

Abstract
Single domain antibodies (sdAbs) correspond to the antigen-binding domains of camelid antibodies. They have the same antigen-binding properties and specificity as monoclonal antibodies (mAbs) but are easier and cheaper to produce. We report here the development of sdAbs targeting human PCSK9 (proprotein convertase subtilisin/kexin type 9) as an alternative to anti-PCSK9 mAbs. After immunizing a llama with human PCSK9, we selected four sdAbs that bind PCSK9 with a high affinity and produced them as fusion proteins with a mouse Fc. All four sdAb-Fcs recognize the C-terminal Cys-His-rich domain of PCSK9. We performed multiple cellular assays and demonstrated that the selected sdAbs efficiently blocked PCSK9-mediated low density lipoprotein receptor (LDLR) degradation in cell lines, in human hepatocytes, and in mouse primary hepatocytes. We further showed that the sdAb-Fcs do not affect binding of PCSK9 to the LDLR but rather block its induced cellular LDLR degradation. Pcsk9 knock-out mice expressing a human bacterial artificial chromosome (BAC) transgene were generated, resulting in plasma levels of ∼300 ng/ml human PCSK9. Mice were singly or doubly injected with the best sdAb-Fc and analyzed at day 4 or 11, respectively. After 4 days, mice exhibited a 32 and 44% decrease in the levels of total cholesterol and apolipoprotein B and ∼1.8-fold higher liver LDLR protein levels. At 11 days, the equivalent values were 24 and 46% and ∼2.3-fold higher LDLR proteins. These data constitute a proof-of-principle for the future usage of sdAbs as PCSK9-targeting drugs that can efficiently reduce LDL-cholesterol, and as tools to study the Cys-His-rich domain-dependent sorting the PCSK9-LDLR complex to lysosomes.
Keywords: cholesterol regulation; inhibitor; low-density lipoprotein (LDL); proprotein convertase subtilisin/kexin type 9 (PCSK9); single-domain antibody (sdAb,nanobody); Familial Hypercholesterolemia; LDL Receptor; cysteine-histidine-rich-domain (CHRD)
Introduction
For over 30 years, a large number of clinical trials have firmly consolidated the importance of lowering LDL-cholesterol (LDLc)3 in the prevention of cardiovascular diseases (CVD) and its associated devastating sequelae (1). Healthy diets and exercise are highly recommended to lower LDLc in patients with high baseline levels. However, many individuals, including those suffering from familial hypercholesterolemia (FH), cannot reach the recommended LDLc levels to prevent cardiovascular complications. With an overall incidence of ∼1:200, FH is a common inherited disease that affects at least 30 million people, of whom ≤1% have been diagnosed. It is characterized by plasma LDLc levels greater that the 95th percentile, which result in tendon xanthomas, xanthelasmas, corneal arcus, and premature atherosclerosis, leading to premature ischemic vascular disease and mortality if left untreated. In most cases, FH subjects exhibit mutations in the LDL receptor (LDLR; 67%) and its ligand apolipoprotein B (apoB; 14%), hampering LDL clearance from the circulation (2). In 2003, merging biological studies with human genetics led to the discovery of PCSK9, the 9th and last member of the family of proprotein convertases related to subtilisin and kexin (3), and the demonstration that the PCSK9 gene represents the 3rd locus of autosomal dominant hypercholesterolemia (4).
PCSK9 is a serine protease first synthesized as a zymogen that autocatalytically cleaves itself in the endoplasmic reticulum (ER) to excise its N-terminal prodomain (3), which acts as a chaperone and a potent inhibitor. However, different from all other convertases (5), PCSK9 is secreted as an enzymatically inactive non-covalent complex with its inhibitory prodomain tightly bound to the catalytic subunit of mature PCSK9 (6). Thus, PCSK9 has no substrate other than itself. Rather, it binds to specific cell-surface receptors and escorts them toward intracellular acidic endosome/lysosome degradation compartments (7, 8). A schematic diagram of PCSK9's primary structure and its domains (prodomain; catalytic domain; hinge; Cys- and His-rich domain (CHRD)) is shown in Fig. 1A. The crystal structure of PCSK9 revealed that the CHRD is composed of three distinct Cys/His-rich modules, denoted M1, M2, and M3 (6).
FIGURE 1.

Generation of llama PCSK9-specific sdAbs, PCSK9 binding to sdAb, effect of the sdAb on LDLR binding. The primary structure of PCSK9 and its deletants are shown: aa, amino acid; sp, signal peptide; pro, prodomain; h, hinge domain; CHRD, Cys/His-rich domain and its modules M1, M2, and M3 (A). Flow chart of the steps for llama sdAb selection (B). Phylogenetic tree deduced from the alignment of the full protein sequences of 10 selected sdAbs (C). The media of HEK293 transiently expressing PCSK9 or its deletants lacking either the pro- and catalytic domains (CHRD) or the CHRD (L455X) and the sdAb P1.40 were mixed and immunoprecipitated with TALON metal affinity resin. The input (conditioned media containing the different PCSK9 variants) and pellets were analyzed by WB with mAb-V5 antibody after SDS-PAGE on 12% Tris-glycine gels (D). Similar experiments were performed on PCSK9 variants lacking the CHRD modules M2 alone (ΔM2), M2 and M1 (ΔM1M2) or M2 and M3 (ΔM2M3) (E). D and E are representative of three independent experiments. Schematic of the representative fusion of P1.40 with a mouse Fc comprising the hinge (h), CH2, and CH3 domains (F). Purified sdAb-Fcs (2 μg) were separated by SDS-PAGE in the presence (+) or absence (−) of DTT and revealed by Coomassie staining (G). The four sdAb-Fcs were incubated with PCSK9 and tested for their ability to prevent binding of PCSK9 to the LDLR EGF-AB coated on a binding assay plate (H). This assay is an average of three technical replicates.
The first PCSK9 target that was identified is the LDLR at the surface of hepatocytes (9–11). The catalytic subunit of PCSK9 was shown to bind the LDLR through its EGF-A domain (8, 12), as well as the LDLR superfamily members VLDLR, ApoER2 (13, 14), and LRP1 (15). Upon LDL binding to cell surface LDLR, the complex is internalized into the cell within heavy chain clathrin-coated vesicles that fuse with early endosomes. Herein, the acidic pH causes conformational changes driving LDL release, the subsequent recycling of the LDLR to the cell surface, and the sorting of LDL to lysosomes for cholesterol recovery and distribution in the cell (16). The PCSK9-LDLR complex also enters the cell via clathrin-coated vesicles (7, 17). However, the acidic pH enhances the affinity of PCSK9 for the LDLR (6) and, through some unknown mechanism requiring the CHRD (7, 13, 18), favors its ability to escort the LDLR to late endosomes/lysosomes for degradation by as yet undefined proteases (7, 10).
The rare PCSK9 mutations identified in FH patients result in a gain-of-function (GOF), i.e. an increased potency of PCSK9 to promote LDLR degradation, with ensuing higher circulating LDLc levels (4, 10). The most dramatic GOF D374Y mutation increases ∼10-fold the affinity of PCSK9 for the LDLR (6) and results in ∼4-fold higher LDLc levels (∼10 mmol/liter), as well as early death due to CVD (19). Loss-of-function PCSK9 mutations were also identified. Two non-sense mutations Y142X and C679X found in ∼2% of black Africans were associated with an ∼40% decrease in LDLc and an ∼88% reduction in the risk of CVD (20, 21). This provided a proof-of-principle that PCSK9 inhibition may be safe and represents a promising approach to treat hypercholesterolemia and prevent CVD (22, 23).
Accordingly, PCSK9 monoclonal antibodies (mAbs) blocking its interaction with the LDLR were developed and are presently prescribed in clinics to patients suffering from severe hypercholesterolemia, who are statin-resistant and/or cannot reach target LDLc using available drugs. Such subcutaneously injected mAbs every 2 or 4 weeks result in a sustained ∼60% reduction in LDLc (5, 22, 24) and thus represent a powerful drug against heart disease that is superior to statins. Over the past 30 years, mAbs became established as effective medicines for several serious diseases (25–27). However, their high molecular mass (∼150 kDa) require large amounts to be injected to reach efficacy (e.g. 150 mg/14 days), and their high cost limits their wide applicability. The mAbs targeting PCSK9 cost ∼$14,000/year/patient (28), likely making them the most costly class of medications marketed so far. This definitely restricts their use to high risk patients not reaching LDLc target levels despite maximal doses of statins (29). Thus, there is an unmet need for cheaper and more accessible inhibitory molecules.
Camelid single domain antibodies (sdAbs), also known as nanobodies, were first discovered in 1993 (30). Different from conventional antibodies, up to 75% of camelid antibodies are devoid of light chains. They are made of two heavy chains (hcAbs), each comprising an antigen-binding domain (VHH or sdAb), followed by two constant domains CH2 and CH3 (31). Although 10-fold smaller (∼13 kDa) than conventional IgGs, sdAbs bind antigen targets with equivalent specificity, affinity, and low toxicity (32) and show enhanced tissue penetration. Importantly, sdAbs can be produced from recombinant bacteria and are thus expected to be cheaper and easier to manufacture. They can also easily be engineered to achieve high potency by affinity maturation and can be humanized for pre-clinical studies in non-human primates and in humans (31, 33). Therefore, they constitute an attractive alternative to mAbs (34–36).
In this study, following llama injections of full-length human PCSK9, we isolated a number of sdAbs that inhibit the function of PCSK9 on LDLR degradation in various cell lines and primary hepatocytes. The validation of a selected sdAb called P1.40 was performed in Pcsk9 knock-out (KO) mice carrying a 67.5-kb human bacterial artificial chromosome (BAC) transgene that results in the expression of human PCSK9 under its own promoter.
Results
Inhibitory PCSK9 sdAbs
We obtained sdAbs directed against human PCSK9 by llama immunization and screening of an immune phage display library (Fig. 1B). The cDNAs of 10 selected sdAbs that bind PCSK9 efficiently were sequenced. Alignment of their corresponding protein sequences revealed that they belong to distinct sub-families (Fig. 1C). A member of each family was selected to favor sdAb diversity. P1.40, PKE9, PKF8, and P2.57 sdAb cDNAs were then subcloned in an expression vector for production in Escherichia coli and purified by His-affinity chromatography.
To define the recognition domains within PCSK9, each sdAb carrying the N-terminal V5 and the C-terminal hexa-His tags was separately expressed in HEK293 cells. The conditioned media were then mixed with those containing different forms of PCSK9 as follows: the full-length protein harboring a C-terminal V5 tag (PCSK9); the CHRD alone; and the C-terminally truncated L455X mutant lacking the CHRD (Fig. 1D) (7, 10). The sdAb-PCSK9 complexes were then immunoprecipitated using a hexa-His-binding resin, resolved by SDS-PAGE, and analyzed by Western blotting (WB) using mAb-V5. As expected, all sdAbs interacted with full-length PCSK9, but surprisingly, all four sdAbs best recognized the CHRD as indicated by the very efficient immunoprecipitation of this domain (Fig. 1D). In comparison, only traces of the L455X mutant (Fig. 1D) were observed. Here, we only show the results for P1.40, as all other sdAbs gave similar data (data not shown). To define which module(s) in the CHRD is/are recognized by the sdAbs, we performed similar experiments with PCSK9 variants lacking the CHRD modules M2 alone (ΔM2), M2 and M1 (ΔM1M2) or M2 and M3 (ΔM2M3). We could only use the above deletants as PCSK9 ΔM1, ΔM3, or ΔM1M3 are not secreted (37). The data showed that P1.40 does not recognize the M2 module (45% of the ΔM2 input bound by P1.40), but the presence of both M1 and M3 is needed for optimal recognition, as only 18% of ΔM1M2 and 14% of ΔM2M3 inputs were immunoprecipitated with P1.40 (Fig. 1E). The three other sdAbs also predominantly recognize ΔM2 (data not shown).
Chimeric sdAbs were then fused to a mouse IgG2a Fc domain, known to increase the half-life of sdAbs in plasma (38), followed by a C-terminal V5 tag (Fig. 1F). Each chimeric protein was produced in HEK293 cells and purified from 600 ml of spent media. The purity of each preparation was assessed by Coomassie staining of an SDS gel run under reducing (+DTT) or non-reducing (−DTT) conditions. All sdAb-Fcs have an apparent molecular mass of ∼42 kDa as monomers under reducing conditions (+DTT) and of ∼85 kDa as disulfide-linked dimers under non-reducing conditions (−DTT) (Fig. 1G). The estimated molecular masses of the dimeric chimeras using MALDI mass spectral analysis were as expected from theoretical calculations, i.e. ∼89 kDa for P1.40; ∼85 kDa for PKE9 and PKF8; and ∼87 kDa for P2.57.
We next used enhanced surface plasmon resonance to estimate the binding constants (Kd) of each sdAb-Fc to PCSK9. The rank orders of binding constants were PKF8 (5.6 nm) > P2.57 (9.8 nm) > PKE9 (37 nm) > P1.40 (49 nm). However, as will be shown later, the above in vitro binding affinities do not necessarily correlate with the most active sdAb-Fc to inhibit the PCSK9-enhanced cellular degradation of the LDLR.
Although these four sdAb-Fcs interact with the CHRD M1/M3 modules (Fig. 1, D and E), we assessed their ability to disrupt the PCSK9 interaction to the EGF-AB domains of the LDLR in an in vitro binding assay (Fig. 1H). The data show that the sdAb-Fcs did not decrease PCSK9 binding to LDLR, demonstrating that none of the sdAbs interferes with the ability of PCSK9 to bind the EGF-AB domain of the LDLR. Surprisingly, almost 2-fold higher PCSK9 binding was observed with all sdAb-Fcs except for P2.57. The doubling of PCSK9 levels bound to EGF-AB is likely due to the fact that sdAb-Fcs are homodimers (Fig. 1G) and hence could conceptually bind up to two PCSK9 molecules.
Effect of sdAb-Fcs on DiI-LDL Uptake and Cell Surface LDLR Levels in HepG2 Cells
To assess the ability of each sdAb-Fc to inhibit the PCSK9-mediated degradation of the LDLR, we pre-incubated purified PCSK9 (0.08 μm) with increasing concentrations of sdAb-Fcs (0–1 μm) for 1 h and applied these media onto HepG2 cells for a further 5 h incubation. PCSK9 alone resulted in an ∼60% decrease of the DiI-LDL uptake, reflecting the reduced levels of functional LDLR at the cell surface (1st two rows in Fig. 2, A and B). All sdAb-Fcs except P2.57 exhibited a dose-dependent inhibitory effect, with the P1.40 being the most potent, giving an ∼80% inhibition at 0.1 μm and 100% at 1 μm (Fig. 2A). In a separate experiment, we also showed that the mAb evolocumab gave a similar ∼80% inhibition between 0.04 and 0.1 μm and a 100% inhibition at 1 μm (Fig. 2B).
FIGURE 2.
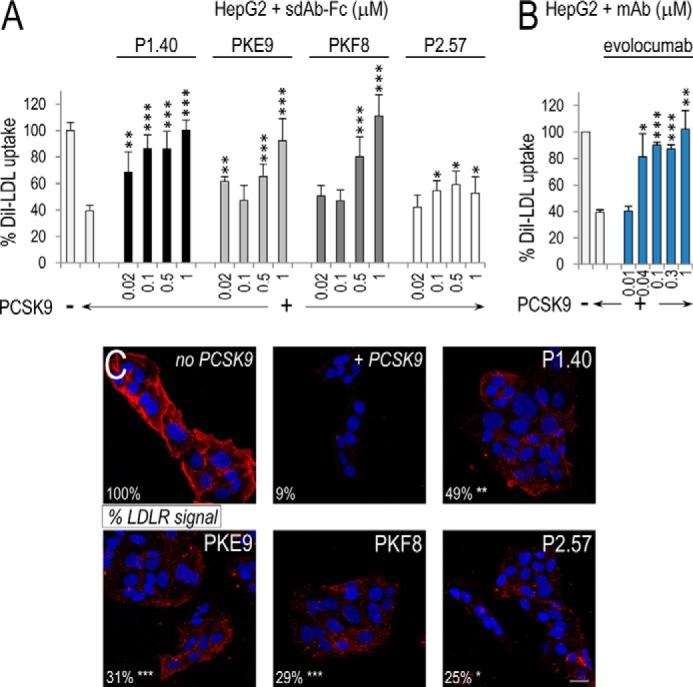
Effect of sdAb-Fcs on DiI-LDL uptake by HepG2 cells and their LDLR cell surface levels. HepG2 cells were incubated for 5 h in the absence or presence of 0.08 μm (5 μg/ml) PCSK9 mixed with increasing concentrations of sdAb-Fcs. DiI-LDL (5 μg/ml final) was added during the last 2 h of incubation. The % DiI-LDL uptake shown here is normalized to the number of cells per well and calibrated to the % uptake in control cells (in absence of PCSK9) (A). A similar experiment was performed with evolocumab (B). In both A and B, the data represent averaged values ± S.E. for at least three independent experiments, with each comprising three independent samples per condition. HepG2 cells were incubated for 4 h without (no PCSK9) or with 0.05 μm (3 μg/ml) PCSK9 (+PCSK9) in the absence or presence of each sdAb-Fc (1.2 μm). LDLR immunocytochemistry with Alexa 555-labeled secondary antibodies was performed, and nuclei were stained with DAPI. Scale bar, 20 μm (C). In two separate experiments, LDLR labeling was quantified based on the average pixel values of 10 fields calibrated to the number of nuclei per field. The numbers (bottom left corner) are expressed as % of the value obtained in the absence of PCSK9 (no PCSK9) and represent averaged pixel values of the two independent experiments. *, p < 0.05; **, p < 0.005; ***, p < 0.0005. p values were obtained from Student's t tests, except for some conditions in A, for which a two-way ANOVA test was more appropriate.
We also tested by immunofluorescence the ability of each sdAb-Fc to rescue the LDLR cell surface labeling of HepG2 cells. PCSK9 (0.05 μm) and the sdAb-Fcs (1.2 μm) were pre-incubated as above and applied onto cells for 4 h (Fig. 2C). LDLR labeling was quantified by confocal microscopy and expressed as a percentage of that in control cells not exposed to PCSK9. All four sdAb-Fcs were able to enhance the levels of immunoreactive LDLR, proving their inhibitory potential, but could not completely rescue the LDLR signal in this assay. Again, P1.40 seemed the most potent sdAb-Fc with ∼50% rescue of immunoreactive cell surface LDLR.
Effect of sdAb-Fcs on Total LDLR Levels in Hepatocytes
The inhibitory effect of sdAb-Fcs was then analyzed in mouse primary hepatocytes, which are closer to an in vivo model than HepG2 cells. The latter were isolated from PCSK9-deficient mice (39, 40) and used to test the sdAb activity toward human PCSK9 (in the absence of endogenous mouse Pcsk9) and to maximize LDLR levels, which were reported to increase by ∼3-fold in KO versus WT mice (39, 40). Notably, human PCSK9 acts similarly on both human and mouse LDLR (41). HepatoZYME media control (Fig. 3A, upper panel) or containing 0.08 μm PCSK9 and 0–1.2 μm of each sdAb-Fc were pre-incubated for 1 h and then incubated with mouse primary hepatocytes for 24 h. Normalized Western blot analyses of mouse LDLR levels revealed that P1.40 did not affect the levels of endogenous LDLR in the absence of PCSK9 (Fig. 3A, upper panel), demonstrating the PCSK9 specificity for the inhibitory effect. In addition, in the presence of extracellular PCSK9 only P1.40 was able to totally rescue LDLR levels, whereas P2.57 had no effect (Fig. 3A, lower panels). PKF8 and PKE9 led to a partial LDLR rescue at 1.2 μm, with relative levels of 0.7 and 0.6, respectively, versus maximal 1.1 with P1.40. The above conclusions were confirmed in the IHH cell line. Indeed, incubation with the three sdAb-Fcs PKE9, PKF8, and P2.57 (data not shown) showed low to good efficiency, whereas P1.40 showed the highest potency. Thus, the presented WB data only depict the results for P1.40 (Fig. 3B), and all the rest of our studies will be conducted with P1.40.
FIGURE 3.
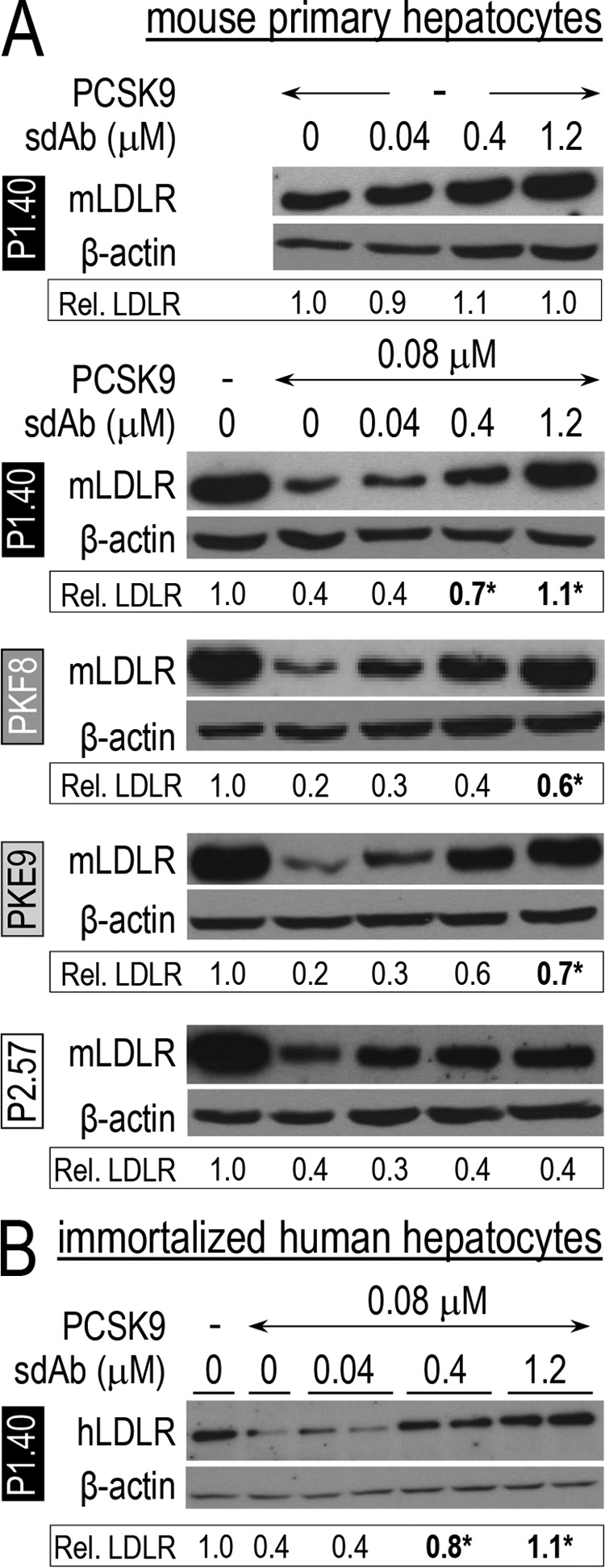
Effect of sdAb-Fcs on total LDLR levels in hepatocytes. Primary hepatocytes isolated from PCSK9-deficient mice (A) or immortalized human hepatocytes (IHH; B) were incubated for 24 h in the absence (−) or presence of 0.08 μm (5 μg/ml) WT PCSK9 and increasing concentrations of sdAb-Fc. Cell lysates were subjected to WB analysis of LDLR and β-actin levels after separation by SDS-PAGE on 8% Tris-glycine gels. LDLR signals were calibrated to β-actin ones and were normalized to the values obtained in the absence of sdAb-Fc. The blots shown are representative of at least five independent experiments, each run as one biological sample per condition. The averaged normalized LDLR values are indicated below each blot. *, p < 0.05 (Student's t test).
Inhibitory Effect of P1.40 on WT and D374Y PCSK9
The inhibitory properties of P1.40 toward WT PCSK9 or its GOF mutant D374Y, which has a >10-fold higher affinity for the LDLR (6), was examined in naive HepG2 cells (Fig. 4A) or HepG2 cells stably expressing PCSK9 (Fig. 4B) (42). Note that the LDLR appears as a doublet consisting of an LDLR that is not O-glycosylated (110 kDa) and that is fully mature and O-glycosylated (150 kDa) (43). Unexpectedly, in both cases, the strongest recovery of the LDLR signal was obtained in the presence of D374Y PCSK9, suggesting that P1.40 neutralized the activity of this mutant form of PCSK9 with a higher efficacy. Indeed, the inhibition of PCSK9 activity at 1.2 μm was estimated at 93% for D374Y PCSK9 versus ∼50% for WT PCSK9 (Fig. 4A). As expected, upon stable PCSK9 expression, the LDLR signal was more efficiently reduced by the D374Y mutant. However, P1.40 incubation of D374Y PCSK9 stably expressing cells significantly increased the LDLR signal by a maximal 2.7-fold, whereas cells expressing the WT PCSK9 showed a more modest 1.7-fold increase obtained at all concentrations, revealing that the best effect was obtained at 0.04 μm P1.40 sdAb-Fc on cells stably expressing WT PCSK9 (Fig. 4B).
FIGURE 4.

Inhibitory effect of P1.40 on WT and D374Y PCSK9. HepG2 cells were incubated 24 h with media lacking PCSK9 or containing 0.08 μm (5 μg/ml) WT PCSK9 or 0.01 μm (0.7 μg/ml) D374Y PCSK9 and 0–1.2 μm sdAb P1.40 (A). The LDLR signal (% LDLR) was calibrated to that of β-actin levels and expressed in percentage of the difference between the 1st lane (no PCSK9) and 2nd lane (+PCSK9). Stably transfected HepG2 cells that expressed ∼300 ng/ml/24 h of WT or D374Y PCSK9 were incubated 24 h with increasing concentrations of the sdAb P1.40 (B). HepG2 cells were incubated 24 h with media lacking PCSK9 or containing 0.08 μm (5 μg/ml) WT PCSK9 and 0–1.2 μm sdAb P1.40 or evolocumab (C). Stably transfected HepG2 cells that expressed ∼300 ng/ml/24 h of D374Y PCSK9 were incubated 24 h with increasing concentrations of the sdAb P1.40 or evolocumab (D). The blots shown are representative of at least two independent experiments consisting of one (A), two (B), and three (C and D) biological samples per condition. In both C and D, we also quantified the total levels of cellular LDLR using an ELISA on pooled cell lysates. The calibrated LDLR signals were normalized to that of the 1st lane (no sdAb). The averaged normalized LDLR values are indicated below each blot. *, p < 0.05; **, p < 0.005; ***, p < 0.0005 (Student's t test).
To compare the efficacies of P1.40 to the mAb evolocumab, we performed similar experiments with WT PCSK9 added extracellularly to HepG2 cells (Fig. 4C) and with HepG2 cells stably expressing D374Y PCSK9 (Fig. 4D). WB analysis of the lysates revealed an ∼90% recovery of the LDLR with 1.2 μm P1.40, whereas 150% was recovered with evolocumab at this concentration (Fig. 4C), suggesting that in this WB assay evolocumab was more active than P1.40. Similarly, in HepG2 cells expressing D374Y PCSK9, evolocumab was more active that P1.40, as evidenced by an ∼3-fold increase in LDLR with P1.40 versus ∼6-fold with evolocumab at 1.2 μm (Fig. 4D). Finally, these conclusions were supported by an LDLR ELISA performed on a pool of cell lysates obtained from experiments in Fig. 4, C and D, which also revealed the higher efficacy of evolocumab compared with P1.40, especially using the D374Y PCSK9 (Fig. 4, C and D, lower sections).
Effect of ER-localized sdAb-Fcs on PCSK9 Secretion and Activity
We next evaluated the ability of intracellular P1.40 to effectively retain PCSK9 within the ER, and hence block the PCSK9 activity. Accordingly, because addition of a C-terminal KDEL motif retains efficiently secretory proteins in the ER (44), we fused a KDEL retention signal to the C terminus of P1.40-Fc-V5. Thus, P1.40 ± KDEL forms were co-expressed in HEK293 cells with PCSK9 or its D374Y mutant, and the LDLR levels were assessed by WB analysis (Fig. 5, A and B). The data demonstrated that P1.40-KDEL was efficiently retained in the ER, as only traces of this protein were observed in the media (Fig. 5, A and B, lanes 3, 6, and 10). Neither P1.40 nor P1.40-KDEL had an impact on LDLR levels (Fig. 5A, lanes 2 and 3), likely due to the quasi-absence of PCSK9 expression in HEK293 cells (mRNAs are 10,000-fold lower than in HepG2 cells). As expected, LDLR levels were reduced significantly (70%) upon expression of D374Y or WT PCSK9, respectively (Fig. 5, A and B, lanes 4 and 8). The ER retention of P1.40-KDEL clearly increased LDLR levels in cells expressing WT or D374Y PCSK9 (Fig. 5, A and B, lanes 4–6 versus 8–10). Note that the inhibitory potency of P1.40 and P1.40-KDEL is almost similar (Fig. 5B, lanes 9 and 10), possibly related to the high luminal concentrations of P1.40 attained during its transit through the Golgi, such that most of the intracellular PCSK9 activity is inhibited. However, higher levels of ER-retained WT PCSK9 by P1.40-KDEL had no effect on LDLR recovery above that of P1.40 alone (0.8 and 0.9; Fig. 5B, lanes 9 and 10), although higher intracellular levels of D374Y PCSK9 significantly increased LDLR levels (0.6 and 0.9; Fig. 5A, lanes 5 and 6).
FIGURE 5.
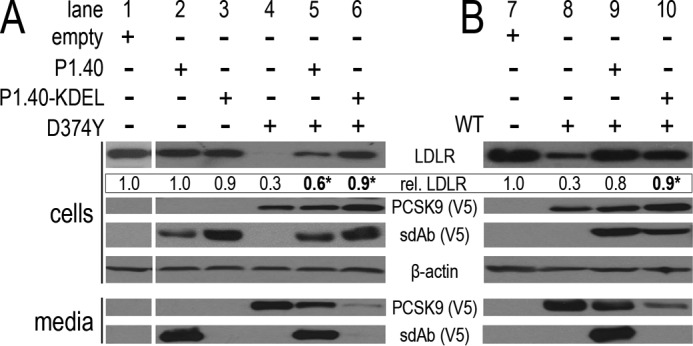
ER retention of the sdAb P1.40 via a C-terminal KDEL motif. P1.40-V5 ± KDEL and PCSK9-V5 mutant D374Y (A) or WT (B) were co-expressed in HEK293 cells. Lysates and media were then analyzed by Western blotting using LDLR, β-actin, and V5 antibodies. These results are representative of at least two independent experiments run as a single biological sample per condition. The averaged normalized LDLR values are indicated below the LDLR blots.
To assess the subcellular localization of the intracellular forms of PCSK9 and LDLR, the above cell lysates were digested with endoglycosidases H (endoH) and F (endoF). As reported previously, most of the overexpressed PCSK9 found in cells are endoH- and endoF-sensitive, suggesting it is in the ER (3, 10). However, in the above cell lysates, the LDLR is always endoH-resistant but endoF-sensitive, even the lower 110-kDa form (data not shown). This suggests that although P1.40 KDEL prevents the secretion of PCSK9, it does not result in the retention of the LDLR in the ER. This is very likely due to the recent discovery that LDLR does not bind efficiently to PCSK9 in the ER because of the presence of a competitive chaperone-like protein GRP94 that prevents such ER interaction (45).
It was shown previously that PCSK9 can induce the degradation of the LDLR by distinct intracellular and extracellular pathways and that cellular overexpression of WT PCSK9 mostly reflects the intracellular pathway, whereas expression of the ∼10-fold more potent GOF D374Y PCSK9 affects both pathways (37, 46). Accordingly, the above data demonstrate that retention of PCSK9 in the ER by P1.40-KDEL has a bigger effect on the inhibition of the D374Y activity than that of WT PCSK9 (Fig. 5, A, lanes 4–6, versus B, lanes 8–10), likely due to the elimination of both pathways by P1.40-KDEL.
PCSK9 Inhibition by P1.40 Versus EGF-AB Domains from the LDLR
Because PCSK9 and LDLR interact essentially through the EGF-A domain of the LDLR (8), we compared the potency of P1.40 (sdAb-Fc) and the chimera consisting of the EGF-AB domains, mimicking the LDLR-binding site to PCSK9, fused to a human Fc (EGFs-Fc). HepG2 cells were incubated for 4 h in the absence or presence of PCSK9 preincubated with the Fc alone as a control or its binding proteins P1.40 and EGFs-Fc (Fig. 6). LDLR levels at the cell surface were then quantified by FACS. At 4 h post-incubation, cell surface LDLR levels were reduced by ∼60% in the presence of 0.2 μm PCSK9. A high concentration of Fc alone (7 μm) had no effect, whereas 0.04 μm P1.40 doubled the LDLR levels. In contrast, a 100-fold higher concentration of EGFs-Fc (4 μm) was required to obtain a similar effect. This suggests that P1.40 is between 10- and 100-fold more potent on a molar basis than the EGFs-Fc protein.
FIGURE 6.
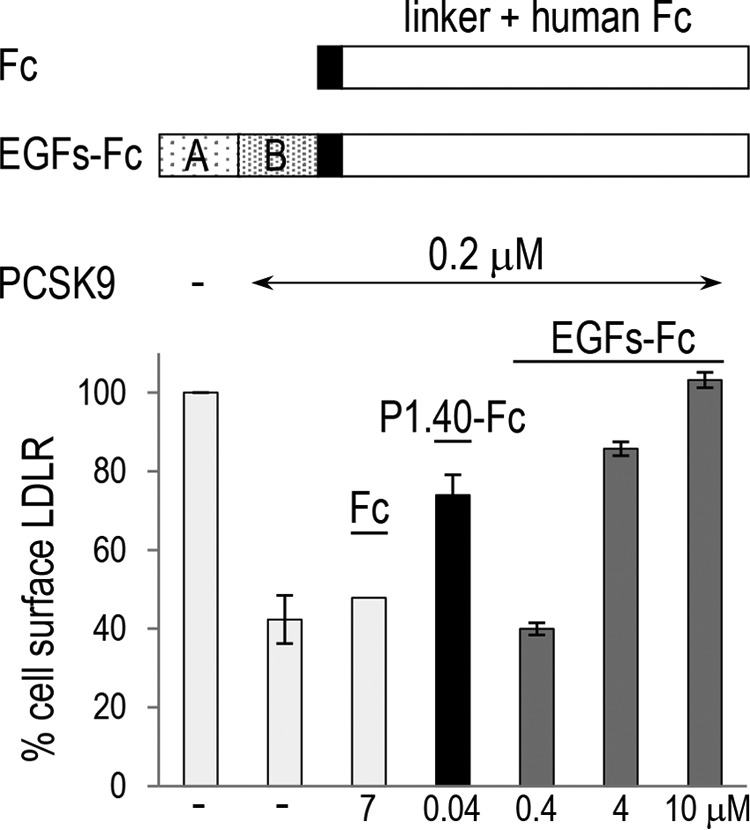
Comparison of the inhibitory activity of the sdAb P1.40 and EGF-AB fused to Fc domains. The primary structure of the Fc alone or EGF-AB fusion is shown. HepG2 cells were incubated for 4 h in the absence (−) or presence of 0.2 μm (15 μg/ml) WT PCSK9 mixed with 7 μm control linker + Fc (Fc), 0.04 μm sdAb P1.40, or 0.4–10 μm of EGFs + Fc. Cells were then analyzed by FACS for cell surface LDLR levels. The average values of two independent experiments are shown, except for Fc (n = 1).
In Vivo Efficacy of sdAb-Fc P1.40
We next validated the activity of P1.40 in vivo. Pcsk9 KO transgenic mice carrying a 67.5-kb fragment of the human chromosome 1p32 containing the PCSK9 gene (Tg+) were obtained. These transgenic mice (Tg+) express exclusively human PCSK9 under the control of its own promoter, resulting in circulating PCSK9 levels of ∼300 ng/ml, which are ∼2-fold higher than endogenous levels of WT mice (47, 48). These Tg+ mice exhibited ∼1.7- and ∼2.9-fold higher levels of circulating total cholesterol and apoB, respectively, as compared with transgene-negative (Tg−) Pcsk9 KO mice (Tg+ PBS versus Tg− PBS; Fig. 7). Tg− and Tg+ male mice were injected with equivalent volumes (∼150 μl) of PBS or PBS containing P1.40 (10 mg/kg). Mice were bled 10 days pre-injection, and 1 h and 2 and 4 days post-injection. After the last bleed, livers were collected and homogenized, and total LDLR protein levels were estimated by WB (Fig. 7, lower panel). As expected, Tg+ mice exhibited lower LDLR levels than Tg− mice (0.4 versus 1.0). P1.40-Fc had no effect in Tg− mice but led to ∼50% recovery of LDLR levels in Tg+ mice (0.7 versus 0.4). Moreover, 32 and 44% reductions were observed in circulating total cholesterol and apoB levels 4 days post-injection (Fig. 7, upper panels). In conclusion, these data indicate that P1.40 neutralized 66 and 71% of the total cholesterol, and apoB increases resulting from the expression of human PCSK9 in KO mice.
FIGURE 7.
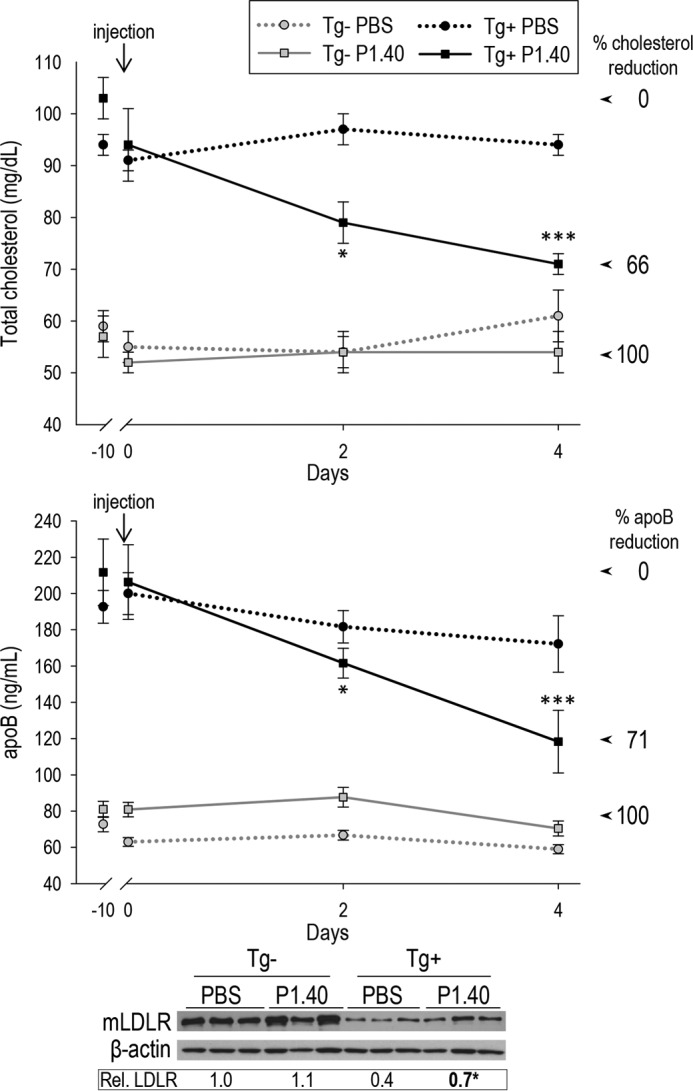
In vivo efficacy of the sdAb P1.40 after a single injection to mice. Plasma from Tg− and Tg+ that received PBS or P1.40 was collected before injection (−10 days) or 1 h and 2 and 4 days post-injection and were analyzed for their cholesterol or apoB contents. The 0% reduction was fixed as the cholesterol or apoB levels in Tg+ mice before P1.40 injection (highest levels), and 100% reduction was given by the average levels observed in Tg− mice (lacking PCSK9) receiving the same treatment. Liver LDLR levels were analyzed by WB, calibrated to β-actin ones, and normalized to the value obtained for Tg− mice injected with PBS. A representative blot is shown, and the averaged normalized LDLR values were obtained from 4 to 9 mice per group. *, p < 0.05; ***, p < 0.0005 (Student's t test).
We next performed a similar experiment to test the possible advantage of a double injection at days 0 and 7 (Fig. 8). Mice were bled 10 days pre-injection, and 1 h and 2, 4, 7, 9, and 11 days following the first injection. After the last bleed, LDLR protein levels were estimated by WB (Fig. 8, lower panel). The data showed that P1.40 led to ∼80% recovery of LDLR levels in Tg+ mice (0.9 versus 0.4). In addition, 24 and 46% reductions were observed in circulating total cholesterol and apoB levels at 11 days after the first injection (Fig. 8, upper panels). These data indicate that two injections of P1.40 resulted in the neutralization of 44 and 100% of the total cholesterol and apoB increases resulting from the expression of human PCSK9 in KO mice.
FIGURE 8.
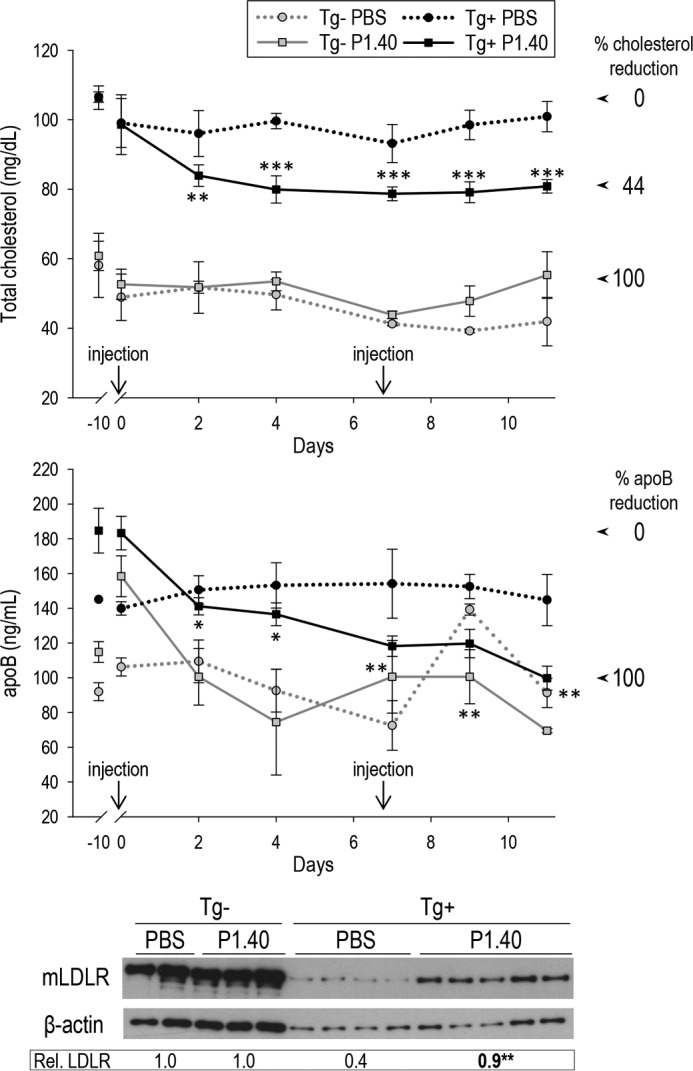
In vivo efficacy of the sdAb P1.40 after two injections to mice. Mice were injected at day 0 and then at day 7. Plasma from Tg− and Tg+ that received PBS or P1.40 was collected before injection (−10 days) or 1 h and 2, 4, 7, 9, and 11 days following the first injection and were analyzed for their cholesterol or apoB contents. The 0% reduction was fixed as the cholesterol or apoB levels in Tg+ mice before P1.40 injection (highest levels), whereas the 100% reduction was given by the average levels observed in Tg− mice (lacking PCSK9) receiving the same treatment. Liver LDLR levels were analyzed by WB, calibrated to β-actin ones, and normalized to the value obtained for Tg− mice injected with PBS. A representative blot is shown, and the averaged normalized LDLR values were obtained from 2 to 5 mice per group. *, p < 0.05; **, p < 0.005; ***, p < 0.0005 (Student's t test).
In conclusion, compared with a single injection, two injections of P1.40 spaced by 7 days resulted in a more efficacious increase in hepatic LDLR and reduction in apoB levels, close to the ones seen in absence of PCSK9.
Discussion
In this work, we report the generation of four PCSK9 sdAbs with nanomolar affinity against the CHRD (Fig. 1). As sdAb-Fc fusion proteins, they efficiently inhibited PCSK9-induced LDLR degradation, although they did not prevent the binding of PCSK9 to LDLR (Fig. 1). In HepG2 cells, IHH cells, and/or primary hepatocytes, they restored LDL uptake, as well as cell surface and total LDLR levels (Figs. 2–4). Even though P1.40 proved to be the most active sdAb candidate in our cell-based assays, it was surprising to find that in vitro it bound the CHRD of PCSK9 with an ∼5-fold lower Kd value than P2.57, which has minimal inhibitory activity. Although this may not be the case in all functional sdAbs, it emphasizes the notion that a lack of correlation may exist between the binding affinity and functional inhibitory effect of PCSK9 sdAbs recognizing the CHRD.
Herein, we focused on the most promising sdAb-Fc, P1.40, and showed that its retention in the ER blocked PCSK9 secretion (Fig. 5). We also showed that P1.40 was more potent than an EGF-AB-Fc fusion chimera (Fig. 6). Finally, 4 and 11 days following a single/double injection, respectively, in transgenic mice expressing exclusively human PCSK9, P1.40 led to 32/24% and 44/46% lower circulating cholesterol and apoB levels, respectively, and to 1.8-/2.3-fold higher levels of hepatic LDLR (Figs. 7 and 8).
The mAbs presently used in the clinic, e.g. evolocumab, bind the catalytic domain of PCSK9 and sterically hinder its binding to the LDLR (49). Although the four sdAbs analyzed in this study differed in their primary sequences (Fig. 1C), they all bind the CHRD modules M1/M3 of PCSK9 (Fig. 1, D and E). It is important to note that all the other proprotein convertases (5) seem poorly immunogenic, as only for PC1 and PC2 do the available antibodies allow the detection of endogenous proteins in tissue extracts or by immunohistochemistry (50). Thus, the CHRD may be the most antigenic domain of PCSK9. Although this domain is not involved in LDLR binding (6), it is required for the PCSK9-induced LDLR degradation. Human loss-of-function mutations are found in the CHRD, and PCSK9 lacking this domain cannot target the LDLR for degradation (7, 13, 18, 22, 46). In agreement, CHRD mAbs that do not block the LDLR interaction can reduce PCSK9 activity, albeit only partially (50–75% versus 100% for clinical mAbs) (51, 52). This likely occurs through prevention of the cellular internalization and/or lysosomal targeting of the PCSK9-LDLR complex (53). Similarly, the four tested sdAb-Fcs did not sterically hinder PCSK9 binding to the LDLR (Fig. 1H), suggesting a similar mode of action as the CHRD-specific mAbs. Future analysis of sdAb-PCSK9 complexes by x-ray crystallography should reveal their respective epitopes. Their degree of proximity to the catalytic domain will allow us to define the inhibitory mechanism of the corresponding sdAbs, and will possibly shed light on the better inhibition of D374Y than WT PCSK9 by P1.40 (Fig. 4A). Whether any antibody targeting the CHRD may achieve a total PCSK9 inhibition, for example by interfering in PCSK9 binding to the LDLR, remains unknown. Comparison of the potency of P1.40 to the mAb evolocumab that recognizes the catalytic domain of PCSK9 (49) revealed that at 1.2 μm the latter more efficiently inhibits LDLR degradation by WT and especially D374Y PCSK9 (Fig. 4, C and D).
Upon its ER localization via the addition of a C-terminal KDEL motif, P1.40-KDEL reduced PCSK9 secretion (by ∼90%) and restored LDLR levels in cells expressing WT PCSK9 and more so D374Y PCSK9 (Fig. 5). The potency of P1.40 was compared with that of a similar construct comprising the LDLR EGF-like domains A and B (EGFs-Fc) and was found to be between 10- and 100-fold higher with an EC50 ∼0.04 μm versus 0.4–4 μm (Fig. 6). In agreement with the above values, fusions containing EGF-A only or its improved form EGF66 exhibited EC50 of 11 and 1.6 μm, respectively, in the same HepG2 cells (54).
Mice lacking endogenous Pcsk9 and expressing human PCSK9 gene under its own promoter exhibited higher total cholesterol and apoB levels than Pcsk9 KO Tg− mice. Single or double injections of P1.40 neutralized the increase in apoB levels after 4 days by ∼70% and after 11 days by 100%, respectively (Figs. 7 and 8). In a “pioneer” experiment, the total cholesterol of WT mice was reduced by a PCSK9 mAb evolocumab injection by 26 and 28% after 3 and 6 days, respectively (49). In our study, we found that P1.40 can reduce total cholesterol by 32 and 24% at 4 or 11 days following P1.40 injections (Figs. 7 and 8), which is similar to the results obtained with evolocumab. Plasma PCSK9 remained quite stable at the different time points. However, when P1.40, which does not interfere with our in-house ELISA, was immunoprecipitated from plasma obtained from the single injection experiment, we found that ∼50% of PCSK9 existed as a complex with P1.40 at day 4, indicating that no major clearance took place in this delay (data not shown). Interestingly, a similar proportion of PCSK9 was captured by a human mAb recognizing the catalytic subunit causing a 40% reduction in LDLc in humanized CETP/LDLR-hemi mice (55). In another approach, the injection of a PCSK9 inhibitory “adnectin” conjugated to polyethylene glycol in a similar mouse model led to a drastic reduction of free PCSK9 in the first 24 h post-injection, but it did not affect circulating cholesterol levels (56), suggesting a higher ability of adnectin to bind PCSK9 than to inhibit its activity toward LDLR. Although the molecular mass of our sdAb-Fc fusion protein is only 40 kDa versus ∼50 kDa for conjugated adnectin, its dimerization (Fig. 1G) may slow down its clearance.
Other proposed approaches to lower PCSK9 levels are based on antisense oligonucleotides. In mice, they were shown to reduce PCSK9 mRNA levels and total cholesterol and/or LDLc by ∼50%, generally after several weeks of treatment (57–60). Specifically, lipidoid nanoparticles achieving liver-specific RNAi silencing of PCSK9 (60) are currently in phase II and have a potential for bi-annual dosing (Alnylam Pharmaceuticals). PCSK9 vaccines, peptide- or virus-like particle-based, were also developed and achieved ∼50% lower total and/or LDLc levels for up to 1 year in rodents and/or primates (61, 62).
Since their discovery in 1993 (32, 33), sdAbs have been studied extensively as new therapeutic tools. The first evidence of their in vivo efficacy was obtained in 2002 in a study showing that tumor growth in mice was inhibited by lysozyme sdAbs (63). Although no sdAb has yet been approved for clinical use, many of them are in clinical trials. For example, the sdAb caplacizumab targets von Willebrand factor (34) and is used to treat acquired thrombotic thrombocytopenic purpura in a phase III clinical trial (Ablynx). Another advanced sdAb is ozoralizumab. It is directed against TNFα and fused to serum albumin, and it is evaluated in a phase II clinical trial for its efficacy in the treatment of rheumatoid arthritis (35). Finally, sdAbs neutralizing the respiratory syncytial (Ablynx) or rota-virus (36) are currently in phase II clinical trials.
In this study, we developed the first sdAb that targets human PCSK9. This sdAb was shown to reduce PCSK9-mediated LDLR degradation in vivo and therefore to increase hepatic LDLR and decrease total cholesterol and apoB levels. Further studies will be necessary to validate its therapeutic potential. Meanwhile, the P1.40 PCSK9 sdAb constitutes a precious tool to better define the mechanism underlying LDLR targeting to lysosomes and thus to identify new PCSK9 partners. Biochemical, genetic, and epidemiological studies revealed that loss of PCSK9 reduces the incidence of atherosclerosis (64), myocardial infarction (65), stroke (66), inflammation (22), sepsis (67), and tumor-associated metastasis (68). Because different domains of PCSK9 may be implicated in these processes, various inhibitory antibodies, including the present one that binds the CHRD and not the catalytic domain, may allow a fine tuning of PCSK9 inhibition.
Experimental Procedures
Generation of Anti-PCSK9 Single Domain Antibodies
Human PCSK9 carrying a C-terminal His tag was purified to homogeneity from the media of baculovirus-infected High Five cells (69). A male llama (Lama glana) was immunized with 80 μg of PCSK9 on day 1 and with 20 μg on days 21, 36, 50, and 64. Complete Freund's adjuvant (Sigma) was used for the primary immunization, and incomplete Freund's adjuvant was used for immunizations 2–5. Leukocytes were isolated from the last bleed at day 71 post-immunization. Total RNA was then isolated, reverse-transcribed, and used to produce an immune VHH library as described previously (70). Briefly, the cDNAs encoding VHH domains were amplified using specific sense (MJ1, 5′-GCCCAGCCGGCCATGGCCSMKGTGCAGCTGGTGGAKTCTGGGGGA-3′; MJ2, 5′-CAGCCGGCCATGGCCCAGGTAAAGCTGGAGGAGTCTGGGGGA-3′; and MJ3, 5′-GCCCAGCCGGCCATGGCCCAGGCTCAGGTACAGCTGGTGGAGTCT-3′) and antisense (CH2, 5′-CGCCATCAAGGTACCAGTTGA-3′; and CH2b3, 5′-GGTACCTGTCATCCACGGACCAGCTGA-3′) primers for VHH and CH2 domains, respectively, and cloned into the phagemid pMED1 vector. The size of the library was measured as 2.3 × 108 independent transformants, greatly exceeding the number of leukocytes used for library construction. Exponentially growing E. coli expressing the phagemid library were infected with helper phages to “rescue” the phage particle auto-assembly and grown overnight for phage production. Isolation of PCSK9-specific sdAbs was performed by phage display. Using PCSK9-coated 96-well plates, four rounds of panning were performed, with each round including steps of phage binding to PCSK9, washes, phage elution, and amplification. In a last step, the phages produced by 45 individual clones were tested for their ability to bind PCSK9, and the 10 strongest binders were selected. Corresponding sdAbs were sequenced and subcloned in a pSJF2H vector comprising an OmpA signal peptide that targets the protein to the periplasmic space of E. coli, and a C-terminal hexa-His tag that allowed sdAb purification by an automated procedure using a KingFisher Flex (Thermo Scientific).
Co-immunoprecipitation
HEK293 cells were plated in 100-mm2 plates and transiently transfected using JetPRIME (Polyplus-transfection®) with 10 μg of vectors (7, 10) coding for various PCSK9 constructs or each of the four sdAbs. The sdAb conditioned media were incubated with those containing PCSK9 variants (Fig. 1A) and immunoprecipitated with TALON Metal Affinity Resin (Clontech) overnight at 4 °C. The following day, the samples were washed four times with radioimmunoprecipitation assay buffer (50 mm Tris-HCl, pH 8.0, 1% (v/v) Nonidet P-40, 0.5% sodium deoxycholate, 150 mm NaCl, and 0.1% (v/v) SDS), and lastly with PBS. The immunoprecipitates were then resolved on a 12% Tris-glycine SDS-PAGE and revealed by a V5-HRP antibody (Invitrogen).
PCSK9-sdAb Complex Binding to the LDLR
The ability of sdAbs to prevent the interaction of PCSK9 with the EGF-A domain of LDLR was assessed using a PCSK9-LDLR in vitro binding assay kit (Circulex).
Purification of sdAb-Fc Fusion Proteins
Four selected sdAbs (P1.40, PKF8, PKE9, and P2.57) were fused through their C terminus to a mouse IgG2a Fc domain carrying a C-terminal V5 tag. Chimeric proteins were expressed in modified proprietary HEK293 cells and purified to homogeneity with a yield of 80–100 mg per 600 ml of medium (National Research Council Canada, Biotechnology Research Institute).
sdAb-Fcs Affinity Measurements to PCSK9
Affinity measurements were performed by the XTAL Biostructures, Inc. Briefly, PCSK9 carrying a C-terminal His tag was captured on a NiHC1000M chip non-covalently at various low densities to determine kinetic binding parameters of the four different sdAb-Fcs. All binding experiments were performed at 25 °C on a BiOptix 404pi enhanced surface plasmon resonance instrument. A 7-point, 3-fold dilution of sdAbs starting at 200 nm was injected over each of three PCSK9 densities, and the sensorgrams were globally fitted with a floating Rmax using Scrubber2 software. Fits across all three ligand densities matched well, suggesting little influence from avidity due to bivalency of the antibodies to the calculated binding parameters.
Cell Culture
Human hepatocellular carcinoma HepG2 cells from the American Type Culture Collection (ATCC) were grown in Eagle's minimal essential medium (Wisent). Cells were stably transfected with either wild type (WT) or D374Y GOF PCSK9 using the FuGENE HD transfection reagent (Promega) and grown with 600 μg/ml G418 (Wisent) for selection of cells stably expressing the cDNA coding for PCSK9. IHH were generously provided by Dr. H. Moshage from the University Hospital Groningen, the Netherlands (71), and were grown in William's E medium (Wisent). HEK293 cells were grown in DMEM. All cells were maintained at 37 °C under 5% CO2.
sdAb-Fc Assays
Purified PCSK9 (69) or conditioned media from HEK293 cells overexpressing either WT or D374Y PCSK9 were preincubated for 1 h at 37 °C with various amounts of purified sdAb-Fcs. The mixture was then applied onto cells for 4–24 h. For experiments with HepG2 cells stably overexpressing WT or D374Y PCSK9, sdAb-Fcs were diluted in the culture medium and directly added to cells. In separate experiments we also incubated the above cells with various concentrations of the inhibitory mAb evolocumab, originally denoted as Amgen's AMG-145 (49), kindly supplied by Dr. Robert Dufour (Institut de Recherches Cliniques de Montréal).
DiI-LDL Uptake Assay
HepG2 cells were plated at a density of 25,000 cells/well in 96-well plates (Corning) in Eagle's minimal essential medium containing 10% FBS. After 18–20 h, cells were washed and switched to serum-free medium (SFM) containing 5% lipoprotein-deficient serum for 24 h. Purified PCSK9 and sdAb-Fcs were pre-incubated at 37 °C in the same medium and added onto the cells (three wells/condition). After 3 h at 37 °C, 10 μl of DiI-LDL (Biomedical Technologies) were added to the cell medium (5 μg/ml), and cells were further incubated for 2 h. After three washes in D-PBS (without calcium and magnesium; Wisent), plates were scanned on a SpectraMax i3 Multi-Mode Detection Platform (Molecular Devices). DiI-LDL uptake was measured in each well as an average fluorescence intensity. Plates were then frozen overnight (−80 °C) and used the next day to perform a CyQuant cell assay (Invitrogen) for normalizing the DiI-LDL uptake to the number of cells in each well.
Immunofluorescence
About 70,000 HepG2 cells/well were seeded in 24-well plates containing coverslips (Fisher) coated with polylysine (Sigma) and cultured overnight in complete medium and in SFM containing 5% lipoprotein-deficient serum for the following 24 h. Following a 4-h incubation with PCSK9 (3 μg/ml) alone or in combination with each sdAb-Fc (1.2 μm), cells were rinsed with cold PBS, incubated 10 min on ice with LDLR antibody (1:200 in SFM; mouse, R&D Systems), rinsed twice with cold PBS, and finally fixed with 4% paraformaldehyde for 10 min on ice and 5 min at room temperature. After two washes in PBS at room temperature and a 30-min incubation with PBS containing 1% BSA, LDLR-antibody complexes were revealed by a 5-min incubation with anti-mouse Alexa Fluor 555-tagged antibody (1:200; goat, Molecular Probes). Cells were then rinsed twice with PBS, and nuclei were stained with DAPI during the final mounting step (ProLong Gold Antifade Reagent with DAPI, Life Technologies, Inc.). Immunofluorescence analyses were performed with a confocal microscope (Zeiss LSM-710) and the Volocity software (x64, PerkinElmer Life Sciences). Because the parameters remained unchanged throughout image acquisition, the values obtained were proportional to LDLR levels (47).
Western Blotting
Cells were washed twice with ice-cold PBS and lysed 40 min on ice with radioimmunoprecipitation assay buffer supplemented with 1× complete protease inhibitor mixture (Roche Applied Science). The cell lysates were then subjected to 8% Tris-glycine SDS-PAGE. The gels were transferred overnight on PVDF membranes (Millipore) that were blocked for 1 h at room temperature in TBST (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.1% Tween 20) containing 5% nonfat dry milk. Membranes were then incubated overnight at 4 °C in 5% milk/TBST with human (goat, R&D Systems) or mouse (goat, Novus Biologicals) LDLR antibody (1:1,000) and human β-actin antibody (mouse, Sigma; 1:2,500) for normalization. After incubation with secondary antibodies coupled to horseradish peroxidase (HRP) at 1:10,000 in 5% milk/TBST, immunoreactive bands were revealed by chemiluminescence using the ECL Plus kit (GE Healthcare). Quantification was performed with ImageJ software or using ChemiDoc MP imaging system (Bio-Rad).
Mouse Primary Hepatocytes
Primary hepatocytes were isolated from the liver of 6–14-week-old mice using a two-step collagenase perfusion method as described previously (40). Fibronectin-coated (0.5 mg/ml, Sigma) plates (24-well) were seeded with ∼105 cells/well in Williams' medium E (Wisent) containing 10% FBS. After 2 h, the medium was replaced with HepatoZYME medium (Gibco) for 12 h prior to treatment.
LDLR ELISA
Cells were lysed for 40 min on ice at 4 °C with ice-cold non-denaturing cell lysis buffer (20 mm Tris-HCl, pH 8, 137 mm NaCl, 2 mm Na2EDTA, 1% Nonidet P-40, 10% glycerol, 4% protease inhibitor mixture without EDTA). Cell lysates were pooled for each experiment; total human LDLR was measured by ELISA (human LDLR DuoSet ELISA Development kit, R&D Systems) and protein levels by a protein assay (Bio-Rad DC). Briefly, a high binding 96-well plate was coated and incubated overnight at room temperature with mouse anti-human LDLR antibody (4 μg/ml) diluted in PBS. The next day, the plate washed four times with PBS, 0.05% Tween 20, and blocking was carried out with PBS, 1% + BSA, 0.05% + Tween 20, 0.01% sodium azide for 1.5 h at room temperature. Following four washes, standards (7 points, 2-fold serial dilutions starting at 8 ng/ml, and a zero point) and pooled cell lysates (5-fold dilution in PBS/1% BSA) were added to the plate and incubated for 2 h at room temperature. After four washes, detection was carried out with biotinylated goat anti-human LDLR antibody (0.4 μg/ml, diluted in ELISA buffer: 20 mm Tris-HCl, pH 7.2–7.4, 137 mm NaCl, 0.05% Tween 20, 0.1% BSA) for 2 h at room temperature. Following four washes, streptavidin-HRP diluted in ELISA buffer (1:200) was added and incubated for 20 min at room temperature, protected from direct light. After four washes, substrate solution containing a 1:1 mixture of H2O2 and tetramethylbenzidine (R&D Systems) was added to the wells and incubated in the dark for 20 min at room temperature. The reaction was stopped with 2 n sulfuric acid (R&D Systems). The optical density of the yellow-colored product was determined using a SpectraMax i3 plate reader (Molecular Devices) set to 450 nm, from which readings at 540 nm were subtracted to correct for any plate imperfections. The total amount of LDLR was normalized to the quantity of total protein measured in each cell lysate.
Expression of sdAb-Fc Fusion Proteins Carrying a KDEL C-terminal Tag
The pIRES2-EGFP vector expressing the sdAb-Fc fusion protein carrying a KDEL ER-retention signal at the C terminus of the protein, downstream of the V5 tag, was obtained as described previously (7). HEK293 cells (∼300,000 cells/well) were seeded in 12-well plates in complete media, transfected overnight with various constructs using FuGENE HD, and switched to SFM for another 24 h.
Expression of EGFs-Fc Fusion Proteins
A DNA sequence encoding amino acids 314–393 of GenBankTM accession number P01130 (the epidermal growth factor-like repeat AB domain (EGF-AB) of human LDLR) was synthesized by GeneArt (Thermo Fisher). Both sequences were ligated between a signal peptide and the sequence of human IgG1 Fc in the plant expression/transformation vector pTRAkc (72). The clones were transformed into Agrobacterium tumefaciens, which was then used for transient expression in Nicotiana benthamiana (73). The recombinant proteins were purified by protein A affinity chromatography.
Fluorescence-associated Cell Sorting (FACS) Analysis
About 500,000 HepG2 cells/well were seeded in 12-well plates in complete media overnight and in SFM for 24 h. Cells were then incubated with PCSK9 (15 μg/ml) for 4 h alone, with sdAb-Fc (0.04 μm), with increasing concentrations of EGF-AB-Fc (Planet Biotechnology Inc.), or their Fc control. Cells were rinsed in FACS buffer (D-PBS containing 0.5% BSA and 1 g/liter glucose (Wisent)), then in 2.5 mm EDTA-2Na-2H2O (Bioshop), and incubated for 20 min at 37 °C in 0.5 ml of EDTA-2Na-2H2O. Detached cells were collected, centrifuged for 5 min at 900 rpm at 4 °C, re-suspended in 0.5 ml of FACS buffer containing 1:250 of human LDLR antibody (R&D Systems), and incubated for 10 min on ice. Cells were then washed with 5 ml of FACS buffer, centrifuged, resuspended in 0.5 ml of FACS buffer containing 1:500 of goat Alexa Fluor 647-labeled anti-mouse antibody (Molecular Probes), and incubated for 5 min. Cells were washed again, re-suspended in 0.3 ml of FACS buffer containing1.67 μg/ml propidium iodide (Sigma), and finally analyzed by FACS using a CyAn flow cytometer (Beckman Coulter).
Animal Studies
The RPCI human BAC library 11 was screened by PCR for the presence of the PCSK9 gene, and the clone 55M23 was identified. A 67.5-kb fragment of the BAC was injected into B6C3F1 fertilized eggs. Mice were then backcrossed to Pcsk9 KO mice in C57BL/6J background for five generations. Thus, these mice only express human PCSK9 and no mouse orthologue. Mice were injected in the tail vein with vehicle (PBS) or 10 mg/kg sdAb-Fc. For single injections mice were analyzed 4 days post-injection, and for double injections, mice were injected at day 0 and 7 and sacrificed at day 11. In both cases blood samples and livers were collected. Total cholesterol levels were determined by the colorimetric assay Infinity (Thermo Scientific) and apoB levels by ELISA (Cloud-Clone Corp.). Liver LDLR levels were measured by Western blot using a mouse LDLR antibody, as for primary hepatocytes.
Statistics
Two-tailed Student's t test was performed to assess the statistical significance of the data sets. For the DiI-LDL uptake assay, a two-way ANOVA was performed when triplicates were repeated more than once.
Author Contributions
E. W. conducted most of the experiments and analyzed the results. D. S. R. conducted several early experiments, including those on DiI-LDL uptake. R. E. prepared the primary hepatocytes and conceived the mouse model. J. H. did most of the cloning work, and M. C. A. contributed to cell culture. Y. A. performed one of the co-immunoprecipitations. K. L. W. provided the EGFs-Fc proteins. J. Z. produced and screened the llama library. A. P. analyzed the data and wrote the manuscript with E. W. and N. G. S., who also conceived and followed the project. S. N. provided the original screen for the best sdAbs that bind PCSK9 from the llama library and their functional analysis in cell lines.
Acknowledgments
We are grateful to all the members of the Seidah laboratory for technical support and discussions. We are grateful to Suzanne Benjannet, who helped us with some experiments. We thank Drs. Rex Parker and Franck Duclos (Bristol Myers Squibb) for the donation of pure human PCSK9; Manon Laprise, Anna Roubtsova, and Jadwiga Marcinkiewicz for animal experimentation; and Dr. Dominic Filion for help in confocal imaging analysis. We also thank Shenghua Li for sdAb production in Dr. Zhang's laboratory, and Drs. Archana Belle and James Maclean from Planet Biotechnology who worked on the development of EGFs+Fc. Finally, we acknowledge the precious help of Dr. Robert Dufour from the Institut de Recherches Cliniques de Montréal for the generous gift of evolocumab.
This work was supported by Canadian Institutes of Health Research Grants PP2-125775 and MOP 102741, Pfizer cardiovascular research awards, a grant from Amorchem, Canada Research Chair 216684, and Fondation Leducq Grant 13CVD03.
- LDLc
- LDL-cholesterol
- apoB
- apolipoprotein B
- CHRD
- cysteine- and histidine-rich domain
- CVD
- cardiovascular disease
- ER
- endoplasmic reticulum
- FH
- familial hypercholesterolemia
- GOF
- gain-of-function
- LDLR
- low density lipoprotein receptor
- sdAb
- single domain antibody
- SFM
- serum-free media
- Tg
- transgene
- endo
- endoglycosidase
- WB
- Western blotting
- IHH
- immortalized human primary hepatocyte
- DiI
- 1,1′-dioctadecyl 3,3,3′,3′-tetramethylindocarbocyanine perchlorate fluorescent carbocyanide dye
- BAC
- bacterial artificial chromosome.
References
- 1. Cholesterol Treatment Trialists' (CTT) Collaboration, Baigent C., Blackwell L., Emberson J., Holland L. E., Reith C., Bhala N., Peto R., Barnes E. H., Keech A., Simes J., and Collins R. (2010) Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 376, 1670–1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hobbs H. H., Brown M. S., and Goldstein J. L. (1992) Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum. Mutat. 1, 445–466 [DOI] [PubMed] [Google Scholar]
- 3. Seidah N. G., Benjannet S., Wickham L., Marcinkiewicz J., Jasmin S. B., Stifani S., Basak A., Prat A., and Chretien M. (2003) The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc. Natl. Acad. Sci. U.S.A. 100, 928–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Abifadel M., Varret M., Rabs J. P., Allard D., Ouguerram K., Devillers M., Cruaud C., Benjannet S., Wickham L., Erlich D., Derré A., Villéger L., Farnier M., Beucler I., Bruckert E., Chambaz J., Chanu B., Lecerf J. M., Luc G., Moulin P., Weissenbach J., Prat A., Krempf M., Junien C., Seidah N. G., and Boileau C. (2003) Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 34, 154–156 [DOI] [PubMed] [Google Scholar]
- 5. Seidah N. G., and Prat A. (2012) The biology and therapeutic targeting of the proprotein convertases. Nat. Rev. Drug. Discov. 11, 367–383 [DOI] [PubMed] [Google Scholar]
- 6. Cunningham D., Danley D. E., Geoghegan K. F., Griffor M. C., Hawkins J. L., Subashi T. A., Varghese A. H., Ammirati M. J., Culp J. S., Hoth L. R., Mansour M. N., McGrath K. M., Seddon A. P., Shenolikar S., Stutzman-Engwall K. J., et al. (2007) Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat. Struct. Mol. Biol. 14, 413–419 [DOI] [PubMed] [Google Scholar]
- 7. Nassoury N., Blasiole D. A., Tebon Oler A., Benjannet S., Hamelin J., Poupon V., McPherson P. S., Attie A. D., Prat A., and Seidah N. G. (2007) The cellular trafficking of the secretory proprotein convertase PCSK9 and its dependence on the LDLR. Traffic 8, 718–732 [DOI] [PubMed] [Google Scholar]
- 8. Zhang D. W., Lagace T. A., Garuti R., Zhao Z., McDonald M., Horton J. D., Cohen J. C., and Hobbs H. H. (2007) Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat a of low density lipoprotein receptor decreases receptor recycling and increases degradation. J. Biol. Chem. 282, 18602–18612 [DOI] [PubMed] [Google Scholar]
- 9. Maxwell K. N., and Breslow J. L. (2004) Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc. Natl. Acad. Sci. U.S.A. 101, 7100–7105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Benjannet S., Rhainds D., Essalmani R., Mayne J., Wickham L., Jin W., Asselin M. C., Hamelin J., Varret M., Allard D., Trillard M., Abifadel M., Tebon A., Attie A. D., Rader D. J., et al. (2004) NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J. Biol. Chem. 279, 48865–48875 [DOI] [PubMed] [Google Scholar]
- 11. Park S. W., Moon Y. A., and Horton J. D. (2004) Post-transcriptional regulation of low density lipoprotein receptor protein by proprotein convertase subtilisin/kexin type 9a in mouse liver. J. Biol. Chem. 279, 50630–50638 [DOI] [PubMed] [Google Scholar]
- 12. Lo Surdo P., Bottomley M. J., Calzetta A., Settembre E. C., Cirillo A., Pandit S., Ni Y. G., Hubbard B., Sitlani A., and Carfí A. (2011) Mechanistic implications for LDL receptor degradation from the PCSK9/LDLR structure at neutral pH. EMBO Rep. 12, 1300–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Poirier S., Mayer G., Benjannet S., Bergeron E., Marcinkiewicz J., Nassoury N., Mayer H., Nimpf J., Prat A., and Seidah N. G. (2008) The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. J. Biol. Chem. 283, 2363–2372 [DOI] [PubMed] [Google Scholar]
- 14. Shan L., Pang L., Zhang R., Murgolo N. J., Lan H., and Hedrick J. A. (2008) PCSK9 binds to multiple receptors and can be functionally inhibited by an EGF-A peptide. Biochem. Biophys. Res. Commun. 375, 69–73 [DOI] [PubMed] [Google Scholar]
- 15. Canuel M., Sun X., Asselin M.-C., Paramithiotis E., Prat A., and Seidah N. G. (2013) Proprotein convertase subtilisin/kexin type 9 (PCSK9) can mediate degradation of the low density lipoprotein receptor-related protein 1 (LRP-1). PLoS ONE 8, e64145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang S., Henry L., Ho Y. K., Pownall H. J., and Rudenko G. (2010) Mechanism of LDL binding and release probed by structure-based mutagenesis of the LDL receptor. J. Lipid Res. 51, 297–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Holla Ø. L., Cameron J., Berge K. E., Ranheim T., and Leren T. P. (2007) Degradation of the LDL receptors by PCSK9 is not mediated by a secreted protein acted upon by PCSK9 extracellularly. BMC Cell Biol. 8, 9–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang D. W., Garuti R., Tang W. J., Cohen J. C., and Hobbs H. H. (2008) Structural requirements for PCSK9-mediated degradation of the low-density lipoprotein receptor. Proc. Natl. Acad. Sci. U.S.A. 105, 13045–13050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Timms K. M., Wagner S., Samuels M. E., Forbey K., Goldfine H., Jammulapati S., Skolnick M. H., Hopkins P. N., Hunt S. C., and Shattuck D. M. (2004) A mutation in PCSK9 causing autosomal-dominant hypercholesterolemia in a Utah pedigree. Hum. Genet. 114, 349–353 [DOI] [PubMed] [Google Scholar]
- 20. Cohen J., Pertsemlidis A., Kotowski I. K., Graham R., Garcia C. K., and Hobbs H. H. (2005) Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 37, 161–165 [DOI] [PubMed] [Google Scholar]
- 21. Kotowski I. K., Pertsemlidis A., Luke A., Cooper R. S., Vega G. L., Cohen J. C., and Hobbs H. H. (2006) A spectrum of PCSK9 alleles contributes to plasma levels of low-density lipoprotein cholesterol. Am. J. Hum. Genet. 78, 410–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Seidah N. G., Awan Z., Chrétien M., and Mbikay M. (2014) PCSK9: a key modulator of cardiovascular health. Circ. Res. 114, 1022–1036 [DOI] [PubMed] [Google Scholar]
- 23. Seidah N. G. (2009) PCSK9 as a therapeutic target of dyslipidemia. Expert Opin. Therapeutic Targets 13, 19–28 [DOI] [PubMed] [Google Scholar]
- 24. Stein E. A., and Raal F. (2014) Reduction of low-density lipoprotein cholesterol by monoclonal antibody inhibition of PCSK9. Annu. Rev. Med. 65, 417–431 [DOI] [PubMed] [Google Scholar]
- 25. McLaughlin P., Grillo-López A. J., Link B. K., Levy R., Czuczman M. S., Williams M. E., Heyman M. R., Bence-Bruckler I., White C. A., Cabanillas F., Jain V., Ho A. D., Lister J., Wey K., Shen D., and Dallaire B. K. (1998) Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J. Clin. Oncol. 16, 2825–2833 [DOI] [PubMed] [Google Scholar]
- 26. Lundin J., Kimby E., Björkholm M., Broliden P. A., Celsing F., Hjalmar V., Möllgård L., Rebello P., Hale G., Waldmann H., Mellstedt H., and Osterborg A. (2002) Phase II trial of subcutaneous anti-CD52 monoclonal antibody alemtuzumab (Campath-1H) as first-line treatment for patients with B-cell chronic lymphocytic leukemia (B-CLL). Blood 100, 768–773 [DOI] [PubMed] [Google Scholar]
- 27. Cobleigh M. A., Vogel C. L., Tripathy D., Robert N. J., Scholl S., Fehrenbacher L., Wolter J. M., Paton V., Shak S., Lieberman G., and Slamon D. J. (1999) Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J. Clin. Oncol. 17, 2639–2648 [DOI] [PubMed] [Google Scholar]
- 28. Shrank W. H., Barlow J. F., and Brennan T. A. (2015) New therapies in the treatment of high cholesterol: an argument to return to goal-based lipid guidelines. JAMA 314, 1443–1444 [DOI] [PubMed] [Google Scholar]
- 29. Genest J., McPherson R., Frohlich J., Anderson T., Campbell N., Carpentier A., Couture P., Dufour R., Fodor G., Francis G. A., Grover S., Gupta M., Hegele R. A., Lau D. C., Leiter L., et al. (2009) 2009 Canadian Cardiovascular Society/Canadian guidelines for the diagnosis and treatment of dyslipidemia and prevention of cardiovascular disease in the adult- 2009 recommendations. Can. J. Cardiol. 25, 567–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hamers-Casterman C., Atarhouch T., Muyldermans S., Robinson G., Hamers C., Songa E. B., Bendahman N., and Hamers R. (1993) Naturally occurring antibodies devoid of light chains. Nature 363, 446–448 [DOI] [PubMed] [Google Scholar]
- 31. Muyldermans S. (2013) Nanobodies: natural single-domain antibodies. Annu. Rev. Biochem. 82, 775–797 [DOI] [PubMed] [Google Scholar]
- 32. van der Linden R., de Geus B., Stok W., Bos W., van Wassenaar D., Verrips T., and Frenken L. (2000) Induction of immune responses and molecular cloning of the heavy chain antibody repertoire of Lama glama. J. Immunol. Methods 240, 185–195 [DOI] [PubMed] [Google Scholar]
- 33. Vincke C., Loris R., Saerens D., Martinez-Rodriguez S., Muyldermans S., and Conrath K. (2009) General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J. Biol. Chem. 284, 3273–3284 [DOI] [PubMed] [Google Scholar]
- 34. Bartunek J., Barbato E., Heyndrickx G., Vanderheyden M., Wijns W., and Holz J. B. (2013) Novel antiplatelet agents: ALX-0081, a Nanobody directed towards von Willebrand factor. J. Cardiovasc Transl Res. 6, 355–363 [DOI] [PubMed] [Google Scholar]
- 35. Kratz F., and Elsadek B. (2012) Clinical impact of serum proteins on drug delivery. J. Control Release 161, 429–445 [DOI] [PubMed] [Google Scholar]
- 36. Sarker S. A., Jäkel M., Sultana S., Alam N. H., Bardhan P. K., Chisti M. J., Salam M. A., Theis W., Hammarström L., and Frenken L. G. (2013) Anti-rotavirus protein reduces stool output in infants with diarrhea: a randomized placebo-controlled trial. Gastroenterology 145, 740–748 [DOI] [PubMed] [Google Scholar]
- 37. Saavedra Y. G., Day R., and Seidah N. G. (2012) The M2 module of the Cys-His-rich domain (CHRD) of PCSK9 is needed for the extracellular low density lipoprotein receptor (LDLR) degradation pathway. J. Biol. Chem. 287, 43492–43501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Powers D. B., Amersdorfer P., Poul M., Nielsen U. B., Shalaby M. R., Adams G. P., Weiner L. M., and Marks J. D. (2001) Expression of single-chain Fv-Fc fusions in Pichia pastoris. J. Immunol. Methods 251, 123–135 [DOI] [PubMed] [Google Scholar]
- 39. Zaid A., Roubtsova A., Essalmani R., Marcinkiewicz J., Chamberland A., Hamelin J., Tremblay M., Jacques H., Jin W., Davignon J., Seidah N. G., and Prat A. (2008) Proprotein convertase subtilisin/kexin type 9 (PCSK9): Hepatocyte-specific low-density lipoprotein receptor degradation and critical role in mouse liver regeneration. Hepatology 48, 646–654 [DOI] [PubMed] [Google Scholar]
- 40. Essalmani R., Susan-Resiga D., Chamberland A., Abifadel M., Creemers J. W., Boileau C., Seidah N. G., and Prat A. (2011) In vivo evidence that furin from hepatocytes inactivates PCSK9. J. Biol. Chem. 286, 4257–4263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ly K., Saavedra Y. G., Canuel M., Routhier S., Desjardins R., Hamelin J., Mayne J., Lazure C., Seidah N. G., and Day R. (2014) Annexin A2 reduces PCSK9 protein levels via a translational mechanism and interacts with the M1 and M2 domains of PCSK9. J. Biol. Chem. 289, 17732–17746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Benjannet S., Hamelin J., Chrétien M., and Seidah N. G. (2012) Loss- and gain-of-function PCSK9 variants: cleavage specificity, dominant negative effects, and low density lipoprotein receptor (LDLR) degradation. J. Biol. Chem. 287, 33745–33755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pedersen N. B., Wang S., Narimatsu Y., Yang Z., Halim A., Schjoldager K. T., Madsen T. D., Seidah N. G., Bennett E. P., Levery S. B., and Clausen H. (2014) Low density lipoprotein receptor class A repeats are O-glycosylated in linker regions. J. Biol. Chem. 289, 17312–17324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pelham H. R. (1988) Evidence that luminal ER proteins are sorted from secreted proteins in a post-ER compartment. EMBO J. 7, 913–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Poirier S., Mamarbachi M., Chen W. T., Lee A. S., and Mayer G. (2015) GRP94 Regulates circulating cholesterol levels through blockade of PCSK9-induced LDLR degradation. Cell Rep. 13, 2064–2071 [DOI] [PubMed] [Google Scholar]
- 46. Poirier S., Mayer G., Poupon V., McPherson P. S., Desjardins R., Ly K., Asselin M. C., Day R., Duclos F. J., Witmer M., Parker R., Prat A., and Seidah N. G. (2009) Dissection of the endogenous cellular pathways of PCSK9-induced LDLR degradation: evidence for an intracellular route. J. Biol. Chem. 284, 28856–28864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Roubtsova A., Munkonda M. N., Awan Z., Marcinkiewicz J., Chamberland A., Lazure C., Cianflone K., Seidah N. G., and Prat A. (2011) Circulating proprotein convertase subtilisin/kexin 9 (PCSK9) regulates VLDLR protein and triglyceride accumulation in visceral adipose tissue. Arterioscler. Thromb. Vasc. Biol. 31, 785–791 [DOI] [PubMed] [Google Scholar]
- 48. Roubtsova A., Chamberland A., Marcinkiewicz J., Essalmani R., Fazel A., Bergeron J. J., Seidah N. G., and Prat A. (2015) PCSK9 deficiency unmasks a sex/tissue-specific subcellular distribution of the LDL and VLDL receptors in mice. J. Lipid Res. 56, 2133–2142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chan J. C., Piper D. E., Cao Q., Liu D., King C., Wang W., Tang J., Liu Q., Higbee J., Xia Z., Di Y., Shetterly S., Arimura Z., Salomonis H., Romanow W. G., et al. (2009) A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates. Proc. Natl. Acad. Sci. U.S.A. 106, 9820–9825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Marcinkiewicz M., Ramla D., Seidah N. G., and Chrétien M. (1994) Developmental expression of the prohormone convertases PC1 and PC2 in mouse pancreatic islets. Endocrinology 135, 1651–1660 [DOI] [PubMed] [Google Scholar]
- 51. Ni Y. G., Condra J. H., Orsatti L., Shen X., Di Marco S., Pandit S., Bottomley M. J., Ruggeri L., Cummings R. T., Cubbon R. M., Santoro J. C., Ehrhardt A., Lewis D., Fisher T. S., Ha S., et al. (2010) A proprotein convertase subtilisin-like/kexin type 9 (PCSK9) C-terminal domain antibody antigen-binding fragment inhibits PCSK9 internalization and restores low density lipoprotein uptake. J. Biol. Chem. 285, 12882–12891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schiele F., Park J., Redemann N., Luippold G., and Nar H. (2014) An antibody against the C-terminal domain of PCSK9 lowers LDL cholesterol levels in vivo. J. Mol. Biol. 426, 843–852 [DOI] [PubMed] [Google Scholar]
- 53. Butkinaree C., Canuel M., Essalmani R., Poirier S., Benjannet S., Asselin M.-C., Roubtsova A., Hamelin J., Marcinkiewicz J., Chamberland A., Guillemot J., Mayer G., Sisodia S. S., Jacob Y., Prat A., and Seidah N. G. (2015) Amyloid precursor-like protein 2 and sortilin do not regulate the PCSK9-mediated low density lipoprotein receptor degradation but interact with each other. J. Biol. Chem. 290, 18609–18620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang Y., Zhou L., Kong-Beltran M., Li W., Moran P., Wang J., Quan C., Tom J., Kolumam G., Elliott J. M., Skelton N. J., Peterson A. S., and Kirchhofer D. (2012) Calcium-independent inhibition of PCSK9 by affinity-improved variants of the LDL receptor EGF(A) domain. J. Mol. Biol. 422, 685–696 [DOI] [PubMed] [Google Scholar]
- 55. Ni Y. G., Di Marco S., Condra J. H., Peterson L. B., Wang W., Wang F., Pandit S., Hammond H. A., Rosa R., Cummings R. T., Wood D. D., Liu X., Bottomley M. J., Shen X., Cubbon R. M., et al. (2011) A PCSK9-binding antibody that structurally mimics the EGF(A) domain of LDL-receptor reduces LDL cholesterol in vivo. J. Lipid Res. 52, 78–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mitchell T., Chao G., Sitkoff D., Lo F., Monshizadegan H., Meyers D., Low S., Russo K., DiBella R., Denhez F., Gao M., Myers J., Duke G., Witmer M., Miao B., et al. (2014) Pharmacologic profile of the adnectin BMS-962476, a small protein biologic alternative to PCSK9 antibodies for low-density lipoprotein lowering. J. Pharmacol. Exp. Ther. 350, 412–424 [DOI] [PubMed] [Google Scholar]
- 57. Graham M. J., Lemonidis K. M., Whipple C. P., Subramaniam A., Monia B. P., Crooke S. T., and Crooke R. M. (2007) Antisense inhibition of proprotein convertase subtilisin/kexin type 9 reduces serum LDL in hyperlipidemic mice. J. Lipid Res. 48, 763–767 [DOI] [PubMed] [Google Scholar]
- 58. Gupta N., Fisker N., Asselin M. C., Lindholm M., Rosenbohm C., Ørum H., Elmén J., Seidah N. G., and Straarup E. M. (2010) A locked nucleic acid antisense oligonucleotide (LNA) silences PCSK9 and enhances LDLR expression in vitro and in vivo. PLoS ONE 5, e10682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yamamoto T., Harada-Shiba M., Nakatani M., Wada S., Yasuhara H., Narukawa K., Sasaki K., Shibata M. A., Torigoe H., Yamaoka T., Imanishi T., and Obika S. (2012) Cholesterol-lowering action of BNA-based antisense oligonucleotides targeting PCSK9 in atherogenic diet-induced hypercholesterolemic mice. Mol. Ther. Nucleic Acids 1, e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Frank-Kamenetsky M., Grefhorst A., Anderson N. N., Racie T. S., Bramlage B., Akinc A., Butler D., Charisse K., Dorkin R., Fan Y., Gamba-Vitalo C., Hadwiger P., Jayaraman M., John M., Jayaprakash K. N., et al. (2008) Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc. Natl. Acad. Sci. U.S.A. 105, 11915–11920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Galabova G., Brunner S., Winsauer G., Juno C., Wanko B., Mairhofer A., Lührs P., Schneeberger A., von Bonin A., Mattner F., Schmidt W., and Staffler G. (2014) Peptide-based anti-PCSK9 vaccines- an approach for long-term LDLc management. PLoS ONE 9, e114469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Crossey E., Amar M. J., Sampson M., Peabody J., Schiller J. T., Chackerian B., and Remaley A. T. (2015) A cholesterol-lowering VLP vaccine that targets PCSK9. Vaccine 33, 5747–5755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cortez-Retamozo V., Lauwereys M., Hassanzadeh Gh G., Gobert M., Conrath K., Muyldermans S., De Baetselier P., and Revets H. (2002) Efficient tumor targeting by single-domain antibody fragments of camels. Int. J. Cancer 98, 456–462 [DOI] [PubMed] [Google Scholar]
- 64. Denis M., Marcinkiewicz J., Zaid A., Gauthier D., Poirier S., Lazure C., Seidah N. G., and Prat A. (2012) Gene inactivation of PCSK9 reduces atherosclerosis in mice. Circulation 125, 894–901 [DOI] [PubMed] [Google Scholar]
- 65. Guella I., Asselta R., Ardissino D., Merlini P. A., Peyvandi F., Kathiresan S., Mannucci P. M., Tubaro M., and Duga S. (2010) Effects of PCSK9 genetic variants on plasma LDL cholesterol levels and risk of premature myocardial infarction in the Italian population. J. Lipid Res. 51, 3342–3349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Huang Y., Ballinger D. G., Stokowski R., Beilharz E., Robinson J. G., Liu S., Robinson R. D., Henderson V. W., Rossouw J. E., and Prentice R. L. (2012) Exploring the interaction between SNP genotype and postmenopausal hormone therapy effects on stroke risk. Genome Med. 4, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. dos Santos C., and Marshall J. C. (2014) Bridging lipid metabolism and innate host defense. Sci. Transl. Med. 6, 258fs41. [DOI] [PubMed] [Google Scholar]
- 68. Sun X., Essalmani R, Day R, Khatib A. M., Seidah N. G., and Prat A. (2012) Proprotein convertase subtilisin/kexin type 9 deficiency reduces melanoma metastasis in liver. Neoplasia 14, 1122–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Benjannet S., Saavedra Y. G., Hamelin J., Asselin M. C., Essalmani R., Pasquato A., Lemaire P., Duke G., Miao B., Duclos F., Parker R., Mayer G., and Seidah N. G. (2010) Effects of the prosegment and pH on the activity of PCSK9: evidence for additional processing events. J. Biol. Chem. 285, 40965–40978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hussack G., Arbabi-Ghahroudi M., van Faassen H., Songer J. G., Ng K. K., MacKenzie R., and Tanha J. (2011) Neutralization of Clostridium difficile toxin A with single-domain antibodies targeting the cell receptor binding domain. J. Biol. Chem. 286, 8961–8976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Schippers I. J., Moshage H., Roelofsen H., Müller M., Heymans H. S., Ruiters M., and Kuipers F. (1997) Immortalized human hepatocytes as a tool for the study of hepatocytic (de-)differentiation. Cell Biol. Toxicol. 13, 375–386 [DOI] [PubMed] [Google Scholar]
- 72. Maclean J., Koekemoer M., Olivier A. J., Stewart D., Hitzeroth I. I., Rademacher T., Fischer R., Williamson A. L., and Rybicki E. P. (2007) Optimization of human papillomavirus type 16 (HPV-16) L1 expression in plants: comparison of the suitability of different HPV-16 L1 gene variants and different cell-compartment localization. J. Gen. Virol. 88, 1460–1469 [DOI] [PubMed] [Google Scholar]
- 73. Voinnet O., Rivas S., Mestre P., and Baulcombe D. (2003) An enhanced transient expression system in plants based on suppression of gene silencing by the p19 protein of tomato bushy stunt virus. Plant J. 33, 949–956 [DOI] [PubMed] [Google Scholar]