

Abstract
LRP1 (LDL receptor-related protein-1) is a ubiquitous receptor with both cell signaling and ligand endocytosis properties. In the liver, LRP1 serves as a chylomicron remnant receptor and also participates in the transport of extracellular cathepsin D to the lysosome for prosaposin activation. The current study showed that in comparison with wild type mice, hepatocyte-specific LRP1 knock-out (hLrp1−/−) mice were more susceptible to fasting-induced lipid accumulation in the liver. Primary hepatocytes isolated from hLrp1−/− mice also accumulated more intracellular lipids and experienced higher levels of endoplasmic reticulum (ER) stress after palmitate treatment compared with similarly treated hLrp1+/+ hepatocytes. Palmitate-treated hLrp1−/− hepatocytes displayed similar LC3-II levels, but the levels of p62 were elevated in comparison with palmitate-treated hLrp1+/+ hepatocytes, suggesting that the elevated lipid accumulation in LRP1-defective hepatocytes was not due to defects in autophagosome formation but was due to impairment of lipophagic lipid hydrolysis in the lysosome. Additional studies showed increased palmitate-induced oxidative stress, mitochondrial and lysosomal permeability, and cell death in hLrp1−/− hepatocytes. Importantly, the elevated cell death and ER stress observed in hLrp1−/− hepatocytes were abrogated by E64D treatment, whereas inhibiting ER stress diminished cell death but not lysosomal permeabilization. Taken together, these results documented that LRP1 deficiency in hepatocytes promotes lipid accumulation and lipotoxicity through lysosomal-mitochondrial permeabilization and ER stress that ultimately result in cell death. Hence, LRP1 dysfunction may be a major risk factor in fatty liver disease progression.
Keywords: hepatocyte, lipoprotein receptor, lipoprotein receptor-related protein (LPR), lipotoxicity, lysosome, hepatosteatosis
Introduction
LRP1 (LDL receptor-related protein-1) is a ubiquitously expressed 600-kDa receptor that was originally identified as an endocytic receptor responsible for cellular uptake and plasma clearance of macromolecules such as apolipoprotein E-containing lipoproteins and protease-protease inhibitor complexes. Subsequent studies also established LRP1 as an integrator of signal transduction events in cell regulation (1). Hence, normal and abnormal functions of this receptor impact the development and progression of a wide spectrum of metabolic diseases, spanning from cardiovascular disease to obesity/diabetes, neurodegenerative disorders, and tumor invasion and metastasis.
The mechanism underlying LRP1 modulation of disease risk varies in a tissue-specific manner (2). For example, LRP1 is a signaling receptor in smooth muscle cells, and its defect causes aortic aneurysm and accelerates vascular occlusive diseases through activation of the PDGF receptor-β signaling cascade (3, 4). Additional studies revealed that LRP1 also regulates PI3K and TGF-β signaling in smooth muscle cells, with broad implications for its role in Marfan syndrome manifestation (5). In macrophages, LRP1 is also a signaling receptor in modulating inflammatory response and cell migration (6). Defective expression of LRP1 in macrophages impairs Akt phosphorylation, leading to robust inflammation, defective efferocytosis, and accelerated atherosclerosis (7–9). In adipocytes, LRP1 participates in pre-adipocyte differentiation to mature adipocytes via modulation of PPARγ expression and canonical Wnt-5a signaling, both of which control regulation of the adipogenic program (10, 11). However, defective LRP1 in mature adipocytes reduces adiposity due to impaired triglyceride-rich lipoprotein uptake (12). Thus, LRP1 has dual signal transduction and cargo endocytosis functions in adipocyte biology. In hepatocytes, in addition to chylomicron remnants and protease complex clearance, LRP1 is also required to complement the mannose 6-phosphate receptor for appropriate lysosomal enzyme trafficking, and its defect impairs prosaposin activation, leading to reduced ABCA1 cell surface translocation and lower HDL secretion (13).
The lysosome is an important intracellular organelle responsible for processing and degradation of macromolecules derived from secretory, endocytic, and autophagic pathways (14). In particular, the lysosome regulates intracellular lipid mobilization via the autophagic-lysosomal machinery known as lipophagy (15, 16). Hence, the impairment of lysosomal function may lead to excessive lipid accumulation (steatosis) and cell death (17). Accordingly, the participation of LRP1 in lysosomal enzyme trafficking and prosaposin activation in hepatocytes suggests an important role of LRP1 in modulating hepatosteatosis and lipoapoptosis. This study was undertaken to test this hypothesis.
Experimental Procedures
Animals and Tissue Analysis
Liver-specific LRP1-null (hLrp1−/−) mice were generated by cross-breeding Lrp1flox/flox mice (in C57BL/6J background) with albumin promoter-driven cre recombinase transgenic mice in C57BL/6 background (Jackson Laboratory, Bar Harbor, ME) as described previously (13). The animals were maintained under controlled environmental conditions with free access to food and water. All experimental procedures were performed with protocols approved by the University of Cincinnati Institutional Animal Use and Care Committee. The animals were euthanized during the fed state (fed ad libitum) or after a 24-h fast. Livers were removed after perfusion with ice-cold phosphate-buffered saline through the hepatic portal vein. The livers were fixed by immersion in neutral buffered formalin (10%), dehydrated in ethanol, and then transferred to xylene solution for embedding in paraffin. Five-micron sections were stained with hematoxylin-eosin, and images were analyzed by microscopy. Lipid accumulation was identified microscopically as lipid vacuoles after hematoxylin-eosin staining (18). The livers were also homogenized for lipid extraction with chloroform/methanol (2:1), and amounts of triglycerides in each sample were determined by colorimetric assay kit (Life Technologies, Inc.) as described (13).
Primary Hepatocyte Isolation and Culture
Wild type and hLrp1−/− mice were anesthetized at 20 weeks of age by isoflurane inhalation. Primary hepatocytes were collected by perfusion with Krebs-Henseleit buffer containing 0.5 mm EGTA and then infused with a digestion solution of Krebs-Henseleit buffer containing 125 units/ml collagenase (Sigma) and 2% BSA at a rate of 6 ml/min/g of liver. The hepatocytes were washed in William's E media (Life Technologies) with penicillin, streptomycin, and l-glutamine followed by a final wash in Percoll solution (GE Healthcare) diluted in PBS. The cells were allowed to incubate in William's E media overnight before treatment. For experiments, the William's E media were removed, and cells were rinsed with PBS. The hepatocytes were incubated for 6 h in HepatoZYME (Life Technologies) containing penicillin and streptomycin with fatty acid-free albumin or palmitate-albumin complex. In selected experiments, the cell-permeable cysteine protease inhibitor E64D (Sigma), tauroursodeoxycholate (Sigma), or thapsigargin (Sigma) dissolved in DMSO was also included during the incubation period. A similar volume of DMSO without inhibitors served as the control for the latter experiments.
Hepatocyte Lipid Accumulation in Vitro
The hLrp1+/+ and hLrp1−/− hepatocytes were plated in 12-well dishes and incubated for 6 h with VLDL or palmitate-BSA complexes, using fatty acid-free BSA as a control. The cells were washed in PBS, fixed in 4% formalin solution, and then permeabilized with 60% isopropyl alcohol before staining with Oil Red O and counterstained with Gill's hematoxylin (Sigma). Microscopic images were obtained on an Olympus IX71 microscope and analyzed using ImageJ software. Intracellular lipids were extracted with isopropyl alcohol, and Oil Red O staining was quantified by absorbance at 510 nm and normalized to protein content determined by a bicinchoninic assay (Thermo Fisher Scientific).
Oxidant Stress and Mitochondrial Permeability Determinations
Primary hepatocytes were incubated with 10 μm 2′,7′-dichlorofluorescin diacetate (Invitrogen) for 1 h, washed with DAPI diluted in PBS, and then fixed for visualization by fluorescent microscopy on an Olympus IX71 microscope with excitation at 495 nm and emission at 529 nm. Changes in mitochondrial membrane potential were determined by incubating cells with 10 mm tetramethylrhodamine methyl ester (Life Technologies) in DMSO for 30 min at 37 °C. For cytochrome c leakage determinations, the hepatocytes were fixed in ice-cold methanol and then permeabilized using 0.1% Triton X-100 in PBS. Cells were then blocked in 10% BSA in PBS, incubated with antibody against cytochrome c (Cell Signaling) overnight at 4 °C, followed by a 1-h incubation with anti-rabbit fluorescently linked secondary antibody diluted in 10% BSA in PBS. For the latter two experiments, cells were also counterstained with DAPI before visualization with excitation at 495 nm and emission at 525 nm, followed by excitation at 358 nm and emission at 461 nm. Images of 200 cells were obtained at ×200 magnification for each experiment, and the data were analyzed using ImageJ software.
Cell Viability, Nuclear Fragmentation, Lysosomal Permeability, and Autophagy Assays
Viability of hLrp1+/+ and hLrp1−/− hepatocytes after incubation with or without fatty acids was determined by dye exclusion after staining for 15 min with 100 μg/ml ethidium bromide (Sigma) and 100 μg/ml acridine orange (Sigma) in PBS. Cell lysates were also prepared after incubation and used to measure caspase-3 activation using the Caspase-3 Activation Kit from Life Technologies (catalog no. 13184). The number of cells with nuclear fragmentation and permeabilized lysosomes was determined after incubation with 4.64 μm acridine orange in sterile Earle's balanced salt solution for 15 min as described (19). Images were obtained at ×400 magnification with excitation at 495 nm and emission at 519 nm and then excitation at 585 nm and emission at 570 nm as described (20). Hepatocyte apoptosis was assessed by TUNEL staining using the protocol supplied by the manufacturer (Roche Diagnostics). Before mounting on slides, the hepatocytes were washed with DAPI diluted in PBS. Images were obtained at ×200 magnification with DAPI staining visualized by excitation at 330/390 nm and emission at 450/600 nm. TUNEL staining was visualized by excitation at 495 nm and emission at 519 nm. All microscopic images were obtained on an Olympus IX71 microscope and analyzed using ImageJ software. Confocal microscopic images were obtained on a Zeiss Laser Scanning Microscope 710 after immunostaining of formalin-fixed and 1% Triton-permeabilized cells with antibodies against LC3 (catalog no. 2775, Cell Signaling) or p62 (catalog no. 610832, BD Biosciences) and detected with fluorescent secondary antibodies (catalog no. Z25002, Thermo Fisher Scientific) with DAPI counterstain.
Western Blotting Analysis
Primary hepatocytes were washed extensively with PBS after incubation before lysis in ice-cold radioimmune precipitation assay buffer (Thermo Fisher Scientific) containing protease and phosphatase inhibitor mixture (Roche Diagnostics). Proteins in the cell lysate were resolved by SDS-polyacrylamide gel electrophoresis and then transferred to polyvinylidene difluoride membranes (Bio-Rad). The membranes were blocked in Odyssey blocking buffer (LI-COR) with 0.1% Tween 20 for 1 h at 4 °C and then incubated for 90 min with a 1:1000 dilution of primary antibodies. Primary antibodies used in this study include α-tubulin antibody (catalog no. MS-581-P1, Thermo Fisher Scientific), p62 antibody (catalog no. 5114, Cell Signaling), LC3 antibody (catalog no. 2775, Cell Signaling), ATF6 (activating transcription factor 6; catalog no. ab11909, Abcam), CHOP2 (catalog no. 2895, Cell Signaling), phospho-IREα (catalog no. PA1-16927, Thermo Fisher Scientific), IREα (catalog no. ab48187, Abcam), and phosphorylated and total PERK (catalog nos. 3179 and 3192, Cell Signaling). The membranes were washed and then incubated with fluorescent secondary antibodies for 45 min and then examined using an Odyssey LI-COR scanner. Densitometry was completed using ImageJ software.
Statistical Analysis
Statistical analysis was performed with Sigma Plot version 12. Values were expressed as mean ± S.D. Multiple comparisons were first tested by Student's t test or analysis of variance. When analysis of variance demonstrated significant differences, individual mean differences were analyzed with the Student-Newman-Keuls test. A value of p < 0.05 was considered to be statistically significant.
Results
Hepatic LRP1 Deficiency Exacerbates Fasting-induced Lipid Accumulation in the Liver
The role of LRP1 in modulating intracellular lipid metabolism in the liver was explored initially by comparing intracellular lipid accumulation in the liver of wild type (hLrp1+/+) and hepatocyte-specific Lrp1 knock-out (hLrp1−/−) mice. Liver morphology and histology were similar between hLrp1+/+ and hLrp1−/− mice, and minimal lipid accumulation was observed in the livers of these animals when fed ad libitum (data not shown). However, when the animals were fasted for 24 h, significant amounts of vacuoles indicative of lipid accumulation were detected in liver sections prepared from both hLrp1+/+ and hLrp1−/− mice (Fig. 1A). Quantification of lipids extracted from the livers of these animals revealed a ∼3-fold increase in triglyceride content in the livers of hLrp1−/− mice compared with that observed in hLrp1+/+ mice (Fig. 1B).
FIGURE 1.
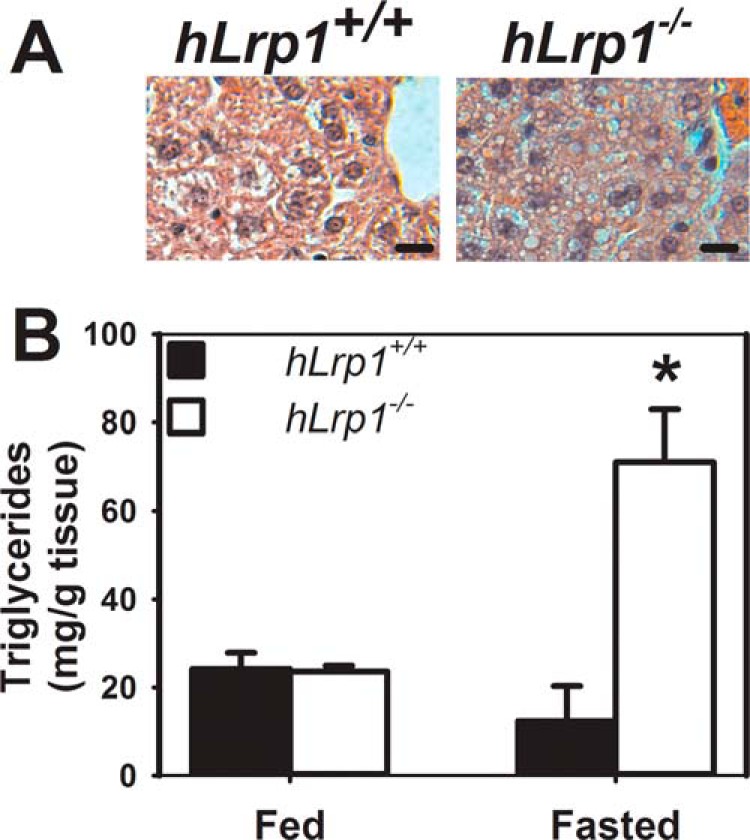
Triglyceride levels in hLrp1+/+ and hLrp1−/− mice. A, representative microscopic images of hematoxylin-eosin stained liver sections from 20-week-old chow-fed hLrp1+/+ and hLrp1−/− mice after a 24-h fast. B, livers were obtained from fasted hLrp1+/+ (filled bars) and hLrp1−/− (open bars) mice fed ad libitum or after a 24-h fast. Lipids were extracted from the tissues for triglyceride determinations. The data represent mean ± S.E. (error bars) from two experiments each, each performed with 3 mice/group. *, significant difference from hLrp1+/+ mice at p < 0.05.
Fatty Acid-dependent Lipid Accumulation in Hepatocytes
Triglyceride accumulation observed in hLrp1−/− mice with defective expression of a triglyceride-rich lipoprotein receptor in the liver was a surprised finding. Indeed, when primary hepatocytes isolated from hLrp1+/+ and hLrp1−/− mice were incubated with or without VLDL, significantly less lipid accumulation was observed in hepatocytes without LRP1 expression (Fig. 2A). We recognized that nonesterified fatty acids released from adipose tissues may also be transported to the liver, especially under fasting conditions, to serve as nutrients and substrates for lipoprotein biosynthesis (21, 22). Thus, differences in hepatic lipid accumulation between hLrp1+/+ and hLrp1−/− mice under fasting conditions suggest that LRP1 deficiency may influence fatty acid processing in hepatocytes. To test this hypothesis, primary hepatocytes were isolated from fasting hLrp1+/+ and hLrp1−/− mice and then incubated with increasing concentrations of palmitate. Lipid accumulation was assessed by Oil Red O staining of the hepatocytes in culture and triglyceride quantification after lipid extraction. Consistent with results reported previously by others (23), palmitate incubation effectively induced lipid accumulation in hepatocytes. Interestingly, significantly more lipids were accumulated in palmitate-treated hLrp1−/− hepatocytes compared with hLrp1+/+ hepatocytes (Fig. 2B).
FIGURE 2.

VLDL and fatty acid-induced lipid accumulation in primary hepatocytes. Primary hepatocytes isolated from chow-fed hLrp1+/+ (filled bars) and hLrp1−/− (open bars) mice were incubated with VLDL (A) or increasing concentrations of palmitate (B) for 6 h. Representative images of hepatocytes after incubation and staining with Oil Red O are shown in the top panels. Scale bar, 100 μm. Lipid accumulation in hepatocytes was quantified by triglyceride assay of cell extracts. The data represent mean ± S.E. (error bars) from three experiments, each performed with four hepatocyte preparations in each group. * and **, differences from the hLrp1+/+ group at p < 0.05 and p < 0.001, respectively.
Impaired Lipophagy with Defective LRP1 Expression in Hepatocytes
The next set of experiments was undertaken to explore the mechanism underlying the increased lipid accumulation observed in palmitate-treated hLrp1−/− hepatocytes. Consideration was given to previous reports that autophagy-mediated lipid transport to lysosomes is required for intracellular lipid droplet breakdown (24) and our observation that defective LRP1 expression in hepatocytes reduces intracellular cathepsin D content and prosaposin activation (13), both of which have been linked to impairment of autophagic degradation (25, 26). Thus, we explored the possibility that the elevated lipid accumulation observed in hLrp1−/− hepatocytes may be due to defective lipophagic degradation of lipid droplets stored intracellularly in response to palmitate treatment. Western blotting analysis of lysates prepared from control- and palmitate-treated hLrp1+/+ and hLrp1−/− hepatocytes showed a slight albeit not significant increase of LC3-II levels (Fig. 3A). In contrast, significant elevation of p62 accumulation was noted in palmitate-treated hLrp1−/− hepatocytes compared with that observed in hLrp1+/+ hepatocytes (Fig. 3B). Analysis of confocal microscopy images revealed the formation of LC3 punctae in both hLrp1+/+ and hLrp1−/− hepatocytes, with a significantly higher number of p62-positive punctae in LRP1-deficient hepatocytes (Fig. 3, C and D). These results indicate that LRP1 deficiency has no direct influence on autophagosome formation, but defective lysosomal degradation of lipid droplets in autophagolysosomes is responsible for the elevated lipid accumulation in hLrp1−/− hepatocytes.
FIGURE 3.
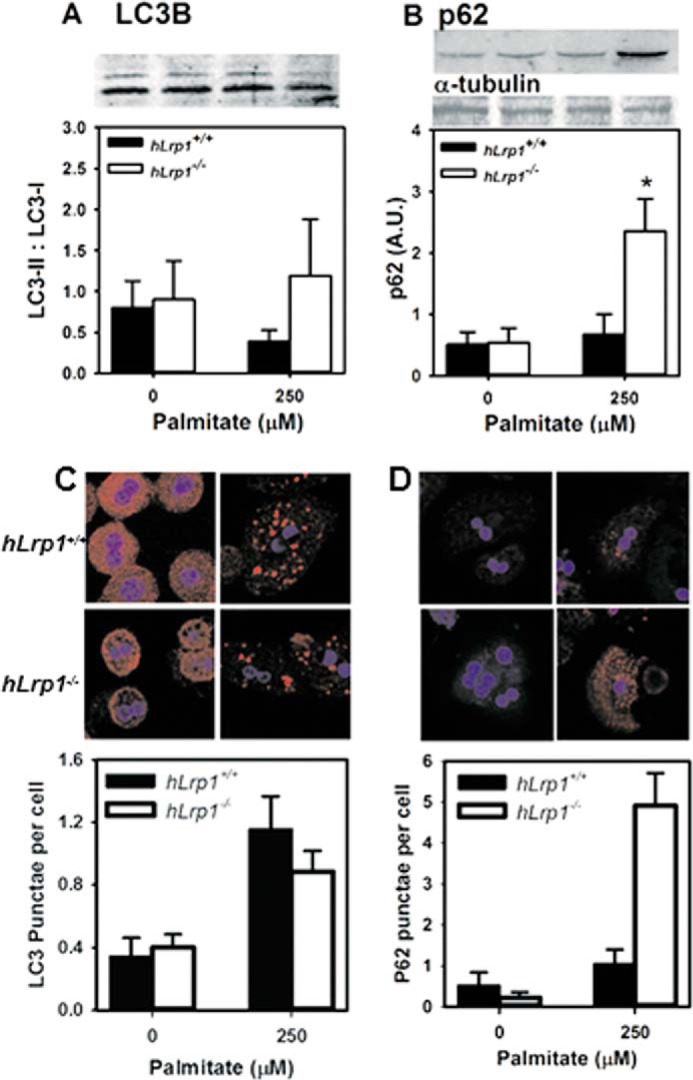
Hepatic LRP1 deficiency impairs lipophagy. Cell lysates prepared from hLrp1+/+ (filled bars) and hLrp1−/− (open bars) hepatocytes after incubation with or without 250 μm palmitate were subjected to Western blotting with antibodies against LC3B (A) to detect LC3-I and LC3-II and p62 (B). Expression levels were determined by densitometry and normalized to the α-tubulin levels as arbitrary units (A.U.). Representative images of the blots are shown at the top of A and B. The hepatocytes after incubation were also examined by confocal microscopy, and representative images with quantification of LC3 and p62 punctae are shown in C and D. The data are presented as mean ± S.E. (error bars) of three independent experiments, each performed in duplicate. *, significant difference from the hLrp1+/+ group at p < 0.01.
Hepatic LRP1 Deficiency Exacerbates Palmitate-induced Cell Death
The overaccumulation of intracellular lipids due to defective lipophagic degradation of lipid droplets may also lead to ER and oxidative stress as well as mitochondrial dysfunction that ultimately results in cell death (27, 28). Indeed, a significantly higher level of CHOP expression was observed in palmitate-treated hLrp1−/− hepatocytes compared with hLrp1+/+ hepatocytes (Fig. 4A). The elevated expression of this apoptotic cell marker was concomitant with induction of ER stress through specific activation of ATF6 (Fig. 4B). Interestingly, LRP1 inactivation has no effect on the IRE1α (inositol-requiring 1α) and PERK arms of the ER stress pathway (Fig. 4, C and D). Consistent with elevated ER stress observed in LRP1-deficient hepatocytes after palmitate treatment, the determination of cellular reactive oxygen species with the cell-permeable fluorescent dye 2′,7′-dichlorofluorescin diacetate also revealed a significantly higher number of hLrp1−/− hepatocytes displaying oxidative stress after incubation with palmitate in comparison with similarly treated hLrp1+/+ hepatocytes (Fig. 5A). We also observed an increased number of hLrp1−/− hepatocytes exhibiting mitochondrial depolarization, as indicated by the reduced tetramethylrhodamine methyl ester orange/red fluorescence (29), compared with hLrp1+/+ hepatocytes after incubation with 250 μm palmitate (Fig. 5B). Finally, significantly more hLrp1−/− hepatocytes also displayed a disperse pattern of cytochrome c immunofluorescence indicative of increased mitochondrial membrane permeability compared with hLrp1+/+ hepatocytes after incubation with palmitate (Fig. 5C).
FIGURE 4.

Hepatic LRP1 deficiency activates ATF6 cleavage. Cell lysates prepared from hLrp1+/+ (filled bars) and hLrp1−/− (open bars) hepatocytes after incubation with or without 250 μm palmitate were subjected to Western blotting with antibodies against CHOP (A), cleaved ATF6 (B), phosphorylated and total IRE1α (C), and phosphorylated and total PERK (D). Anti-GADPH blots for GADPH levels were used as loading controls. Representative images of the blots are shown at the top of C and D, and the data are presented as mean ± S.E. (error bars) of three independent experiments, each performed with three biological replicates in each group. *, significant difference from the hLrp1+/+ group at p < 0.01.
FIGURE 5.

Palmitate-induced oxidative stress and mitochondrial dysfunction in hLrp1+/+ and hLrp1−/− hepatocytes. Primary hepatocytes obtained from hLrp1+/+ and hLrp1−/− mice were plated on coverslips and treated with or without palmitate for 6 h before incubation with 10 μm 2′,7′-dichlorofluorescin diacetate (DCFDA) to visualize reactive oxygen species (A), tetramethylrhodamine methyl ester (TMRM) to visualize mitochondrial membrane potential changes (B), and anti-cytochrome c (Cyt C) to visualize cytochrome c release from the mitochondria (C). The top panels show representative images corresponding to the results shown in the bottom panels. The data represent mean ± S.E. obtained by counting 200 cells in three separate experiments with duplicate cultures. *, significant difference from the hLrp1+/+ group at p < 0.01.
The increase of CHOP expression and mitochondrial membrane permeability observed in hLrp1−/− hepatocytes after palmitate treatment is consistent with cell death. This interpretation was supported by additional experiments that showed that cell viability, as determined by ethidium bromide and acridine orange staining, was dramatically lower in hLrp1−/− hepatocytes compared with hLrp1+/+ hepatocytes after palmitate incubation (Fig. 6A). A higher number of hLrp1−/− hepatocytes compared with hLrp1+/+ hepatocytes also exhibited nuclear fragmentation and apoptosis characteristics after palmitate treatment (Fig. 6, B and C). The increased sensitivity of hLrp1−/− hepatocytes to palmitate-induced apoptosis was further confirmed by elevated caspase-3 activation (Fig. 6D). Taken together, these results indicate that LRP1 deficiency in hepatocytes increased their sensitivity to palmitate-induced cell death, through mechanisms related to ER and oxidative stress and mitochondrial dysfunction.
FIGURE 6.

Palmitate-induced death of hLrp1+/+ and hLrp1−/− hepatocytes. Primary hepatocytes obtained from hLrp1+/+ and hLrp1−/− mice plated on coverslips were incubated with or without palmitate for 6 h. Cell viability was determined by dye exclusion after staining with ethidium bromide and acridine orange (A); nuclear fragmentation was examined visually after incubation with low concentration of acridine orange (B); and cells undergoing apoptosis were determined by staining with TUNEL reagents and a caspase-3 activation assay (C and D). The data in each panel represent mean ± S.E. (error bars) of results obtained by counting 200 cells in three separate experiments with duplicate cultures. * and **, significant difference from the hLrp1+/+ group at p < 0.05 and p < 0.01, respectively.
Hepatic LRP1 Deficiency Increases Sensitivity to Palmitate-induced Lysosomal Permeabilization
One mechanism that links saturated fatty acid-induced mitochondrial dysfunction and cellular stress with hepatotoxicity is lysosomal disruption (30). Therefore, additional experiments were performed to compare lysosomal permeability between hLrp1+/+ and hLrp1−/− hepatocytes. Acridine orange staining of hLrp1+/+ and hLrp1−/− hepatocytes incubated in the absence of palmitate revealed a distinct punctate staining pattern indicative of intact acidic lysosomal environments in both cell types. The acridine orange staining became more diffuse with increasing concentrations of palmitate in the incubation media, indicative of lysosomal permeabilization. Importantly, when incubated with equal concentrations of palmitate, more hLrp1−/− hepatocytes displayed lysosomal permeabilization compared with hLrp1+/+ hepatocytes (Fig. 7). Thus, these data revealed the importance of LRP1 expression to preserve lysosomal integrity and protect against palmitate-induced lysosomal leakage.
FIGURE 7.
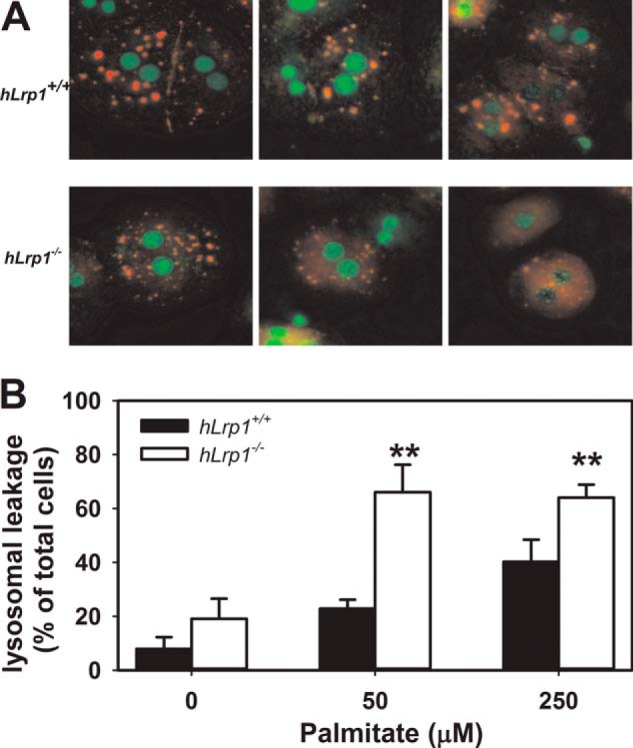
Palmitate-induced lysosomal permeability in hLrp1+/+ and hLrp1−/− hepatocytes. Primary hepatocytes obtained from hLrp1+/+ and hLrp1−/− mice plated on coverslips were treated with or without palmitate for 6 h and then stained with 4.64 μm acridine orange for 15 min. A, representative images with nuclei identified by green staining, intact lysosomes identified by punctate orange staining, and lysosomal leakage demonstrated with a diffuse orange staining pattern. B, mean ± S.E. (error bars) of data obtained by counting 200 cells in three separate experiments with duplicate cultures. **, significant difference from the hLrp1+/+ group at p < 0.01.
Cysteine Protease Inhibitor Attenuates Lipotoxicity, Steatosis, and ER Stress in Palmitate-treated hLrp1−/− Hepatocytes
The relationship between elevated ER stress and lysosomal permeabilization in increased hLrp1−/− hepatocyte death was explored by comparing the effectiveness of the ER stress inhibitor tauroursodeoxycholate and the cysteine protease inhibitor E64D to attenuate palmitate-induced hepatotoxicity and steatosis. Results showed that E64D treatment reduced ATF6 cleavage and reduced lipid accumulation concomitant with increased viability of palmitate-treated hLrp1−/− hepatocytes to levels observed with hLrp1+/+ hepatocytes (Fig. 8). Interestingly, reducing ER stress with tauroursodeoxycholate treatment improved the viability of palmitate-treated hLrp1−/− hepatocytes to levels observed in hLrp1+/+ hepatocytes (Fig. 9, A and B) but failed to correct the lysosomal permeability observed in palmitate-treated hLrp1−/− hepatocytes (Fig. 9C). Moreover, in addition to exacerbating palmitate-induced ER stress and cell death, LRP1 deficiency also increased cell death and apoptosis induced by thapsigargin (Fig. 10). Taken together, these data indicate that the elevated sensitivity of hLrp1−/− hepatocytes to palmitate-induced cell death is caused by increased lysosomal dysfunction and membrane permeability as well as accelerated ER stress.
FIGURE 8.

E64D treatment improves hLrp1−/− hepatocyte ER stress, steatosis, and viability. Primary hepatocytes isolated from hLrp1+/+ and hLrp1−/− mice were treated with or without 10 μm E64D during the 6-h incubation with palmitate. Cell lysates were prepared to assess ATF6 cleavage (A), with representative images of the ATF6 cleavage products (top band) and tubulin control (bottom band) shown in the inset, or lipid accumulation (B). In separate experiments, cells were plated on coverslips and then stained with TUNEL reagents to assess apoptosis (C) or treated with ethidium bromide and acridine orange and analyzed for viable cells by dye exclusion (D). The graphs show the mean ± S.E. (error bars) of data obtained by counting 200 cells in three separate experiments with duplicate cultures. * and **, significant differences from the hLrp1+/+ group at p < 0.05 and p < 0.01, respectively.
FIGURE 9.

Tauroursodeoxycholate treatment improves viability but not lysosomal permeability of palmitate-treated hLrp1−/− hepatocytes. Primary hepatocytes isolated from hLrp1+/+ and hLrp1−/− mice were plated on coverslips and then treated with or without tauroursodeoxycholate (TUDCA) during the 6-h incubation with palmitate. The percentage of viable cells (A), apoptotic cells (B), and cells displaying lysosomal membrane permeability (C) was determined as described in the legends to Figs. 6 and 7. The graphs show the mean ± S.E. (error bars) of data obtained by counting 200 cells in three separate experiments with two biological replicate cultures. *, significant difference from the hLrp1+/+ group at p < 0.05.
FIGURE 10.

Effects of palmitate and thapsigargin on cell viability and lysosomal membrane permeability. Primary hepatocytes isolated from hLrp1+/+ and hLrp1−/− mice were treated with or without 250 μm palmitate for 6 h or with 400 nm thapsigargin for 16 h. The percentages of viable cells (A) and apoptotic cells (B) were determined as described in legends to Figs. 6 and 7. The graphs show the mean ± S.E. (error bars) of data obtained by counting 200 cells in two separate experiments with three biological replicate cultures. *, significant difference from hLrp1+/+ group at p < 0.05.
The heightened sensitivity of LRP1-deficient hepatocytes to palmitate-induced lysosomal dysfunction, lipophagic degradation, and ER stress observed in vitro suggested that the lipids accumulated in the liver of fasted hLrp1−/− mice may be due to similar defects in vivo. Indeed, Western blotting analysis of cell extracts prepared from livers of hLrp1+/+ and hLrp1−/− mice after a 24-h fast showed higher levels of CHOP, ATF6, and p62 in livers of hLrp1−/− mice (Fig. 11). Similar to the results observed with primary hepatocytes in vitro, no difference in IRE1 and PERK phosphorylation or LC3-I/LC3-II ratio were detected in livers of fasted hLrp1+/+ and hLrp1−/− mice (Fig. 11). In contrast to the in vitro experiments, we did not observe any signs of hepatocyte cell death after a 24-h fasting period. Whether prolonged starvation would lead to hepatocyte cell death in LRP1-deficient mice cannot be assessed because of animal care regulatory issues.
FIGURE 11.

Hepatic LRP1 deficiency impairs lipophagy in fasting mice. Liver extracts prepared from hLrp1+/+ (filled bars) and hLrp1−/− (open bars) mice after a 24-h fast were subjected to Western blotting analysis of ER stress and autophagic markers as indicated. The inset shows representative images of the blots with α-tubulin (bottom bands in CHOP, ATF6, and p62 blots), phosphorylated and total IRE1, and phosphorylated and total PERK levels. The data are presented as mean ± S.E. (error bars) of two independent experiments, each performed with four mice in each group. *, significant difference from the hLrp1+/+ group at p < 0.05.
Discussion
The increasing prevalence of obesity and chronic consumption of high fat diets in Western countries has resulted in 17–30% of the population suffering from the pathological condition of nonalcoholic fatty liver disease (31, 32). Although the condition is generally asymptomatic and considered benign, ∼10–20% of nonalcoholic fatty liver disease cases progress to nonalcoholic steatohepatitis, a chronic liver disease characterized by fatty liver degeneration, fibrosis, and inflammation. Nonalcoholic steatohepatitis can also progress to cirrhosis, a state of advanced liver dysfunction that requires immediate medical intervention. Approximately 70% of the cirrhosis cases also proceed to hepatocellular carcinoma, the fifth most common cancer worldwide (31, 32). However, despite being a major health issue, identifying nonalcoholic fatty liver disease patients that are prone to nonalcoholic steatohepatitis, cirrhosis, and hepatocellular carcinoma proves to be difficult because the etiology and mechanism underlying this disease progression remain unclear. Whereas steatosis is a prerequisite, a second hit involving environmental and/or genetic factors appears to be necessary for nonalcoholic fatty liver disease progression to nonalcoholic steatohepatitis and liver cirrhosis (33). A recent study demonstrating the association between low LRP1 expression levels with poor hepatocellular carcinoma prognosis suggested that LRP1 dysfunction may promote liver disease progression (34).
The current study provided mechanistic insights to support the hypothesis that the LPR1 defect promotes liver diseases. Our results documented that LRP1 deficiency exacerbates palmitate-induced hepatosteatosis and cell death. The elevated steatosis observed in the livers of fasted hLrp1−/− mice and palmitate-induced triglyceride accumulation in LRP1-deficient hepatocytes is counterintuitive, considering the well documented role of LRP1 as a chylomicron receptor responsible for the clearance of triglyceride-rich lipoproteins (35). However, it should be noted that LRP1 deficiency in the liver does not impair chylomicron remnant clearance because the LDL receptor also binds to apolipoprotein E-containing lipoproteins and can be utilized for chylomicron remnant internalization into hepatocytes. Thus, defective expression of both LRP1 and LDL receptor in the liver is necessary to exhibit defects in chylomicron remnant catabolism (35). Moreover, and in contrast to fed and postprandial conditions in which triglyceride-rich lipoproteins are the major source of fatty acid delivered to the liver, the main source of lipid nutrients transported to the liver under fasting conditions is nonesterified fatty acids originating from extrahepatic lipid storage tissues, such as the adipocytes (21, 22). The increase in starvation-induced lipid accumulation observed in hLrp1−/− mice indicates that LRP1 does not participate in nonesterified fatty acid uptake by the liver but plays an important role in intracellular lipid processing in hepatocytes after fatty acid internalization.
Fatty acid metabolism in the liver is a dynamic process in which the fatty acids may undergo β-oxidation in the mitochondria or may be used as substrates for membrane and lipoprotein biosynthesis. Excess fatty acids are re-esterified to triglycerides and stored as lipid droplets in the cytosol. Lipid droplet turnover and mobilization are accomplished through the action of cytosolic triglyceride lipases (36) or by lipophagic degradation (16). The latter process requires the sequestration of lipid droplets into autophagosomes, which then fuse with lysosomes to form autophagolysosomes, where the triglycerides can be hydrolyzed by acid lipases in the lysosomes (16). Impairment of the autophagic process has been shown to increase triglyceride storage in lipid droplets (15, 24, 37). In the current study, we found that LRP1 deficiency significantly increases lipid accumulation in response to palmitate incubation. Results showing no difference in LC3-II levels between palmitate-treated hLrp1+/+ and hLrp1−/− hepatocytes indicated that LRP1 deficiency does not influence the autophagosome formation in hepatocytes. However, the higher p62 levels observed in hLrp1−/− hepatocytes suggest that impairment in the final step of autophagy (i.e. degradation of the lipid droplets in autophagolysosomes) is responsible for the elevated lipid accumulation in LRP1-defective hepatocytes. These results are in contrast to a previous report showing that LRP1 is required for LC3-II generation in toxin-induced autophagy of gastric epithelial cells (38). The difference between these studies may be due to cell type-specific differences, which further highlights differences in LRP1 functions among various tissues. Regardless, defective lipophagic degradation in the lysosomes of hLrp1−/− hepatocytes is consistent with our previous report showing that LRP1 deficiency impairs prosaposin activation and reduces the level of the aspartyl protease cathepsin D in hepatic cells (13). Deficiencies in prosaposin activation and reduced cathepsin D levels have been shown previously to alter autophagy, leading to increased intracellular lipid accumulation in a rare variant form of Gaucher disease (26, 39).
In addition to elevated palmitate-induced lipid accumulation, the current study showed that LRP1 deficiency also increases hepatocyte sensitivity to palmitate-induced cell death. It is important to note that no difference in cell viability was observed between hLrp1+/+ and hLrp1−/− hepatocytes under normal culturing conditions. Thus, lipophagy impairment due to LRP1 deficiency may account for the increased sensitivity of hLrp1−/− hepatocytes to palmitate-induced apoptosis. Previous studies have shown that lipophagy protects hepatocytes against oxidant stress and lipotoxicity (40, 41). Thus, our observations that the elevated reactive oxygen species production and reduced viability of palmitate-treated hLrp1−/− hepatocytes, which can be improved by the cysteine protease inhibitor E64D, provided additional support for the interpretation that LRP1 deficiency in hepatocytes impairs lipophagy to promote palmitate-induced lipid accumulation, lysosomal permeabilization, mitochondrial dysfunction, ER stress, and apoptosis. The observation that ER stress inhibition alleviates palmitate-induced cell death but not lysosomal permeabilization in hLrp1−/− hepatocytes indicates that LRP1 deficiency promotes lysosomal dysfunction that leads to mitochondrial depolarization, cytochrome c release, reactive oxygen species generation, and subsequently elevated ER stress with the consequence of accelerated cell death. Taken together, the results of this study indicate that LRP1 deficiency exacerbates diet-induced liver diseases by promoting lipid accumulation and lipotoxicity through the lysosomal-mitochondrial axis (30).
Author Contributions
D. Y. H. conceived and coordinated the study. D. Y. H. and A. N. H. wrote the paper. A. N. H. and J. E. B. designed, performed, and analyzed data for all experiments in this paper. A. J. designed, analyzed, and interpreted the autophagy and ER stress data for the experiments. All authors reviewed the results and approved the final version of the manuscript.
This work was supported by National Institutes of Health Grant RO1 DK074932 (to D. Y. H.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- CHOP
- CCAAT enhancer-binding protein homologous protein
- ER
- endoplasmic reticulum
- PERK
- protein kinase R-like endoplasmic reticulum kinase.
References
- 1. Herz J., and Strickland D. K. (2001) LRP: a multifunctional scavenger and signaling receptor. J. Clin. Invest. 108, 779–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lillis A. P., Van Duyn L. B., Murphy-Ullrich J. E., and Strickland D. K. (2008) LDL receptor-related protein 1: unique tissue-specific functions revealed by selective gene knockout studies. Physiol. Rev. 88, 887–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Boucher P., Gotthardt M., Li W.-P., Anderson R. G. W., and Herz J. (2003) LRP: role in vascular wall integrity and protection from atherosclerosis. Science 300, 329–332 [DOI] [PubMed] [Google Scholar]
- 4. Basford J. E., Moore Z. W. Q., Zhou L., Herz J., and Hui D. Y. (2009) Smooth muscle LDL receptor-related protein-1 inactivation reduces vascular reactivity and promotes injury-induced neointima formation. Arterioscler. Thromb. Vasc. Biol. 29, 1772–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhou L., Takayama Y., Boucher P., Tallquist M. D., and Herz J. (2009) LRP1 regulates architecture of the vascular wall by controlling PDGFRb-dependent phosphatidylinositol 3-kinase activation. PLoS One 4, e6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cao C., Lawrence D. A., Li Y., Von Arnim C. A. F., Herz J., Su E. J., Makarova A., Hyman B. T., Strickland D. K., and Zhang L. (2006) Endocytic receptor LRP together with tPA and PAI-I coordinates Mac-1-dependent macrophage migration. EMBO J. 25, 1860–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hu L., Boesten L. S. M., May P., Herz J., Bovenschen N., Huisman M. V., Berbée J. F. P., Havekes L. M., van Vlijmen B. J. M., and Tamsma J. T. (2006) Macrophage low density lipoprotein receptor related protein deficiency enhances atherosclerosis in apoE/LDLR double knockout mice. Arterioscler. Thromb. Vasc. Biol. 26, 2710–2715 [DOI] [PubMed] [Google Scholar]
- 8. Overton C. D., Yancey P. G., Major A. S., Linton M. F., and Fazio S. (2007) Deletion of macrophage LDL receptor-related protein increases atherogenesis in the mouse. Circ. Res. 100, 670–677 [DOI] [PubMed] [Google Scholar]
- 9. Yancey P. G., Blakemore J., Ding L., Fan D., Overton C. D., Zhang Y., Linton M. F., and Fazio S. (2010) Macrophage LRP-1 controls plaque cellularity by regulating efferocytosis and Akt activation. Arterioscler. Thromb. Vasc. Biol. 30, 787–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Terrand J., Bruban V., Zhou L., Gong W., El Asmar Z., May P., Zurhove K., Haffner P., Philippe C., Woldt E., Matz R. L., Gracia C., Metzger D., Auwerx J., Herz J., and Boucher P. (2009) LRP1 controls intracellular cholesterol storage and fatty acid synthesis through modulation of Wnt signaling. J. Biol. Chem. 284, 381–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Masson O., Chavey C., Dray C., Meulle A., Daviaud D., Quilliot D., Muller C., Valet P., and Liaudet-Coopman E. (2009) LRP1 receptor controls adipogenesis and is up-regulated in human and mouse obese adipose tissue. PLoS One 4, e7422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hofmann S. M., Zhou L., Perez-Tilve D., Greer T., Grant E., Wancata L., Thomas A., Pfluger P. T., Basford J. E., Gilham D., Herz J., Tschöp M. H., and Hui D. Y. (2007) Adipocyte LDL receptor-related protein-1 expression modulates postprandial lipid transport and glucose homeostasis in mice. J. Clin. Invest. 117, 3271–3282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Basford J. E., Wancata L., Hofmann S. M., Silva R. A., Davidson W. S., Howles P. N., and Hui D. Y. (2011) Hepatic deficiency of low density lipoprotein receptor related protein-1 reduces high density lipoprotein secretion and plasma levels in mice. J. Biol. Chem. 286, 13079–13087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Luzio J. P., Pryor P. R., and Bright N. A. (2007) Lysosomes: fusion and function. Nat. Rev. Mol. Cell Biol. 8, 622–632 [DOI] [PubMed] [Google Scholar]
- 15. Dong H., and Czaja M. J. (2011) Regulation of lipid droplets by autophagy. Trends Endocrinol. Metab. 22, 234–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu K., and Czaja M. J. (2013) Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ. 20, 3–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Czaja M. J. (2010) Autophagy in health and disease. 2. Regulation of lipid metabolism and storage by autophagy: pathophysiological implications. Am. J. Physiol. Cell Physiol. 298, C973–C978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chatterjee T. K., Basford J. E., Knoll E., Tong W. S., Blanco V., Blomkalns A. L., Rudich S., Lentsch A. B., Hui D. Y., and Weintraub N. L. (2014) HDAC9 knockout mice are protected from adipose tissue dysfunction and systemic metabolic disease during high-fat feeding. Diabetes 63, 176–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Robbins E., and Marcus P. I. (1963) Dynamics of acridine orange-cell interaction. J. Cell Biol. 18, 237–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ribble D., Goldstein N. B., Norris D. A., and Shellman Y. G. (2005) A simple technique for quantifying apoptosis in 96-well plates. BMC Biotechnol. 5, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Steinberg D. (1963) Fatty acid mobilization: mechanisms of regulation and metabolic consequences. Biochem. Soc. Symp. 24, 111–143 [PubMed] [Google Scholar]
- 22. Moller L., Stodkilde-Jorgensen H., Jensen F. T., and Jorgensen J. O. L. (2008) Fasting in healthy subjects is associated with intrahepatic accumulation of lipids as assessed by 1H-magnetic resonance spectroscopy. Clin. Sci. 114, 547–552 [DOI] [PubMed] [Google Scholar]
- 23. Malhi H., Bronk S. F., Werneburg N. W., and Gores G. J. (2006) Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J. Biol. Chem. 281, 12093–12101 [DOI] [PubMed] [Google Scholar]
- 24. Singh R., Kaushik S., Wang Y., Xiang Y., Novak I., Komatsu M., Tanaka K., Cuervo A. M., and Czaja M. J. (2009) Autophagy regulates lipid metabolism. Nature 458, 1131–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tatti M., Motta M., Di Bartolomeo S., Cianfanelli V., and Salvioli R. (2013) Cathepsin-mediated regulation of autophagy in saposin C deficiency. Autophagy 9, 1–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tatti M., Motta M., Di Bartolomeo S., Scarpa S., Cianfanelli V., Cecconi F., and Salvioli R. (2012) Reduced cathepsins B and D cause impaired autophagic degradation that can be almost completely restored by overexpresssion of these two proteases in SapC-deficient fibroblasts. Hum. Mol. Genet. 21, 5159–5173 [DOI] [PubMed] [Google Scholar]
- 27. Caldwell S. H., Chang C. Y., Nakamoto R. K., and Krugner-Higby L. (2004) Mitochondria in nonalcoholic fatty liver disease. Clin. Liver Dis. 8, 595–617, x [DOI] [PubMed] [Google Scholar]
- 28. Leamy A. K., Egnatchik R. A., and Young J. D. (2013) Molecular mechanisms and the role of saturated fatty acids in the progression of non-alcoholic fatty liver disease. Prog. Lipid Res. 52, 165–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Scaduto R. C. Jr., and Grotyohann L. W. (1999) Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys. J. 76, 469–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li Z., Berk M., McIntyre T. M., Gores G. J., and Feldstein A. E. (2008) The lysosomal-motochondrial axis in free fatty acid-induced hepatic lipotoxicity. Hepatology 47, 1495–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kneeman J. M., Misdraji J., and Corey K. E. (2012) Secondary causes of nonalcoholic fatty liver disease. Therap. Adv. Gastroenterol. 5, 199–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Day C. P. (2011) Non-alcoholic fatty liver disease: a massive problem. Clin. Med. 11, 176–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Day C. P., and James O. F. (1998) Steatohepatitis: a tale of two hits. Gastroenterology 114, 842–845 [DOI] [PubMed] [Google Scholar]
- 34. Huang X.-Y., Shi G.-M., Devbhandari R. P., Ke A.-W., Wang Y., Wang X.-Y., Wang Z., Shi Y.-H., Xiao Y.-S., Ding Z.-B., Dai Z., Xu Y., Jia W.-P., Tang Z.-Y., Fan J., and Zhou J. (2012) Low level of low density lipoprotein receptor related protein 1 predicts an unfavorable prognosis of hepatocellular carcinoma after curative resection. PLoS One 7, e32775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rohlmann A., Gotthardt M., Hammer R. E., and Herz J. (1998) Inducible inactivation of hepatic LRP gene by Cre-mediated recombination confirms role of LRP in clearance of chylomicron remnants. J. Clin. Invest. 101, 689–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Quiroga A. D., and Lehner R. (2012) Liver triacylglycerol lipases. Biochim. Biophys. Acta 1821, 762–769 [DOI] [PubMed] [Google Scholar]
- 37. Malhi H., and Gores G. J. (2008) Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin. Liver Dis. 28, 360–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yahiro K., Satoh M., Nakano M., Hisatsune J., Isomoto H., Sap J., Suzuki H., Nomura F., Noda M., Moss J., and Hirayama T. (2012) Low-density lipoprotein receptor-related protein-1 (LRP1) mediates autophagy and apoptosis caused by Helicobacter pylori VacA. J. Biol. Chem. 287, 31104–31115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sun Y., and Grabowski G. A. (2013) Altered autophagy in the mice with a deficiency of saposin A and saposin B. Autophagy 9, 1115–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang Y., Singh R., Xiang Y., and Czaja M. J. (2010) Macroautophagy and chaperone-mediated autophagy are required for hepatocyte resistance to oxidant stress. Hepatology 52, 266–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cai N., Zhao X., Jing Y., Sun K., Jiao S., Chen X., Yang H., Zhou Y., and Wei L. (2014) Autophagy protects against palmitate-induced apoptosis in hepatocytes. Cell Biosci. 4, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]