

Abstract
Interleukin-35 (IL-35) is a newly described member of the IL-12 family. It has been reported to inhibit inflammation and autoimmune inflammatory disease and can increase apoptotic sensitivity. Little is known about the role of IL-35 during viral infection. Herein, high levels of IL-35 were found in peripheral blood mononuclear cells and throat swabs from patients with seasonal influenza A virus (IAV) relative to healthy individuals. IAV infection of human lung epithelial and primary cells increased levels of IL-35 mRNA and protein. Further studies demonstrated that IAV-induced IL-35 transcription is regulated by NF-κB. IL-35 expression was significantly suppressed by selective inhibitors of cyclooxygenase-2 (COX-2) and inducible nitric-oxide synthase, indicating their involvement in IL-35 expression. Interestingly, IL-35 production may have suppressed IAV RNA replication and viral protein synthesis via induction of type I and III interferons (IFN), leading to activation of downstream IFN effectors, including double-stranded RNA-dependent protein kinase, 2′,5′-oligoadenylate synthetase, and myxovirus resistance protein. IL-35 exhibited extensive antiviral activity against the hepatitis B virus, enterovirus 71, and vesicular stomatitis virus. Our results demonstrate that IL-35 is a novel IAV-inducible cytokine, and its production elicits antiviral activity.
Keywords: antiviral agent, host defense, host-pathogen interaction, influenza virus, innate immunity, interferon, interleukin, NF-κB (NF-KB), viral infection
Introduction
Seasonal influenza A virus (IAV)2 is a common human infection, resulting in significant morbidity and mortality (1). It is a negative-sense RNA virus of the Orthomyxoviridae family (2). IAV attaches to membrane receptors and enters epithelial, macrophage, and dendritic cells (3). These cells release large quantities of antiviral and immunostimulatory cytokines during IAV infection. An extensive array of cytokines and chemokines are produced by host cells in response to IAV infection, including interleukin-1(IL-1), IL-2, IL-8, IL-10, IL-15, IL-18, IL-27, IL-32, TNF-α, IFN-α/β/γ, MIP-1α/β (macrophage inflammatory protein-1 α/β) (4–7), and other cytokines and related pathway factors (1, 8). There remain many unknown cytokines induced by and involved in the response to IAV infection. Further investigation of factors activated by IAV infection is critical to better understand host-virus interaction and advance antiviral research.
IL-35 is the newest member of the IL-12 family, which includes IL-12, IL-23, and IL-27 (9). These proteins are composed of an α (p19, p28, p35) and β (p40, Epstein-Barr virus-induced gene 3 (EBI3)) chain (10). IL-35, originally named by Niedbala et al. (11), consists of an IL-12/p35 and IL27/EBI3 chain. EBI3 encodes a 34-kDa glycoprotein homologous to IL-12/p40 (12). EBI3 heterodimerizes with p35, forming a hematoprotein in BJAB B lymphoma, COS7, and human placental trophoblast cells (13). IL-35 signals conduct through either a unique heterodimer of IL-12Rb2 and gp130 or homodimers of each chain (14).
IL-35 has been linked to various disease, including autoimmune encephalomyelitis (15), autoimmune diabetes (16), inflammatory bowel disorder (17), collagen II-induced arthritis (11), airway inflammation (18), allergic asthma (18, 19), multiple sclerosis (20), and chronic and aggressive periodontitis (21). It also impacts colorectal cancer progression and prognosis (22) and is highly expressed in tumor tissue in lung and colon cancer and esophageal, hepatocellular, and cervical carcinomas (23). Regulatory T cells (Tregs) are a critical subpopulation of CD4+ T cells essential to immune response (24, 25). IL-35 can convert naïve T cells into strongly suppressive Tregs (26), promote Treg expansion, and suppress proliferation of conventional T cells (27, 28). It might be specifically produced by Tregs and may be required for maximal suppressive activity (27). Although studies report that IL-35 is expressed in patients infected with the hepatitis B virus (HBV) (29–31), the role of IL-35 during infection, particularly, IAV infection is largely unknown.
Herein, we show that IL-35 expression is induced by IAV infection and is regulated by the transcription factor, NF-κB. The inflammatory factors, cyclooxygenase-2 (COX-2) and inducible nitric-oxide synthase (iNOS), may also be involved in this signaling cascade. IL-35 also activated the IFN pathway and indirectly elicited antiviral activity in response to viral infection. Our results provide a basis for further investigation of the relationship between IAV and IL-35 and suggest that IL-35 might be a potential novel target for antiviral therapies.
Results
IL-35 Expression Is Elevated in Patients Infected with IAV
IL-35 consists of the heterodimeric IL-27/EBI3 chain and the IL-12/p35 chain (11). To investigate induction of IL-35 expression during IAV infection, peripheral blood mononuclear cells (PBMCs) were isolated from IAV-infected patients and healthy individuals (controls). EBI3 mRNA levels were ∼3-fold higher (IAV, 20.21 ± 1.312, n = 12, black box; controls, 6.275 ± 0.9348, n = 12, white box; **, p < 0.01, Fig. 1A, left), and p35 mRNA levels were ∼6-fold higher (IAV, 38.70 ± 12.80, n = 12, black box; controls, 6.638 ± 1.188, n = 12, white box; **, p < 0.01, Fig. 1A, right) in IAV-infected patients compared with controls. These results are similar to those obtained from throat swabs; EBI3 mRNA levels were ∼6-fold higher (IAV, 56.09 ± 10.23, n = 10, black box; controls,10.44 ± 3.803, n = 10, white box; **, p < 0.01, Fig. 1B, left), and p35 mRNA levels were ∼4-fold higher (IAV, 98.30 ± 21.82, n = 10, black box; controls, 26.57 ± 7.483, n = 10, white box; **, p < 0.01, Fig. 1B, right) in IAV-infected patients compared with controls. These data indicate that IL-35 is elevated in IAV-infected patients.
FIGURE 1.

Measurement of IL-35 in IAV-infected patients and healthy individuals. A, total RNA was extracted from freshly isolated PBMCs from healthy individuals (n = 12) or IAV-infected patients (n = 12). EBI3 (left) and p35 (right) mRNA was quantified using qPCR. B, throat swab EBI3 (left) and p35 (right) mRNA levels in controls (n = 10) and patients (n = 10). In A and B, the lowest value from controls was as assigned a value of 1. Data are expressed as -fold induction (folds) relative to the lowest value from controls. Data are expressed as the mean ± S.E. from samples tested in triplicate. Boxplots illustrate medians with 25 and 75%, and error bars for 5 and 95% percentiles (**, p < 0.01). C, correlation analysis between NP mRNA and EBI3 (left, n = 33) and p35 (right, n = 33) mRNA levels in PBMCs from IAV-infected patients. Solid line, linear growth trend; r, correlation coefficient; Pearson correlation analysis was used to determine r values; Student's t test was used to determine p values.
To verify whether IL-35 expression was related to virus infection, IAV NP and IL-35 mRNA levels were analyzed using Pearson's correlation. A statistically significant correlation was observed between NP and IL-35 mRNA levels in PBMCs from IAV-infected patients (NP and IL-35/EBI3: r = 0.70, n = 33; **, p < 0.01, Fig. 1C, left; NP and IL-35/p35: r = 0.73, n = 33; **p < 0. 01, Fig. 1C, right). These data indicate that IL-35 expression positively correlates with IAV infection.
IAV-induced IL-35 Expression in Different Cell Types
Because high levels of IL-35 mRNA were observed in IAV-infected patients, we next assessed in vitro changes in IL-35 expression in response to IAV infection. A549 cells were infected with IAV at various doses. IAV-induced (m.o.i. < 1) IL-35 mRNA expression was positively correlated with the infectious dose of IAV (Fig. 2A). Viral infection induced maximal expression at 1 m.o.i. These data suggest that IAV stimulates IL-35 mRNA expression in a dose-dependent manner (m.o.i. < 1).
FIGURE 2.

Induction of IL-35 by IAV in different cell types. A, A549 cells were infected at the indicated concentrations of IAV for 24 h. EBI3 and p35 mRNA levels were quantified using qRT-PCR. B, A549 cells were infected with IAV (m.o.i. = 1) for the indicated timeframes, and EBI3 and p35 mRNA levels were quantified. C, A549 cells were infected with heat-inactivated IAV (65 °C, 1 h) or IAV (m.o.i. = 1) for 24 h; EBI3, p35, p19, p28, and p40 mRNA were quantified. D, experiments performed as in C, except IL-35 protein was detected using ELISA. E, experiments were performed as in C, except where PBMCs were used. F, experiments were performed as in C, except where AT II cells were used. G, A549 cells were infected with IAV (m.o.i. = 1) or mock for 24 hl IκB and p- IκB were detected by Western blot. Bar graphs represent the mean ± S.D. n = 3. *, p < 0.05; **, p < 0.01).
Expression of the two IL-35 subunits at various time points was measured in A549 cells infected with 1 m.o.i. IAV. IL-35 was significantly up-regulated 12 hpi (Fig. 2B). IL-35/p35 mRNA reached peak expression 24 hpi; IL-35/EBI3 mRNA reached peak expression 48 hpi. Similar results were obtained in IAV-infected PBMCs: both IL-35/EBI3 and IL-35/p35 mRNA reached peak expression 24 hpi (data not shown). Based on these results, 24 hpi was the chosen time point for measurement of IL-35 mRNA expression in subsequent experiments. These results indicate that IAV infection induces IL-35 expression in a time-dependent manner.
Quantitative PCR analysis showed that IAV (m.o.i. = 1) infection of A549 cells significantly increased IL-35 mRNA expression compared with mock infection (Fig. 2C). Moreover, p19 (the subunit of IL-23), p28 (the subunit of IL-27), and p40 (the subunit of IL-12) were up-regulated (Fig. 2C). The results are consistent with previous reports (32–35). In contrast, heat-inactivated IAV did not significantly increase IL-35 expression. An ELISA kit was used to measure IL-35 protein expression in culture supernatants. IL-35 expression was up-regulated ∼2-fold by IAV infection compared with mock infection (Fig. 2D). Similar results were obtained in freshly isolated PBMCs and alveolar type II (AT II) cells (Fig. 2, E and F, respectively), wherein IL-35 mRNA levels were up-regulated by IAV infection. In the meantime, NF-κB was activated by IAV infection with p-IκB (36, 37) levels measured in A549 cells (Fig. 2G). These data indicate that IAV infection can induce IL-35 mRNA and protein expression.
IL-35 Expression Is Induced by IAV at the Transcriptional Level
To investigate the molecular underpinnings of IAV-induced IL-35 up-regulation in A549 cells, promoter reporter assays were performed. As IL-35 consists of EBI3 and p35, luciferase reporter plasmids containing the EBI3 (pIL-35/EBI3-Luc) and p35 (pIL-35/p35-Luc) promoters were constructed. A549 cells were co-transfected with pIL-35-Luc and pRL-TK and infected or not infected with IAV (m.o.i. = 1). IAV infection significantly increased the activities of pIL-35/EBI3-Luc and pIL-35/p35-Luc (Fig. 3A). These results suggest that IAV can induce IL-35 expression at the transcriptional level.
FIGURE 3.

IAV induces IL-35 expression at the transcriptional level. A, A549 cells were transfected with luciferase reporter plasmids containing the IL-35/EBI3 (EBI3-Luc) or IL-35/p35 promoter (p35-Luc) along with pRL-TK and either infected or not infected with IAV (m.o.i. = 1) for 24 h. Luciferase activity was measured after 12 h-serum starvation. B, A549 cells were transfected with EBI3-Luc or a series of truncated and mutated plasmids containing the IL-35/EBI3 promoter along with pRL-TK; cells were then infected with IAV (m.o.i. = 1) for 24 h. Luciferase activity was measured as in A. C, experiments performed as in B, except p35-Luc or a series of truncated and mutated plasmids containing the IL-35/p35 promoter. D, A549 cells were co-transfected pCMV-p50 or pCMV-p65 with EBI3-Luc or p35-Luc along with pRL-TK; cells were then infected with IAV (m.o.i. = 1) for 24 h. Luciferase activity was measured as in A, and the expression of p50 and p65 was detected by Western blot (lower panel). E, experiments were performed as in D, except where shRNA-p50 or shRNA-p65 were used. The silencing efficiencies for p50 and p65 were measured by Western blot (lower panel). F and G, experiments were performed as in D and E, except pCMV-CREB (F) or shRNA-CREB (G) were used. CREB expression was detected by Western blot (lower panel). n.s., not significant. H and I, A549 cells were infected with IAV (m.o.i. =1) for 12 h. ChIP assays were performed using a NF-κB/p65 antibody (Ab, normal IgG as control). Bond DNA fractions on the IL-35/EBI3 (H) or IL-35/p35 promoter (I) were measured using qRT-PCR utilizing the indicated primers. Bar graphs represent the mean ± S.D. n = 3. *, p < 0.05.
Transcription factor binding sites at the IL-35 promoter were predicted by transcription factor assay databases, including JARSPAR DATABASE, ALGGEN, Biobase, Gene regulation, and Transgene. Diagrams showing comprehensive analysis of these sites at the IL-35 promoter are shown in Fig. 3, B and C. To identify potential regulatory sites, a series of truncated IL-35 promoters was constructed. Luciferase activity assays showed that elimination of the −150 to −450 region of pIL-35/EBI3-Luc greatly decreased promoter activity (Fig. 3B). The results further suggest that the −360 to −347 and −233 to −242 NF-κB (p65/p50) binding sites are important for IAV-induced activation of the IL-35/EBI3 promoter. Elimination of the −1110 to −1509 region of pIL-35/p35-Luc significantly decreased IAV-induced IL-35/p35 promoter activity (Fig. 3C). Our results indicate that the −1434 to −1444 NF-κB/p50 binding site and the −1368 to −1378 NF-κB/p65 binding site may be important for IAV-induced activation of the IL-35/p35 promoter.
Four mutant reporters were constructed for the NF-κB binding sites. These reporters encompassed site 1 (−360 to −347), site 2 (−233 to −242), and site 3 (−84 to −97) regions of pIL-35/EBI3-Luc. Site 3 is an important cis-regulatory element binding site in IL-18 and IL-1β-induced IL-27/EBI3 promoter activity (38). Mutant reporters also encompassed the site 4 region (−1368 to −1378) on pIL-35/p35-Luc. Our results suggest that there was a decrease in IAV-induced mutant reporter luciferase activity compared with activity using the wild-type promoter (Fig. 3, B and C).
To confirm whether p50 or p65 regulates IL-35 promoter activity, overexpression and knockdown experiments of p50 or p65 were performed. Overexpression of p65, but not p50, significantly increased IL-35/EBI3 and IL-35/p35 luciferase activity in cells infected with IAV relative to control (Fig. 3D). Knockdown of NF-κB with shRNA-p65, but not shRNA-p50, significantly decreased IAV-induced IL-35/EBI3 and IL-35/p35 luciferase activity relative to the shRNA control (Fig. 3E). These data suggest that NF-κB/p65 might be a vital transcription factor regulating IL-35 promoter activity.
We also investigated whether CREB binding sites on the IL-35 promoter are important for IAV-induced promoter activity. Our results show that overexpression (Fig. 3F) or knockdown of CREB with shRNA-CREB (Fig. 3G) does not influence IL-35 promoter activity. To verify whether NF-κB/p65 binds to the IL-35 promoter, ChIP assays were performed. Results show that three fragments on the IL-35/EBI3 promoter (Fig. 3H) and one on the IL-35/p35 promoter (Fig. 3I) were bound by p65 at much higher levels with IAV infection compared with control. These data suggest that NF-κB/p65 is an important element required for IAV-induced activation of the IL-35 promoter.
COX-2 and iNOS Are Involved in IAV-induced IL-35 Expression
COX-2 and iNOS are important inflammatory factors regulating interleukins expression induced by viral infection (6, 39, 40). We, thus, investigated the roles of COX-2 and iNOS in IAV-induced IL-35 expression. A549 cells were transfected with a COX-2 expression plasmid (pCMV-COX-2) or a vector for 24 h and then infected with IAV. IL-35 mRNA levels increased with COX-2 overexpression (data not shown). A549 cells were then treated with Etoricoxib, a selective COX-2 inhibitor, for 2 h at different doses (10, 50, or 100 μm) before infection with IAV (m.o.i. = 1). DMSO and IAV infection without inhibitor were used as solvent and positive controls, respectively. Our results show that IL-35 mRNA levels decreased in a dose-dependent manner with pretreatment with Etoricoxib (Fig. 4A). A549 cells were transfected with two different COX-2-specific siRNAs that modestly reduced levels of COX-2 mRNA and protein (Fig. 4B). IL-35 expressions were confirmed to be down-regulated by COX-2 silencing with siCOX-2-#2 (Fig. 4C). These data suggest that COX-2 positively regulates IAV-induced IL-35 expression. It is reported that overexpressed COX-2 contributed to prostaglandin (PG) overproduction (41); we incubated A549 cells with the dissolved prostaglandin E2 (PGE2) for 2 h, then infected with IAV (m.o.i. = 1). The results showed that expression of IL-35 mRNA was significantly up-regulated by PGE2 (Fig. 4D).
FIGURE 4.

COX-2 and iNOS are involved in IAV-induced IL-35 expression. A, A549 cells were pretreated with the indicated concentrations of Etoricoxib for 2 h, infected with IAV (m.o.i. = 1) for 24 h, then subjected to qRT-PCR analysis of IL-35 expression (above) and Western blot for COX-2 protein level (below). B, A549 cells were transfected with si-COX-2 or siRNA-control, then subjected to qRT-PCR analysis of COX-2 mRNA level (above) and Western blot for COX-2 protein level (below). C, A549 cells were transfected with si-COX-2 or siRNA-control for 24 h prior to infection with IAV(m.o.i. = 1) for 12 h, then IL-35 expression was analyzed by qRT-PCR. D, experiments performed as in A, except 10 μm PGE2 was dissolved and then added. E and F, relative COX-2 and EBI3 (E) or p35 (F) mRNA levels in PBMCs from IAV-infected patients were subjected to Pearson correlation analysis (n = 20). G, Experiments were performed as in A, except SMT was used. H and I, experiments were performed as in B and C, except si-iNOS was transfected. J, experiments were performed as in D, except 100 μm sodium nitroferricyanide(III) dihydrate (SNP) was used. K and L, relative iNOS and EBI3 (K) or p35 (L) mRNA levels in PBMCs from IAV-infected patients were subjected to Pearson correlation analysis (n = 32). Bar graphs represent the mean ± S.D. n = 3. *, p < 0.05; **, p < 0.01).
We next determined whether IL-35 expression correlates with COX-2 levels in clinical samples. IL-35 and COX-2 mRNA levels were measured in PBMCs from IAV-infected patients then subjected to correlation analysis. There were statistically significant correlations between IL-35 and COX-2 mRNA levels (IL-35/EBI3 and COX-2: r = 0.51; *, p < 0.05, n = 20, Fig. 4E; IL-35/p35 and COX-2: r = 0.47; *, p < 0.05, n = 20, Fig. 4F; Pearson's correlation). Similarly, IL-35 mRNA levels decreased in a dose-dependent manner when A549 cells were treated with S-methylisothiourea sulfate (50, 100 μm; Alexis Biochemicals, Grünberg, Germany), the selective iNOS inhibitor (Fig. 4G), and IL-35 mRNA decreased (Fig. 4I) with si-iNOS-#3 (Fig. 4H) transfected. Meanwhile, IL-35 mRNAs were up-regulated in A549 cells with sodium nitroferricyanide(III) dihydrate (SNP) incubation (Fig. 4J). To verify the relationship between IL-35 and iNOS expression during IAV infection, PBMCs were isolated from IAV-infected patients. IL-35 and iNOS mRNA levels were measured and analyzed using Pearson's correlation. A statistically significant correlation was found between IL-35 and iNOS expression (IL-35/EBI3 and iNOS: r = 0.65; **, p < 0.01, n = 32, Fig. 4K; IL-35/p35 and iNOS: r = 0.68; **, p < 0.01, n = 32, Fig. 4L). Collectively, our results suggest that both COX-2 and iNOS may positively regulate IAV-induced IL-35 expression in A549 cells.
IL-35 Hampers Virus Replication
We sought to determine the biological function of -+IL-35 during virus infection. Recombinant pCMV-IL-35 plasmids were constructed (Fig. 5A), and Western blots with EBI3 and p35 antibodies were used to confirm the cellular overexpression IL-35 (Fig. 5B). However, overexpression of IL-35 in A549 cells did not strongly affect IAV replication (data not shown). Li et al. (42) shows that IL-32γ does not directly affect HBV replication in HepG2.2.15 cell, and subsequently established protocols utilizing collection of supernatants from IL-32γ-treated PBMCs for indirect antiviral assays. Using the method mentioned above, Jurkat cells were first electroporated with pCMV-IL-35; after 36 h, the IL-35 protein level in the supernatants were measured by ELISA (Fig. 5C), and the supernatants were collected for antiviral experiments. A549 cells were incubated with the collected supernatants of Jurkat cells and then infected with IAV (m.o.i. =1). IAV NP RNA levels, including plus-sense RNA and minus-sense RNA were measured 3 hpi via qRT-PCR; supernatant IAV titers were measured using a hemagglutinin assay 24 hpi. This treatment significantly decreased NP RNA levels (Fig. 6A) and significantly decreased IAV titers (Fig. 6B). Our results indicate that the incubation of A549 cells with the supernatants of IL-35-transfected Jurkat cells can inhibit IAV replication effectively.
FIGURE 5.
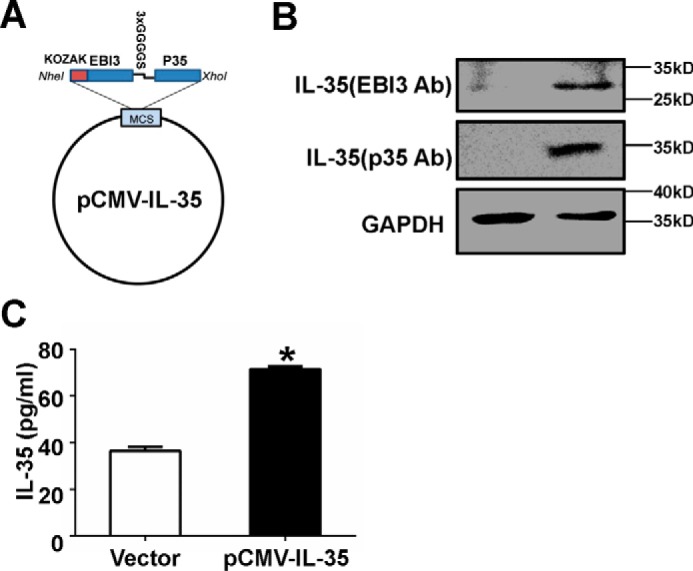
The expression of constructed IL-35 was determined by measuring protein levels. A, IL-35/EBI3 and IL-35/p35, linked by 3xGGGGS, was inserted into the NheI/XhoI sites of pcDNA3.1(−) to generate pCMV-IL-35. B, Western blots were performed to measure IL-35 in 293 cells transfected with pCMV-IL-35 for 48 h. C, Jurkat cells were electroporated with vector or pCMV-IL-35 for 36 h, and IL-35 protein levels in the supernatants were measured using ELISA. Ab, antibody. *, p < 0.05.
FIGURE 6.

The indirect antiviral activity of IL-35. A and B, Jurkat cells were transfected with vector or pCMV-IL-35. Thirty-six hours later the supernatants were collected for indirect antiviral assays. Antiviral assays were performed in 1 m.o.i. IAV-infected A549 cells incubated with the collected supernatants for 24 h. IAV RNAs (A) and IAV production (B) were analyzed using qRT-PCR and hemagglutination assays, respectively. C and D, Jurkat cells were treated with 100 ng/ml rhIL-35 and 2 μg/ml IFNγ. Twenty-four hours later the supernatants were collected, and indirect antiviral assays were performed as in A (C) and B (D). E, Jurkat cells were treated with doses of rhIL-35 or co-treated with 2 μg/ml IFNγ for 24 h, and cell viability was measured using the MTT assay. F, A549 cells were incubated with the supernatants from rhIL-35-treated Jurkat cells, and cell viability were measured using the MTT assay. G and H, EV71-infected RD cells were cultured then incubated with the supernatants described in A (G) or C (H); the supernatants were then collected and used for EV71 copy testing using the absolute value of qRT-PCR. I and J, Huh7 cells transfected with pHBV were incubated with collected supernatants from Jurkat cells described in A (I) or C (J). HBe/s Ag was analyzed using ELISA. K, A549 cells or Vero cells were incubated with collected supernatants from Jurkat cells described in A, then infected with VSV-eGFP (m.o.i. = 1) for 6 h. VSV-eGFP replication was visualized via fluorescence microscopy and measured using flow cytometry for eGFP expression. Bar graphs represent the mean ± S.D. n = 3; *, p < 0.05; **, p < 0.01.
We next investigated whether secreted IL-35 can inhibit IAV replication using commercial rhIL-35. Previous studies demonstrated interactions between interleukins and IFNγ. IL-12 was positively regulated by IFNγ (43, 44), and IL-23 interacted with IL-12, IL-18, and IL-2 to promote IFNγ production in NK cells (45). In turn, IFNγ interacted with IL-27 to induce Treg proliferation, limiting pathology due to infection (46). Evidence also suggested that IFNγ and STAT1-dependent expression of IL-12Rβ2 were crucial for T cell activation (47). In human cancer cell lines, IL-35 expression can be induced after TNF-α and IFNγ stimulation (23). IL-35 not only decreased production of IL-17 but can also increase IFNγ production (11). Jurkat cells were incubated with 100 ng/ml of rhIL-35 (11) and 2 μg/ml IFNγ for 24 h, and the supernatants were collected for antiviral assays. A549 cells were subsequently incubated with these supernatants and then infected with IAV (m.o.i. = 1). IAV NP RNA levels were measured via qRT-PCR (Fig. 6C), and IAV titers were measured using a hemagglutinin assay (Fig. 6D). Jurkat cell viability was not affected by 24-h incubation with rhIL-35 (at different doses) and with or without 2 μg/ml IFNγ (Fig. 6E). The activity of A549 cells was not affected by incubation with supernatants from Jurkat cells treated with 100 ng/ml rhIL-35 with or without 2 μg/ml IFNγ (Fig. 6F). The data suggest that the incubation of A549 cells with the supernatants of rhIL-35 and IFNγ-treated Jurkat cells can restrict IAV infection enormously.
Next, we investigated whether IL-35 has wide-ranging antiviral function. Rhabdomyosarcoma (RD) cells were incubated with the supernatants of Jurkat cells described in Fig. 5, A or C, and then infected with human enterovirus 71 (EV71) (m.o.i. = 1). 12 h later, EV71 VP1 expression was measured using absolute qRT-PCR. Replication of EV71 was inhibited by IL-35 overexpression (Fig. 6G), and EV71 copy numbers were significantly reduced by this treatment of rhIL-35 and IFNγ (Fig. 6H). Next, Huh7 cells were transfected with pHBV and incubated with supernatants from Jurkat cells described in Fig. 5, A or C. Our results show relatively lower HBeAg and HBsAg levels when cells were incubated with Jurkat cell-derived supernatants compared with control (Fig. 6, I and J). We then assessed the effects of the above protocol on the production of recombinant vesicular stomatitis virus carrying enhanced green fluorescent protein (VSV-eGFP) in A549 cells. Consistent with our other results, VSV-eGFP infection was significantly reduced when cells were incubated with Jurkat cell-derived supernatants compared with control (Fig. 6K), and the number of infected cells decreased from 75.94% to 38.97%, as measured by flow cytometry. However, there were no antiviral effects in Vero cells on VSV-eGFP expression (Fig. 6K). Because Vero cells lack functional IFN gene expression (48–50), we inferred that any potential antiviral functions of IL-35 require IFN production.
Similar results utilizing the above protocol were obtained when PBMCs were used. Freshly isolated PBMCs from healthy donors were electroporated with pCMV-IL-35 or vector, and the supernatants were collected for antiviral assays. IAV NP RNA levels and virus titers (data not shown) were reduced in IAV (m.o.i. = 1)-infected A549 cells incubated with the above PBMC-derived supernatants. HBeAg and HBsAg expression was reduced in pHBV-transfected Huh7 cells cultured with the same PBMC-derived supernatants (data not shown). EV71 copy numbers were also reduced in 1 m.o.i. EV71-infected RD cells cultured with these PBMC-derived supernatants (data not shown). These data indicate that treatment with the above PBMC-derived supernatants had antiviral functions not only in IAV infection, but also in VSV, HBV, and EV71 infection. Taking together, IL-35 has extensive antiviral activity to DNA and RNA viruses.
IL-35-induced IFN Production
To investigate whether the effects of IL-35 on antiviral activity depends on the presence of IFNs, Jurkat cells were electroporated with pCMV-IL-35 plasmids for 24 h. Total cellular RNA was extracted, and IFN-α, IFN-β, and IFN-λ1 mRNA levels were quantified using qRT-PCR. IFN-α, IFN-β, and IFN-λ1 mRNA levels significantly increased with pCMV-IL-35 transfection compared with vector transfection (Fig. 7A). Similar results were observed in PBMCs transfected with pCMV-IL-35. IFN-α, IFN-β, and IFN-λ1 mRNA levels were up-regulated in pCMV-IL-35-transfected PBMCs compared with vector transfection (Fig. 7B). Furthermore, IFN-α (Fig. 7C), IFN-β (Fig. 7D), and IFN-λ1 (Fig. 7E) were activated in pCMV-IL-35-transfected Jurkat cells compared with vector transfection. These data suggest that IFN expression can be induced by IL-35.
FIGURE 7.

IL-35-induces IFN expression. A, Jurkat cells were transfected with pCMV-IL-35 or control vector for 36 h before analysis the IFN-α/β/λ1 RNA levels using qRT-PCR. B, freshly isolated PBMCs from healthy controls were transfected with pCMV-IL-35 or control vector for 24 h before analysis the IFN-α/β/λ1 RNA levels using qRT-PCR. C, D, and E, experiments performed as in A, except IFN-α (C), IFN-β (D), or IFN-λ1 (E) protein levels were measured by ELISA. F and G, Jurkat cells were transfected with vector or pCMV-IL-35. Thirty-six hours later the supernatants were collected, and A549 cells were incubated in these supernatants for 24 h. PKR, OAS, and Mx mRNA levels were quantified using qRT-PCR (F), and protein was detected by Western blot (G). H, Jurkat cells were treated with 100 ng/ml rhIL-35 and 2 μg/ml IFNγ. mRNA levels of IFN-α, IFN-β, and IFN-λ1 were measured using qRT-PCR at different time-points. I, A549 cells were incubated with the supernatant from Jurkat cells in H and then infected with IAV (m.o.i. = 1). IFN-stimulated gene protein levels of PKR, OAS, and Mx were detected using Western blots. All experiments were performed at least in triplicate with similar results. J, A549 cells were transfected with shIFNAR1, shIFNLR1, and shRNA-control for 48 h, and IFNAR1 and IFNLR1 were detected by Western blot with indicated antibody. K, the collected supernatants in A were used to incubated with A549 cells for 24 h followed by IAV (m.o.i. = 1) infection for 12 h. IAV NP plus-sense and minus-sense RNAs were measured by qRT-PCR. Bar graphs represent the mean ± S.D., n = 3 (*, p < 0.05; **, p < 0.01).
We next explored whether expression of downstream IFN effectors can be induced by IL-35. Jurkat cells were transfected with pCMV-IL-35, and the supernatants were collected after 36 h. A549 cells were subsequently incubated with these Jurkat cell-derived supernatants and infected with 1 m.o.i. IAV. qRT-PCR analysis and Western blots revealed that intracellular RNA-dependent protein kinase (PKR), 2′,5′-oligoadenylate synthetase (OAS), myxovirus resistance protein (Mx) mRNA, and protein levels increased with this treatment (Fig. 7, F and G). Similar results were obtained using rhIL-35. The expression of IFN-α, IFN-β, and IFN-λ1 were all induced by rhIL-35 in a time-dependent manner (Fig. 7H). The PKR, OAS, and Mx were also up-regulated (Fig. 7I).
To investigate whether the IFNs mediated IL-35 anti-viral action, IFNAR1 or IFNLR1 were knocked down by shIFNAR1 or shIFNLR1 (Fig. 7J), and the anti-viral action of IL-35 was not detected in the presence of either shIFNAR1 or shIFNLR1 (Fig. 7K). Collectively, these data demonstrate that IL-35 can induce IFN production and stimulate expression of downstream IFN effectors, and the induced IFNs mediate IL-35 anti-viral activity.
Discussion
Herein, we investigated IL-35 expression during IAV infection. IAV is one of the most common causes of infection in humans (51) and can lead to high morbidity and significant mortality. Host cells secrete a variety of cytokines and chemokines in response to IAV infection (1, 4–7). We present the first evidence from clinical samples that IL-35 expression may change in response to IAV infection, consistent with additional findings that IL-35 mRNA levels were positively correlated with IAV NP mRNA levels.
We used various cell lines in this study for different purposes. Lung epithelial cells are the primary targets of IAV infection (52). AT II cells constitute ∼60% of alveolar epithelial cells and ∼15% of all lung parenchymal cells. A549 cells are adenocarcinomic human alveolar basal epithelial cells commonly used in IAV research. PBMCs are widely used to investigate immune response to viral and microbial infections (53, 54). Our findings suggest that IL-35 is significantly up-regulated in these three cell types during IAV infection in a time- and dose-dependent manner.
Our results suggest that IAV infection can induce IL-35 expression via the transcription factor, NF-kB/p65, and the inflammatory factors COX-2 and iNOS. IL-35 can also activate the IFN pathway and may have potential antiviral activity. Consistent with these findings, others have shown that HBV infection can induce IL-35 expression. One report shows that IL-35 mRNA and protein are detectable in CD4+ T cells in patients with chronic hepatitis B (55). Likewise, in patients chronically infected with HBV, CD4+ T cells have significantly higher levels of EBI3 mRNA and protein compared with healthy individuals and patients in whom HBV infection had resolved. Furthermore, IL-35 has also been shown to suppress proliferation of HBV antigen-specific cytotoxic T-lymphocytes and IFN-γ production in vitro (56).
NF-κB is inactive in the cytoplasm due to IκB masking its nuclear localization sequence (57). Upon viral infection, phosphorylated IKK can phosphorylate IκBα, which is then ubiquitinated (58, 59), allowing NF-κB to enter the nucleus (39). We show that binding of NF-κB/p65 to the p65 transcription binding site initiates IL-35 transcription during IAV infection.
COX-2 is critical to the inflammatory response (60) and can be activated by IAV infection in A549 cells (61). It can regulate protein expression, including that of CRC3, IL-10, and IL-17 (62). iNOS is an inducible and calcium-independent isoform of NO synthetase. It plays a critical modulatory role in immune response, chronic inflammation, and carcinogenesis and can be stimulated by viral infection (6, 63, 64). Our findings suggest that COX-2 and iNOS may be important factors in IAV-induced IL-35 expression. Furthermore, both COX-2 and iNOS mRNA levels are positively correlated with IL-35 mRNA levels in IAV-infected blood samples.
IL-35 is detectable in peripheral CD4+ T cells and is believed to play important functions in inhibition of the immune response during chronic HBV infection (56), suggesting it may be a potential therapeutic target to control HBV infection (29). Thus, we investigated the biological function of virus-induced IL-35 expression. Using the indirect approach, we demonstrate that overexpression of IL-35 and incubation with rhIL-35 may suppress IAV, EV71, VSV, and HBV replication and viral production.
IFN-α/β is the host's central innate immune response to viral infection (65), although IFN-λ1 has also been shown to inhibit replication of a number of viruses (66). In this study, the indirect antiviral effect of IL-35 was abolished in IFN-deficient Vero cells (48), suggesting that IL-35 function during viral infection depends on expression of IFN. Our findings show that IL-35 can induce IFN-α, IFN-β, and IFN-λ1 mRNA expression and increases PKR, OAS, and Mx mRNA and protein expression. And we found up-regulated expression of IL-35 receptors (14), including gp130 and IL-12Rb2, concurrent with IFN production (data not shown). Previous reports show that signaling through the IL-35 receptor requires STAT1 and STAT4 (14). Here, IL-35 may have stimulated IFN expression via activating IL-35 receptors, thereby presumably stimulating the Jak-STAT pathway, leading to phosphorylation of STAT1 and STAT4 and subsequent up-regulation of IFN.
In conclusion, we propose a hypothetical model of IAV-induced IL-35 production and its biological function (Fig. 8). IAV infection stimulates IL-35 expression via activation of its promoter by the transcription factor, NF-κB/p65, and through activation of COX-2 and iNOS pathways. IL-35 then activates IFN and its downstream effectors, leading to inhibition of viral replication and production. Further studies are required to better understand the IL-35-related complex regulatory mechanisms of antiviral host response during virus infection. However, our findings provide evidence of a distinct role for IL-35 in this process and point to potential novel clinical uses for IL-35 in antiviral therapy.
FIGURE 8.
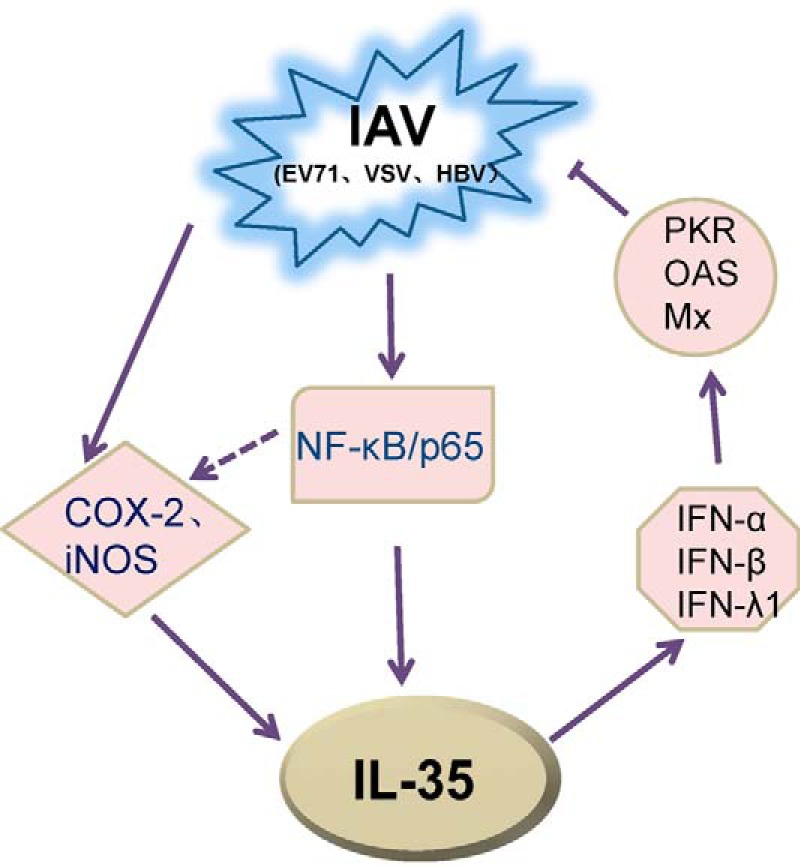
Hypothetical model of IAV-induced IL-35 production and potential antiviral activity of IL-35. Solid arrows represent signaling pathways identified in this study. IAV-induced IL-35 expression via NF-κB/p65 at the transcriptional level and via COX-2 and iNOS. Enhanced IL-35 production activated IFN and IFN-stimulated genes, leading to inhibition of viral (IAV, EV71, VSV, HBV) replication.
Experimental Procedures
Ethics Statement
Peripheral blood samples and throat swabs were obtained from IAV-infected patients and healthy individuals. Clinical samples were collected in accordance to the Declaration of Helsinki from patients admitted to the Hubei Provincial Center for Disease Control and Prevention and were approved by the Institutional Review Board of the College of Life Sciences of Wuhan University in accordance to its guidelines for protection of human subjects. Written informed consent was obtained from all participants.
Cell and Virus
Human lung epithelial cells (A549) were cultured in F12K medium (Gibco). Human T cell lymphoblast-like cell (Jurkat) lines were cultured in RPMI 1640 medium (Gibco). RD cells were cultured in minimum Eagle's medium (MEM, Gibco). Huh7 cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Gibco). All media were supplemented with 10% fetal bovine serum (FBS, Gibco). All cultures were maintained at 37 °C in a 5% CO2 incubator. The IAV/Hong Kong/498/97 (H3N2) strain used in these studies was provided by the China Center for Type Culture Collection. Recombinant VSV-eGFP was a gift from Mingzhou Chen (Wuhan University). EV71 was obtained from Xiangyang (GenBankTM accession number JN230523.1).
Isolation of PBMCs and Primary AT II Cells
PBMCs were isolated using density centrifugation diluted 1:1 in a solution of human lymphocytes (TBD-Science, Tianjin, China) as previously described (67). PBMCs were washed twice with PBS and cultured in RPMI 1640 medium at 37 °C in a 5% CO2 incubator. Human AT II cells were purchased (Wuhan PriCells Biotechnology & Medicine Co., Ltd, Wuhan, China) and cultured in RPMI 1640 medium (37 °C, 5% CO2).
Transfection
Cells were plated at a density of 4 × 105/plate and grown to 80% confluency before transfection. Cells were transfected with Lipofectamine 2000 reagent (Invitrogen) for 24 h, serum-starved for another 24 h, then harvested for analysis. PBMCs and Jurkat cells were transfected by electroporation. Solution I (20% ATP-disodium salt, 12% MgCl2·6H20) and Solution II (1.2% KH2PO4, 0.12% NaHCO3, 0.04% glucose, pH 7.4) were freshly mixed on ice (1:4, v/v). Cells at a density of 106 were lightly suspended in a 100-μl blended solution with 4 μg of plasmids on ice. The mixture was added into a cuvette (BTX, Taiwan, China) and electroporated using Nucleofector (Amaxa, Lonza Group Ltd., Germany). Cells were washed into 6-well culture plates and cultured in RPMI 1640.
Biological and Chemical Reagents
Coding regions of IL-35 were amplified from templates, including IL-27/EBI3 (32) and p35 cDNA, a gift from the Jiahuai Han laboratory (Xiamen University), by PCR with the following primers: IL-35/EBI3, 5′-TTAATAGCTAGCGCGGCCGCCACCATGGGGAGGAAAGGGCCCCCAGCA-3′ (sense), 5′-AGAACCACCACCACCAGAACCACCACCACCAGAACCACCACCACCCTTGCCCAGGCTCAT-3′ (antisense); IL-35/p35, 5′-GGTGGTGGTGGTTCTGGTGGTGGTGGTTCTGGTGGTGGTGGTTCTAGAAACCTCCCCGTG-3′ (sense), 5′-CCGCTCGAGTCAGGAAGCATTCAGATAGCT-3′ (antisense). The two fragments were linked, generating the IL-35 fragment via the linker, 3×GGGGS. The KOZAK sequence-containing IL-35 fragment was inserted into NheI/XhoI-containing sites of pcDNA3.1 (−) to generate the expression plasmid, pCMV-IL-35, using previously described methods (11). IL-35/EBI3 and IL-35/p35 promoters, the truncates, and mutants were amplified using genomic PCR with the following primers: for IL-35/EBI3 promoter, 5′-TTTGGTACCCTGTCTATCTCCGTGTCCTC-3′ (sense), 5′-TTTAAGCTTCTGCTCTCAGGAGTGGGT-3′ (antisense); for −450 IL-35/EBI3 promoter, 5′-TTTGGTACCTGTCTTCCTTCTGTCTTCTC-3′ (sense); for −250 IL-35/EBI3 promoter, 5′-TTTGGTACCCCACCCTCGGGGCCTT-3′ (sense); for −150 IL-35/EBI3 promoter, 5′-TTTGGTACCCCAGTGAGTCAGACCTGA-3′ (sense); for IL-35/p35 promoter, 5′-GATGGTACCAGATGAGCCACCCAGAA-3′ (sense), 5′-GCTAAGCTTCTTGCGGCGCTTTCGGAT-3′ (antisense); for −1110 IL-35/p35 promoter, 5′-TTTGGTACCTCTTCCCTCTGCTCTACTCCT-3′ (sense); for −710 IL-35/p35 promoter, 5′-TTTGGTACCCTCTAGGTCTTTCCTCCCA-3′ (sense); for −310 IL-35/p35 promoter, 5′-TTTGGTACCGACACGGGGCGTCCGGCTAA-3′ (sense). Target sequence mutations were generated by site-directed mutagenesis using specific primers. The following primer pairs for the mutagenesis of luciferase reporters were: for IL-35/EBI3 promoter site 1 mutant, 5′-TGCCTGGGGTTTTAGCCGCTTCAGGGC-3′ (sense), 5′-GCCCTGAAGCGGCTAAAACCCCAGGCA-3′ (antisense); for IL-35/EBI3 promoter site 2 mutant, 5′-CCCCACCCTCGTTGTTTTCCGAGCA-3′ (sense), 5′-ACCCCTGCTCGGAAAACAACGAGGGT-3′ (antisense); for IL-35/EBI3 promoter site 3 mutant, 5′-TGGGCTGGGCTTTTAGCTGGGCAGGTC-3′ (sense), 5′-GACCTGCCCAGCTAAAAGCCCAGCCCA-3′ (antisense); for IL-35/p35 promoter site 4 mutant, 5′-TGACTAATGCCTAGAGGATTAACAACTGAA-3′ (sense), 5′-TTCAGTTGTTAATCCTCTAGGCATTAGTCA-3′ (antisense). Fragments were inserted into KpnI/HindIII-containing sites of the luciferase vector, pGL3-Basic, to express pIL-35/EBI3-Luc and pIL-35/p35-Luc, respectively. All constructs were confirmed by DNA sequencing.
The following reagents were purchased: antibodies against PKR, OAS, Mx, GAPDH, NF-κB/p65 and p50, IFNAR1, IFNLR1, IκB, p-IκB, and IL-35/EBI3 (Santa Cruz Biotechnology, Santa Cruz, CA); antibodies against IL-35/p35 (R&D Systems, Minneapolis, MN); a human IL-35 ELISA kit (eBioscience, San Diego, CA); recombinant human IL-35 (rhIL-35, Sino Biological Inc., Beijing, China); prostaglandin E2 (PGE2, Sigma); sodium nitroferricyanide(III) dihydrate (SNP, nitric oxide donor, Beyotime, China), and the inhibitor of iNOS, SMT; recombinant human IFNγ (Peprotech, London, UK), si-COX-2,si-iNOS, and si-p35 (Gene Pharma, China). The inhibitor of COX-2, Etoricoxib (Frosst Iberica SA, Madrid, Spain), was purchased from a pharmacy. shIFNAR1 and shIFNLR1 were constructed in this study.
Reverse Transcription and Relative Quantitative Real-time PCR (qPCR)
Total cellular RNA was extracted using TRIzol (Invitrogen). Samples were reverse-transcribed using random primers at 37 °C for 1 h. qPCR was performed using SYBR Select Master Mix (antibody) with primers for the indicated target gene: for GAPDH, 5′-AAGGCTGTGGGCAAGG-3′ (sense), 5′-TGGAGGAGTGGGTGTCG-3′ (antisense); for IL-35/EBI3, 5′-GCCTGCTCCAAACTCCAC-3′ (sense), 5′-CGGGCTTGATGATGTGCT-3′ (antisense); for IL-35/p35, 5′-GTACCAGGTGGAGTTCAA-3′ (sense), 5′-AATAGTCACTGCCCGAAT-3′ (antisense); for p19, 5′-TGGAGATGGCTGTGACCC-3′ (sense), 5′-TGGGACTGAGGCTTGGAATC-3′ (antisense); for p28, 5′-CGCTTTGCGGAATCTCAC-3′ (sense), 5′-GGGCATGGAAGGGCTGAA-3′ (antisense); for p40, 5′-AGGGACATCATCAAACCTGACC-3′ (sense); 5′-GCTGAGGTCTTGTCCGTGAA-3′ (antisense); for COX-2, 5′-TGCATTCTTTGCCCAGCACT-3′ (sense), 5′-AAAGGCGCAGTTTACGCTGT-3′ (antisense); for iNOS, 5′-GTTCTCAAGGCACAGGTCTC-3′ (sense), 5′-GCAGGTCACTTATGTCACTTATC-3′ (antisense); for IFN-α, 5′-TTTCTCCTGCCTGAAGGACAG-3′ (sense), 5′-GCTCATGATTTCTGCTCTGACA-3′ (antisense); for IFN-β, 5′-AAAGAAGCAGCAATTTTCAGC-3′ (sense), 5′-CCTTGGCCTTCAGGTAATGCA-3′ (antisense); for IFNλ1, 5′-CTTCCAAGCCCACCCCAACT-3′ (sense), 5′-GGCCTCCAGGACCTTCAGC-3′ (antisense); for PKR, 5′-AGAGTAACCGTTGGTGACATAACCT-3′ (sense), 5′-GCAGCCTCTGCAGCTCTATGTT-3′ (antisense); for OAS, 5′-AGAAGGCAGCTCACGAAACC-3′(sense), 5′-CCACCACCCAAGTTTCCTGTA-3′ (antisense); for Mx, 5′-GCCGGCTGTGGATATGCTA-3′ (sense), 5′-TTTATCGAAACATCTGTGAAAGCAA-3′ (antisense). EV71 copies were measured using the absolute value of qPCR of viral VP-1with primers: 5′-CCCTTTAGTGGTTAGGATTT-3′ (sense), 5′-CACCAGTTGGTTTAATGGAG-3′ (antisense).
Measurement of IAV Replication
A549 cells were infected with IAV (m.o.i. = 1), as previously described (68). Viral titers were measured using a hemagglutination assay in U-shaped plates at various time points, as described previously (51). Total RNAs were extracted with TRIzol according to the manufacturer's instructions. Then samples were divided equally and reverse-transcribed using the following primers: specific NP minus-sense RNA, 5′-CTCACCGAGTGACATCAACATCATG-3′; NP plus-sense RNA, 5′-AGTAGAAACAAGGGTATTTTTCTTTAATTGTCAT-3′. Relative levels of IAV RNA were measured using qPCR as reported (61). NP qPCR primers were 5′-ATCAGACCGAACGAGAATCCAGC-3′ (sense) and 5′-GGAGGCCCTCTGTTGATTAGTGT-3′ (antisense).
Western Blots
Proteins were extracted from cultured cells via lysis (0.01% EDTA, 0.1% Triton X-100, 10% proteinase inhibitor mixture) on ice followed by sonication and centrifugation. Protein concentrations in the supernatant were quantified using a protein assay kit (Bio-Rad). Western blot analysis was performed with samples normalized to GAPDH or β-actin. Immunoblots were visualized using Chemiluminescent HRP substrate (Millipore, Billerica, MA).
ELISA
IL-35 concentration in culture supernatants was measured using a human IL-35 ELISA kit (88-7357, eBioscience), with an EBI3-specific capture antibody and a p35-specific detection antibody, according to the manufacturer's instructions. HBV expression was detected, as previously described (69). HBsAg and HBeAg levels in culture supernatants 48 h post-transfection were measured using an ELISA and a HBV HBsAg and HBeAg diagnostic kit (Shanghai KeHua Biotech, Shanghai, China).
Chromatin Immunoprecipitation (ChIP) Assay
ChIP of A549 cells infected with IAV was performed using a p65 or IgG antibody as previously described (70). Bond DNA fractions were measured using qPCR, and the primers were: ChIP 1, sense (5′ to 3′) CCCTCCATGTCCCTGAGC, antisense (5′ to 3′) GCTGTGAAAGGGAGAAGTG; ChIP 2, sense (5′ to 3′) CAGCAGGTCTCCACTTCT, antisense (5′ to 3′) GGGAGATGAATTAGGGTG; ChIP 3, sense (5′ to 3′), CCCCACCAGTGAGTCAGA, antisense (5′ to 3′) CGTCGTGGGAGACTTGAG; ChIP 4, sense (5′ to 3′) AGGGCCTGGGTGTAGTCA, antisense (5′ to 3′) TTTGGATGTGCTGGGTTT.
Luciferase Assay
A549 cells were transfected with luciferase reporter plasmids containing the promoter of IL-35 along with pRL-TK infected with IAV. Luciferase activity was measured after 12 h or 24 h of serum starvation and normalized to Renilla luciferase activity. Results are expressed as relative luciferase activity.
MTT Assay
90-μl Jurkat cell suspensions were plated on 96-well plates (104/cm2). rhIL-35 (0, 5, 10, 50, 100, or 500 ng/ml) was added to wells with or without IFNγ (2 μg/ml, total <10 μl). Each group contained 5 wells. Each plate contained a zero-value well with DMSO but without cells. Plates were cultured at 37 °C in a 5% CO2 incubator for 24 h. MTT reagent (20 μl, 5 mg/ml, methylthiazolyldiphenyl-tetrazolium bromide dissolved in PBS, pH 7.4, stirred at constant temperature for 30 min and passed through a 0.22-μm microfiltration membrane, stored at −20 °C) was added to each well, and cells were cultured for another 4 h. Triple solution (100 μl, 10% SDS, 5% isobutyl alcohol, 0.012 m HCl) was added to each well, and plates were then cultured at 37 °C overnight. Absorbance values for each well were determined at 570 nm.
A549 cells were plated on 96-well plates (104/cm2) and cultured in DMEM (200 μl/well with 10% FBS). Cells were treated with Jurkat supernatants previously incubated with 100 ng/ml rhIL-35 with or without 2 μg/ml IFNγ. After 24 h, MTT reagent (20 μl) was added to each well, and cells were cultured for 4 h. Media were removed, and DMSO (150 μl) was added to each well with shaking for 10 min to dissolve formazan. Absorbance values for each well were determined at 490 nm, adjusted to the zero-value well.
Statistical Analysis
Relationships between IL-35 expression and either IAV NP levels in clinical samples, COX-2 mRNA, or iNOS mRNA levels were analyzed using Pearson's correlation. All the experiments were performed in triplicate. Two-group comparisons were performed using Student's t test. Data are expressed as the mean ± S.D., except for the clinical results, shown as mean ± S.E. Results with p < 0.05 (*) and p < 0.01 (**) were considered statistically significant.
Author Contributions
Y. Z. conceived and coordinated the study. L. W. was designed and performed the study and wrote the paper. S. Z. participated in experimental scheme and analyzed the results. G. X. construct the plasmids and analyzed the experiments shown in Figs. 3 and 5. J. F., T. H., and F. Z. analyzed the experiments shown in Figs. 1, 2, and 6, respectively. Y.-L. S. prepared all the experiment materials. S. L. analyzed the data and proofread the manuscript. L. Y. directed the experiments. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgment
We thank Jiahuai Han laboratory of Xiamen University for kindly providing IL-12A cDNA.
This work was supported by research grants from the Major State Basic Research Development Program of China (2013CB911102) and National Natural Science Foundation of China (81461130019, 81271821, 31500149, and 31570870). The authors declare that they have no conflicts of interest with the contents of this article.
- IAV
- influenza A virus
- Treg
- regulatory T cell
- EBI3
- Epstein-Barr virus-induced gene 3
- COX-2
- cyclooxygenase-2
- iNOS
- inducible nitric-oxide synthase
- m.o.i.
- multiplicity of infection
- HBV
- hepatitis B virus
- EV71
- enterovirus 71
- VSV
- vesicular stomatitis virus
- eGFP
- enhanced green fluorescent protein
- NP
- nucleoprotein
- PKR
- double-stranded RNA-activated protein kinase
- OAS
- 2′,5′-oligoadenylate synthetase
- Mx
- myxovirus resistance protein
- AT II
- human alveolar type II
- qPCR
- relative quantitative real-time PCR
- qRT
- quantitative real-time
- PBMC
- peripheral blood mononuclear cell
- CREB
- cAMP-response element-binding protein
- PG
- prostaglandin
- hpi
- hours post infection
- RD
- rhabdomyosarcoma
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- r
- correlation coefficient
- HBeAg
- hepatitis B e antigen
- HBsAg
- hepatitis B s antigen.
References
- 1. Tripathi S., White M. R., and Hartshorn K. L. (2015) The amazing innate immune response to influenza A virus infection. Innate Immun. 21, 73–98 [DOI] [PubMed] [Google Scholar]
- 2. Shaw M. W., Arden N. H., and Maassab H. F. (1992) New aspects of influenza viruses. Clin. Microbiol. Rev. 5, 74–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Londrigan S. L., Tate M. D., Brooks A. G., and Reading P. C. (2012) Cell-surface receptors on macrophages and dendritic cells for attachment and entry of influenza virus. J. Leukoc. Biol. 92, 97–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mogensen T. H., and Paludan S. R. (2001) Molecular pathways in virus-induced cytokine production. Microbiol. Mol. Biol. Rev. 65, 131–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nakamura R., Maeda N., Shibata K., Yamada H., Kase T., and Yoshikai Y. (2010) Interleukin-15 is critical in the pathogenesis of influenza a virus-induced acute lung injury. J. Virol. 84, 5574–5582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li W., Yang F., Liu Y., Gong R., Liu L., Feng Y., Hu P., Sun W., Hao Q., Kang L., Wu J., and Zhu Y. (2009) Negative feedback regulation of IL-32 production by iNOS activation in response to dsRNA or influenza virus infection. Eur. J. Immunol. 39, 1019–1024 [DOI] [PubMed] [Google Scholar]
- 7. Li W., Sun W., Liu L., Yang F., Li Y., Chen Y., Fang J., Zhang W., Wu J., and Zhu Y. (2010) IL-32: a host proinflammatory factor against influenza viral replication is upregulated by aberrant epigenetic modifications during influenza A virus infection. J. Immunol. 185, 5056–5065 [DOI] [PubMed] [Google Scholar]
- 8. Fukuyama S., and Kawaoka Y. (2011) The pathogenesis of influenza virus infections: the contributions of virus and host factors. Curr. Opin. Immunol. 23, 481–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Collison L. W., and Vignali D. A. (2008) Interleukin-35: odd one out or part of the family? Immunol. Rev. 226, 248–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang B., Dai S., Dong Z., Sun Y., Song X., Guo C., Zhu F., Wang Q., and Zhang L. (2014) The modulation of endoplasmic reticulum stress by chemical chaperone upregulates immune negative cytokine IL-35 in apolipoprotein E-deficient mice. PloS One 9, e87787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Niedbala W., Wei X. Q., Cai B., Hueber A. J., Leung B. P., McInnes I. B., and Liew F. Y. (2007) IL-35 is a novel cytokine with therapeutic effects against collagen-induced arthritis through the expansion of regulatory T cells and suppression of Th17 cells. Eur. J. Immunol. 37, 3021–3029 [DOI] [PubMed] [Google Scholar]
- 12. Devergne O., Hummel M., Koeppen H., Le Beau M. M., Nathanson E. C., Kieff E., and Birkenbach M. (1996) A novel interleukin-12 p40-related protein induced by latent Epstein-Barr virus infection in B lymphocytes. J. Virol. 70, 1143–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Devergne O., Birkenbach M., and Kieff E. (1997) Epstein-Barr virus-induced gene 3 and the p35 subunit of interleukin 12 form a novel heterodimeric hematopoietin. Proc. Natl. Acad. Sci. U.S.A. 94, 12041–12046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Collison L. W., Delgoffe G. M., Guy C. S., Vignali K. M., Chaturvedi V., Fairweather D., Satoskar A. R., Garcia K. C., Hunter C. A., Drake C. G., Murray P. J., and Vignali D. A. (2012) The composition and signaling of the IL-35 receptor are unconventional. Nat. Immunol. 13, 290–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu J. Q., Liu Z., Zhang X., Shi Y., Talebian F., Carl J. W. Jr., Yu C., Shi F. D., Whitacre C. C., Trgovcich J., and Bai X. F. (2012) Increased Th17 and regulatory T cell responses in EBV-induced gene 3-deficient mice lead to marginally enhanced development of autoimmune encephalomyelitis. J. Immunol. 188, 3099–3106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bettini M., Castellaw A. H., Lennon G. P., Burton A. R., and Vignali D. A. (2012) Prevention of autoimmune diabetes by ectopic pancreatic β-cell expression of interleukin-35. Diabetes 61, 1519–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wirtz S., Billmeier U., Mchedlidze T., Blumberg R. S., and Neurath M. F. (2011) Interleukin-35 mediates mucosal immune responses that protect against T-cell-dependent colitis. Gastroenterology 141, 1875–1886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang C. H., Loo E. X., Kuo I. C., Soh G. H., Goh D. L., Lee B. W., and Chua K. Y. (2011) Airway inflammation and IgE production induced by dust mite allergen-specific memory/effector Th2 cell line can be effectively attenuated by IL-35. J. Immunol. 187, 462–471 [DOI] [PubMed] [Google Scholar]
- 19. Wong C. K., Leung T. F., Chu I. M., Dong J., Lam Y. Y., and Lam C. W. (2015) Aberrant expression of regulatory cytokine IL-35 and pattern recognition receptor NOD2 in patients with allergic asthma. Inflammation 38, 348–360 [DOI] [PubMed] [Google Scholar]
- 20. Jafarzadeh A., Jamali M., Mahdavi R., Ebrahimi H. A., Hajghani H., Khosravimashizi A., Nemati M., Najafipour H., Sheikhi A., Mohammadi M. M., and Daneshvar H. (2015) Circulating levels of interleukin-35 in patients with multiple sclerosis: evaluation of the influences of FOXP3 gene polymorphism and treatment program. J. Mol. Neurosci. 55, 891–897 [DOI] [PubMed] [Google Scholar]
- 21. Kalburgi N. B., Muley A., Shivaprasad B. M., and Koregol A. C. (2013) Expression profile of IL-35 mRNA in gingiva of chronic periodontitis and aggressive periodontitis patients: a semiquantitative RT-PCR study. Dis. Markers 35, 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zeng J. C., Zhang Z., Li T. Y., Liang Y. F., Wang H. M., Bao J. J., Zhang J. A., Wang W. D., Xiang W. Y., Kong B., Wang Z. Y., Wu B. H., Chen X. D., He L., Zhang S., Wang C. Y., and Xu J. F. (2013) Assessing the role of IL-35 in colorectal cancer progression and prognosis. Int. J. Clin. Exp. Pathol. 6, 1806–1816 [PMC free article] [PubMed] [Google Scholar]
- 23. Long J., Zhang X., Wen M., Kong Q., Lv Z., An Y., and Wei X. Q. (2013) IL-35 over-expression increases apoptosis sensitivity and suppresses cell growth in human cancer cells. Biochem. Biophys. Res. Commun. 430, 364–369 [DOI] [PubMed] [Google Scholar]
- 24. Sakaguchi S., Sakaguchi N., Shimizu J., Yamazaki S., Sakihama T., Itoh M., Kuniyasu Y., Nomura T., Toda M., and Takahashi T. (2001) Immunologic tolerance maintained by CD25+ CD4+ regulatory T cells: their common role in controlling autoimmunity, tumor immunity, and transplantation tolerance. Immunol. Rev. 182, 18–32 [DOI] [PubMed] [Google Scholar]
- 25. Shevach E. M., DiPaolo R. A., Andersson J., Zhao D. M., Stephens G. L., and Thornton A. M. (2006) The lifestyle of naturally occurring CD4+ CD25+ Foxp3+ regulatory T cells. Immunol. Rev. 212, 60–73 [DOI] [PubMed] [Google Scholar]
- 26. Collison L. W., Chaturvedi V., Henderson A. L., Giacomin P. R., Guy C., Bankoti J., Finkelstein D., Forbes K., Workman C. J., Brown S. A., Rehg J. E., Jones M. L., Ni H. T., Artis D., Turk M. J., and Vignali D. A. (2010) IL-35-mediated induction of a potent regulatory T cell population. Nat. Immunol. 11, 1093–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Collison L. W., Workman C. J., Kuo T. T., Boyd K., Wang Y., Vignali K. M., Cross R., Sehy D., Blumberg R. S., and Vignali D. A. (2007) The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 450, 566–569 [DOI] [PubMed] [Google Scholar]
- 28. Yang J., Yang M., Htut T. M., Ouyang X., Hanidu A., Li X., Sellati R., Jiang H., Zhang S., Li H., Zhao J., Ting A. T., Mayer L., Unkeless J. C., Labadia M. E., Hodge M., Li J., and Xiong H. (2008) Epstein-Barr virus-induced gene 3 negatively regulates IL-17, IL-22 and RORγt. Eur. J. Immunol. 38, 1204–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xiang X. G., and Xie Q. (2015) IL-35: a potential therapeutic target for controlling hepatitis B virus infection. J. Dig. Dis. 16, 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu F., Tong F., He Y., and Liu H. (2011) Detectable expression of IL-35 in CD4+ T cells from peripheral blood of chronic hepatitis B patients. Clin. Immunol. 139, 1–5 [DOI] [PubMed] [Google Scholar]
- 31. Shi Y. Y., Dai M. J., Wu G. P., Zhou P. P., Fang Y., and Yan X. B. (2015) Levels of interleukin-35 and its relationship with regulatory T-cells in chronic hepatitis B patients. Viral Immunol. 28, 93–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu L., Cao Z., Chen J., Li R., Cao Y., Zhu C., Wu K., Wu J., Liu F., and Zhu Y. (2012) Influenza A virus induces interleukin-27 through cyclooxygenase-2 and protein kinase A signaling. J. Biol. Chem. 287, 11899–11910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Paich H. A., Sheridan P. A., Handy J., Karlsson E. A., Schultz-Cherry S., Hudgens M. G., Noah T. L., Weir S. S., and Beck M. A. (2013) Overweight and obese adult humans have a defective cellular immune response to pandemic H1N1 influenza A virus. Obesity 21, 2377–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Park M. K., Ngo V., Kwon Y. M., Lee Y. T., Yoo S., Cho Y. H., Hong S. M., Hwang H. S., Ko E. J., Jung Y. J., Moon D. W., Jeong E. J., Kim M. C., Lee Y. N., Jang J. H., Oh J. S., Kim C. H., and Kang S. M. (2013) Lactobacillus plantarum DK119 as a probiotic confers protection against influenza virus by modulating innate immunity. PloS One 8, e75368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pirhonen J., Matikainen S., and Julkunen I. (2002) Regulation of virus-induced IL-12 and IL-23 expression in human macrophages. J. Immunol. 169, 5673–5678 [DOI] [PubMed] [Google Scholar]
- 36. DiDonato J. A., Mercurio F., and Karin M. (2012) NF-κB and the link between inflammation and cancer. Immunol. Rev. 246, 379–400 [DOI] [PubMed] [Google Scholar]
- 37. Razani B., Reichardt A. D., and Cheng G. (2011) Non-canonical NF-κB signaling activation and regulation: principles and perspectives. Immunol. Rev. 244, 44–54 [DOI] [PubMed] [Google Scholar]
- 38. Poleganov M. A., Bachmann M., Pfeilschifter J., and Mühl H. (2008) Genome-wide analysis displays marked induction of EBI3/IL-27B in IL-18-activated AML-derived KG1 cells: critical role of two κB binding sites in the human EBI3 promotor. Mol. Immunol. 45, 2869–2880 [DOI] [PubMed] [Google Scholar]
- 39. Wang Q., Chen X., Feng J., Cao Y., Song Y., Wang H., Zhu C., Liu S., and Zhu Y. (2013) Soluble interleukin-6 receptor-mediated innate immune response to DNA and RNA viruses. J. Virol. 87, 11244–11254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yue X., Yang F., Yang Y., Mu Y., Sun W., Li W., Xu D., Wu J., and Zhu Y. (2011) Induction of cyclooxygenase-2 expression by hepatitis B virus depends on demethylation-associated recruitment of transcription factors to the promoter. Virol. J. 8, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mohammed N. A., Abd El-Aleem S. A., El-Hafiz H. A., and McMahon R. F. (2004) Distribution of constitutive (COX-1) and inducible (COX-2) cyclooxygenase in postviral human liver cirrhosis: a possible role for COX-2 in the pathogenesis of liver cirrhosis. J. Clin. Pathol. 57, 350–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li Y., Xie J., Xu X., Liu L., Wan Y., Liu Y., Zhu C., and Zhu Y. (2013) Inducible interleukin 32 (IL-32) exerts extensive antiviral function via selective stimulation of interferon λ1 (IFN-λ1). J. Biol. Chem. 288, 20927–20941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ma X., Chow J. M., Gri G., Carra G., Gerosa F., Wolf S. F., Dzialo R., and Trinchieri G. (1996) The interleukin 12 p40 gene promoter is primed by interferon γ in monocytic cells. J. Exp. Med. 183, 147–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kriegel M. A., Tretter T., Blank N., Schiller M., Gabler C., Winkler S., Kalden J. R., and Lorenz H. M. (2006) Interleukin-4 supports interleukin-12-induced proliferation and interferon-γ secretion in human activated lymphoblasts and T helper type 1 cells. Immunology 119, 43–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Parham C., Chirica M., Timans J., Vaisberg E., Travis M., Cheung J., Pflanz S., Zhang R., Singh K. P., Vega F., To W., Wagner J., O'Farrell A. M., McClanahan T., Zurawski S., Hannum C., et al. (2002) A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rβ1 and a novel cytokine receptor subunit, IL-23R. J. Immunol. 168, 5699–5708 [DOI] [PubMed] [Google Scholar]
- 46. Hall A. O., Beiting D. P., Tato C., John B., Oldenhove G., Lombana C. G., Pritchard G. H., Silver J. S., Bouladoux N., Stumhofer J. S., Harris T. H., Grainger J., Wojno E. D., Wagage S., Roos D. S., et al. (2012) The cytokines interleukin 27 and interferon-γ promote distinct Treg cell populations required to limit infection-induced pathology. Immunity 37, 511–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Afkarian M., Sedy J. R., Yang J., Jacobson N. G., Cereb N., Yang S. Y., Murphy T. L., and Murphy K. M. (2002) T-bet is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells. Nat. Immunol. 3, 549–557 [DOI] [PubMed] [Google Scholar]
- 48. Emeny J. M., and Morgan M. J. (1979) Regulation of the interferon system: evidence that Vero cells have a genetic defect in interferon production. J. Gen. Virol. 43, 247–252 [DOI] [PubMed] [Google Scholar]
- 49. Pauli E. K., Schmolke M., Wolff T., Viemann D., Roth J., Bode J. G., and Ludwig S. (2008) Influenza A virus inhibits type I IFN signaling via NF-κB-dependent induction of SOCS-3 expression. PLoS Pathog. 4, e1000196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Prescott J., Hall P., Acuna-Retamar M., Ye C., Wathelet M. G., Ebihara H., Feldmann H., and Hjelle B. (2010) New World hantaviruses activate IFNλ production in type I IFN-deficient vero E6 cells. PloS One 5, e11159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mukhtar M. M., Li S., Li W., Wan T., Mu Y., Wei W., Kang L., Rasool S. T., Xiao Y., Zhu Y., and Wu J. (2009) Single-chain intracellular antibodies inhibit influenza virus replication by disrupting interaction of proteins involved in viral replication and transcription. Int. J. Biochem. Cell Biol. 41, 554–560 [DOI] [PubMed] [Google Scholar]
- 52. Short K. R., Veldhuis Kroeze E. J., Reperant L. A., Richard M., and Kuiken T. (2014) Influenza virus and endothelial cells: a species specific relationship. Front. Microbiol. 5, 653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Netea M. G., Azam T., Lewis E. C., Joosten L. A., Wang M., Langenberg D., Meng X., Chan E. D., Yoon D. Y., Ottenhoff T., Kim S. H., and Dinarello C. A. (2006) Mycobacterium tuberculosis induces interleukin-32 production through a caspase- 1/IL-18/interferon-γ-dependent mechanism. PLoS Med. 3, e277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ronni T., Sareneva T., Pirhonen J., and Julkunen I. (1995) Activation of IFN-α, IFN-γ, MxA, and IFN regulatory factor 1 genes in influenza A virus-infected human peripheral blood mononuclear cells. J. Immunol. 154, 2764–2774 [PubMed] [Google Scholar]
- 55. Zhou Y., Zhang H., and Li Y. (2015) IL-35 expression in peripheral blood CD4(+) T cells from chronic hepatitis B virus-infected patients directly correlates with virus load. Cytokine 73, 169–175 [DOI] [PubMed] [Google Scholar]
- 56. Li X., Tian L., Dong Y., Zhu Q., Wang Y., Han W., Liu X., Ni Q., Chen Y., and Li L. (2015) IL-35 inhibits HBV antigen-specific IFN-γ-producing CTLs in vitro. Clin. Sci. 129, 395–404 [DOI] [PubMed] [Google Scholar]
- 57. Li Q., and Verma I. M. (2002) NF-κB regulation in the immune system. Nat. Rev. Immunol. 2, 725–734 [DOI] [PubMed] [Google Scholar]
- 58. Mercurio F., Zhu H., Murray B. W., Shevchenko A., Bennett B. L., Li J., Young D. B., Barbosa M., Mann M., Manning A., and Rao A. (1997) IKK-1 and IKK-2: cytokine-activated IκB kinases essential for NF-κB activation. Science 278, 860–866 [DOI] [PubMed] [Google Scholar]
- 59. Zandi E., Rothwarf D. M., Delhase M., Hayakawa M., and Karin M. (1997) The IκB kinase complex (IKK) contains two kinase subunits, IKKα and IKKβ, necessary for IκB phosphorylation and NF-κB activation. Cell 91, 243–252 [DOI] [PubMed] [Google Scholar]
- 60. Lee S. M., Cheung C. Y., Nicholls J. M., Hui K. P., Leung C. Y., Uiprasertkul M., Tipoe G. L., Lau Y. L., Poon L. L., Ip N. Y., Guan Y., and Peiris J. S. (2008) Hyperinduction of cyclooxygenase-2-mediated proinflammatory cascade: a mechanism for the pathogenesis of avian influenza H5N1 infection. J. Infect. Dis. 198, 525–535 [DOI] [PubMed] [Google Scholar]
- 61. Li W., Liu Y., Mukhtar M. M., Gong R., Pan Y., Rasool S. T., Gao Y., Kang L., Hao Q., Peng G., Chen Y., Chen X., Wu J., and Zhu Y. (2008) Activation of interleukin-32 pro-inflammatory pathway in response to influenza A virus infection. PloS One 3, e1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liu L., Li R., Pan Y., Chen J., Li Y., Wu J., and Zhu Y. (2011) High-throughput screen of protein expression levels induced by cyclooxygenase-2 during influenza a virus infection. Clin. Chim. Acta 412, 1081–1085 [DOI] [PubMed] [Google Scholar]
- 63. Imanishi N., Andoh T., Sakai S., Satoh M., Katada Y., Ueda K., Terasawa K., and Ochiai H. (2005) Induction of inducible nitric oxide (NO) synthase mRNA and NO production in macrophages infected with influenza A/PR/8 virus and stimulated with its ether-split product. Microbiol. Immunol. 49, 41–48 [DOI] [PubMed] [Google Scholar]
- 64. Rasool S. T., Tang H., Wu J., Li W., Mukhtar M. M., Zhang J., Mu Y., Xing H. X., Wu J., and Zhu Y. (2008) Increased level of IL-32 during human immunodeficiency virus infection suppresses HIV replication. Immunol. Lett. 117, 161–167 [DOI] [PubMed] [Google Scholar]
- 65. Barnes B., Lubyova B., and Pitha P. M. (2002) On the role of IRF in host defense. J. Interferon Cytokine Res. 22, 59–71 [DOI] [PubMed] [Google Scholar]
- 66. Robek M. D., Boyd B. S., and Chisari F. V. (2005) λ interferon inhibits hepatitis B and C virus replication. J. Virol. 79, 3851–3854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Choi H. J., Dinarello C. A., and Shapiro L. (2001) Interleukin-18 inhibits human immunodeficiency virus type 1 production in peripheral blood mononuclear cells. J. Infect. Dis. 184, 560–568 [DOI] [PubMed] [Google Scholar]
- 68. Zou F., Liu Y., Liu L., Wu K., Wei W., Zhu Y., and Wu J. (2007) Retinoic acid activates human inducible nitric-oxide synthase gene through binding of RARα/RXRα heterodimer to a novel retinoic acid response element in the promoter. Biochem. Biophys. Res. Commun. 355, 494–500 [DOI] [PubMed] [Google Scholar]
- 69. Pan Y., Wei W., Kang L., Wang Z., Fang J., Zhu Y., and Wu J. (2007) NS5A protein of HCV enhances HBV replication and resistance to interferon response. Biochem. Biophys. Res. Commun. 359, 70–75 [DOI] [PubMed] [Google Scholar]
- 70. Yan X., Hao Q., Mu Y., Timani K. A., Ye L., Zhu Y., and Wu J. (2006) Nucleocapsid protein of SARS-CoV activates the expression of cyclooxygenase-2 by binding directly to regulatory elements for nuclear factor-κ B and CCAAT/enhancer binding protein. Int. J. Biochem. Cell Biol. 38, 1417–1428 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]