prives C. (2010) Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc. Natl. Acad. Sci. U.S.A. 107, 7461–7466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hu W., Zhang C., Wu R., Sun Y., Levine A., and Feng Z. (2010) Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. U.S.A. 107, 7455–7460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang C., Lin M., Wu R., Wang X., Yang B., Levine A. J., Hu W., and Feng Z. (2011) Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc. Natl. Acad. Sci. U.S.A. 108, 16259–16264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bensaad K., Tsuruta A., Selak M. A., Vidal M. N., Nakano K., Bartrons R., Gottlieb E., and Vousden K. H. (2006) TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 126, 107–120 [DOI] [PubMed] [Google Scholar]
- 23. Schwartzenberg-Bar-Yoseph F., Armoni M., and Karnieli E. (2004) The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 64, 2627–2633 [DOI] [PubMed] [Google Scholar]
- 24. Kawauchi K., Araki K., Tobiume K., and Tanaka N. (2008) p53 regulates glucose metabolism through an IKK-NF-κB pathway and inhibits cell transformation. Nat. Cell Biol. 10, 611–618 [DOI] [PubMed] [Google Scholar]
- 25. Heffernan-Stroud L. A., and Obeid L. M. (2011) p53 and regulation of bioactive sphingolipids. Adv Enzyme Regul. 51, 219–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hannun Y. A., and Obeid L. M. (2008) Principles of bioactive lipid signalling: lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 9, 139–150 [DOI] [PubMed] [Google Scholar]
- 27. Maceyka M., and Spiegel S. (2014) Sphingolipid metabolites in inflammatory disease. Nature 510, 58–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hoeferlin L. A., Fekry B., Ogretmen B., Krupenko S. A., and Krupenko N. I. (2013) Folate stress induces apoptosis via p53-dependent de novo ceramide synthesis and up-regulation of ceramide synthase 6. J. Biol. Chem. 288, 12880–12890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Crott J. W., Liu Z., Keyes M. K., Choi S. W., Jang H., Moyer M. P., and Mason J. B. (2008) Moderate folate depletion modulates the expression of selected genes involved in cell cycle, intracellular signaling, and folate uptake in human colonic epithelial cell lines. J. Nutr. Biochem. 19, 328–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Crott J. W., Liu Z., Choi S. W., and Mason J. B. (2007) Folate depletion in human lymphocytes up-regulates p53 expression despite marked induction of strand breaks in exons 5–8 of the gene. Mutat. Res. 626, 171–179 [DOI] [PubMed] [Google Scholar]
- 31. Fekry B., Esmaeilniakooshkghazi A., Krupenko S. A., and Krupenko N. I. (2016) Ceramide synthase 6 is a novel target of methotrexate mediating its antiproliferative effect in a p53-dependent manner. PLoS ONE 11, e0146618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Meyers-Needham M., Ponnusamy S., Gencer S., Jiang W., Thomas R. J., Senkal C. E., and Ogretmen B. (2012) Concerted functions of HDAC1 and microRNA-574-5p repress alternatively spliced ceramide synthase 1 expression in human cancer cells. EMBO Mol. Med. 4, 78–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Oleinik N. V., Krupenko N. I., Priest D. G., and Krupenko S. A. (2005) Cancer cells activate p53 in response to 10-formyltetrahydrofolate dehydrogenase expression. Biochem. J. 391, 503–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Messeguer X., Escudero R., Farré D., Núñez O., Martínez J., and Albà M. M. (2002) PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 18, 333–334 [DOI] [PubMed] [Google Scholar]
- 35. Farré D., Roset R., Huerta M., Adsuara J. E., Roselló L., Albà M. M., and Messeguer X. (2003) Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res. 31, 3651–3653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Krupenko S. A., Horstman D. A., Wagner C., and Cook R. J. (1995) Baculovirus expression and purification of rat 10-formyltetrahydrofolate dehydrogenase. Protein Expr. Purif. 6, 457–464 [DOI] [PubMed] [Google Scholar]
- 37. Choong M. L., Yang H., Lee M. A., and Lane D. P. (2009) Specific activation of the p53 pathway by low dose actinomycin D: a new route to p53 based cyclotherapy. Cell Cycle 8, 2810–2818 [DOI] [PubMed] [Google Scholar]
- 38. Vassilev L. T., Vu B. T., Graves B., Carvajal D., Podlaski F., Filipovic Z., Kong N., Kammlott U., Lukacs C., Klein C., Fotouhi N., and Liu E. A. (2004) In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848 [DOI] [PubMed] [Google Scholar]
- 39. Cremesti A. E., Goni F. M., and Kolesnick R. (2002) Role of sphingomyelinase and ceramide in modulating rafts: do biophysical properties determine biologic outcome? FEBS Lett. 531, 47–53 [DOI] [PubMed] [Google Scholar]
- 40. Colombini M. (2013) Membrane channels formed by ceramide. Handb. Exp. Pharmacol. 215, 109–126 [DOI] [PubMed] [Google Scholar]
- 41. Castro B. M., Prieto M., and Silva L. C. (2014) Ceramide: a simple sphingolipid with unique biophysical properties. Prog. Lipid Res. 54, 53–67 [DOI] [PubMed] [Google Scholar]
- 42. Futerman A. H., and Hannun Y. A. (2004) The complex life of simple sphingolipids. EMBO Rep. 5, 777–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Saddoughi S. A., and Ogretmen B. (2013) Diverse functions of ceramide in cancer cell death and proliferation. Adv. Cancer Res. 117, 37–58 [DOI] [PubMed] [Google Scholar]
- 44. Stiban J., Tidhar R., and Futerman A. H. (2010) Ceramide synthases: roles in cell physiology and signaling. Adv. Exp. Med. Biol 688, 60–71 [DOI] [PubMed] [Google Scholar]
- 45. Park J. W., Park W. J., and Futerman A. H. (2014) Ceramide synthases as potential targets for therapeutic intervention in human diseases. Biochim. Biophys. Acta 1841, 671–681 [DOI] [PubMed] [Google Scholar]
- 46. Mullen T. D., Hannun Y. A., and Obeid L. M. (2012) Ceramide synthases at the centre of sphingolipid metabolism and biology. Biochem. J. 441, 789–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wegner M. S., Wanger R. A., Oertel S., Brachtendorf S., Hartmann D., Schiffmann S., Marschalek R., Schreiber Y., Ferreirós N., Geisslinger G., and Grösch S. (2014) Ceramide synthases CerS4 and CerS5 are upregulated by 17beta-estradiol and GPER1 via AP-1 in human breast cancer cells. Biochem. Pharmacol. 92, 577–589 [DOI] [PubMed] [Google Scholar]
- 48. Panjarian S., Kozhaya L., Arayssi S., Yehia M., Bielawski J., Bielawska A., Usta J., Hannun Y. A., Obeid L. M., and Dbaibo G. S. (2008) De novo N-palmitoylsphingosine synthesis is the major biochemical mechanism of ceramide accumulation following p53 up-regulation. Prostaglandins Other Lipid. Mediat 86, 41–48 [DOI] [PubMed] [Google Scholar]
- 49. Suzuki M., Cao K., Kato S., Komizu Y., Mizutani N., Tanaka K., Arima C., Tai M. C., Yanagisawa K., Togawa N., Shiraishi T., Usami N., Taniguchi T., Fukui T., Yokoi K., et al. (2016) Targeting ceramide synthase 6-dependent metastasis-prone phenotype in lung cancer cells. J. Clin. Invest. 126, 254–265 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 50. Zhang X., Schulz R., Edmunds S., Krüger E., Markert E., Gaedcke J., Cormet-Boyaka E., Ghadimi M., Beissbarth T., Levine A. J., Moll U. M., and Dobbelstein M. (2015) MicroRNA-101 suppresses tumor cell proliferation by acting as an endogenous proteasome inhibitor via targeting the proteasome assembly factor POMP. Mol. Cell 59, 243–257 [DOI] [PubMed] [Google Scholar]
- 51. Wei C. L., Wu Q., Vega V. B., Chiu K. P., Ng P., Zhang T., Shahab A., Yong H. C., Fu Y., Weng Z., Liu J., Zhao X. D., Chew J. L., Lee Y. L., Kuznetsov V. A., et al. (2006) A global map of p53 transcription-factor binding sites in the human genome. Cell 124, 207–219 [DOI] [PubMed] [Google Scholar]
- 52. Oleinik N. V., Krupenko N. I., and Krupenko S. A. (2011) Epigenetic silencing of ALDH1L1, a metabolic regulator of cellular proliferation, in cancers. Genes Cancer 2, 130–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mathupala S. P., Heese C., and Pedersen P. L. (1997) Glucose catabolism in cancer cells: the type II hexokinase promoter contains functionally active response elements for the tumor suppressor p53. J. Biol. Chem. 272, 22776–22780 [DOI] [PubMed] [Google Scholar]
- 54. Sanchez-Macedo N., Feng J., Faubert B., Chang N., Elia A., Rushing E. J., Tsuchihara K., Bungard D., Berger S. L., Jones R. G., Mak T. W., and Zaugg K. (2013) Depletion of the novel p53-target gene carnitine palmitoyltransferase 1C delays tumor growth in the neurofibromatosis type I tumor model. Cell Death Differ. 20, 659–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang S. J., Yu G., Jiang L., Li T., Lin Q., Tang Y., and Gu W. (2013) p53-Dependent regulation of metabolic function through transcriptional activation of pantothenate kinase-1 gene. Cell Cycle 12, 753–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ou Y., Wang S. J., Jiang L., Zheng B., and Gu W. (2015) p53 Protein-mediated regulation of phosphoglycerate dehydrogenase (PHGDH) is crucial for the apoptotic response upon serine starvation. J. Biol. Chem. 290, 457–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jiang L., Kon N., Li T., Wang S. J., Su T., Hibshoosh H., Baer R., and Gu W. (2015) Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hoeferlin L. A., Oleinik N. V., Krupenko N. I., and Krupenko S. A. (2011) Activation of p21-dependent G1/G2 arrest in the absence of DNA damage as an antiapoptotic response to metabolic stress. Genes Cancer 2, 889–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tishler R. B., Calderwood S. K., Coleman C. N., and Price B. D. (1993) Increases in sequence specific DNA binding by p53 following treatment with chemotherapeutic and DNA damaging agents. Cancer Res. 53, 2212–2216 [PubMed] [Google Scholar]