

Abstract
Mutations in the gene encoding parkin, an auto-inhibited E3 ubiquitin ligase that functions in the clearance of damaged mitochondria, are the most common cause of autosomal recessive juvenile Parkinsonism. The mechanism regulating parkin activation remains poorly understood. Here we show, by using isothermal titration calorimetry, solution NMR, and fluorescence spectroscopy, that parkin can bind ubiquitin and phosphomimetic ubiquitin by recognizing the canonical hydrophobic patch and C terminus of ubiquitin. The affinity of parkin for both phosphomimetic and unmodified ubiquitin is markedly enhanced upon removal of the ubiquitin-like (UBL) domain of parkin. This suggests that the agonistic binding of ubiquitin to parkin in trans is counterbalanced by the antagonistic activity of the parkin UBL domain in cis. Intriguingly, UBL binding is enthalpy-driven, whereas ubiquitin binding is driven by an increase in the total entropy of the system. These thermodynamic differences are explained by different chemistry in the ubiquitin- and UBL-binding pockets of parkin and, as shown by molecular dynamics simulations, are not a consequence of changes in protein conformational entropy. Indeed, comparison of conformational fluctuations reveals that the RING1-IBR element becomes considerably more rigid upon complex formation. A model of parkin activation is proposed in which E2∼Ub binding triggers large scale diffusional motion of the RING2 domain toward the ubiquitin-stabilized RING1-IBR assembly to complete formation of the active parkin-E2∼Ub transfer complex. Thus, ubiquitin plays a dual role in parkin activation by competing with the inhibitory UBL domain and stabilizing the active form of parkin.
Keywords: isothermal titration calorimetry (ITC), molecular dynamics, parkin, Parkinson disease, protein-protein interaction, ubiquitin, ubiquitin ligase, protein dynamics
Introduction
Mutations in the PARK2 gene that encodes the protein parkin have been found to cause a familial form of Parkinson disease termed autosomal recessive juvenile parkinsonism (AR-JP)2 (1–3). Parkin is an E3 ubiquitin ligase of the RBR family (Fig. 1A). RBR-type ubiquitin ligases function by first forming a covalent thioester intermediate between a key catalytic cysteine and the C terminus of ubiquitin, and then transferring the activated ubiquitin to their substrate (4). Under basal conditions, parkin resides in the cytosol (5, 6), and cytosolic parkin is thought to be catalytically inactive (5). Accordingly, recently reported crystal structures show parkin in an auto-inhibited conformation; both the interaction platform with the incoming E2 enzyme and the catalytic cysteine residue Cys431 are occluded because of intramolecular interactions (Fig. 1B) (7–9). Autoregulation of E3 activity by the formation of such inhibitory interactions is a common feature of RBR ligases and has been previously described for the ubiquitin ligases HHARI and HOIP (4, 10, 11).
FIGURE 1.

Parkin is an auto-inhibited E3 enzyme that requires structural rearrangement for activation. A, domain architecture of rat parkin, comprising a UBL domain, an ∼70-residue linker, and four domains of the RING family. Parkin is an RBR-type ubiquitin ligase with its catalytic center embedded in the most C-terminal RING2 domain (Cys431 in rat parkin). The RING1 domain serves as an adapter platform for the incoming E2 enzyme. The yellow region represents a short REP, which functions in the auto-inhibition of parkin (7). B, quaternary structure of full-length parkin, drawn from PDB code 4K95 (7). Key sites are indicated. Important auto-inhibitory interactions in parkin include blockade of the catalytic cysteine by the RING0 domain and occlusion of the E2-binding site (blue) by the REP element. C and D, structural comparison of free parkin (C) from R. norvegicus (PDB code 4K95 (7)) and phosphoubiquitin-bound parkin (D) from P. humanus (PDB code 5CAW (19)). In both crystal structures, the catalytic cysteine is occluded by the RING0 domain. Protein representations were generated by PyMOL (Schrödinger, LLC.).
Parkin functions in the ubiquitylation of mitochondrial proteins, an event that initiates the degradation of dysfunctional mitochondria by mitophagy (5, 6); this implies that the latent E3 ubiquitin ligase activity of parkin can be released in response to external factors. At least two events are required to release the latent E3 activity of parkin in cells. It has been shown that upon loss of the electrochemical potential across the inner mitochondrial membrane, the protein kinase PINK1, which is anchored on the outer mitochondrial membrane with the kinase domain facing the cytoplasm, phosphorylates parkin at residue Ser65 of its UBL domain (12–14). Furthermore, it has been reported that PINK1 also phosphorylates ubiquitin, intriguingly at the homologous residue Ser65, which is conserved between ubiquitin and the UBL domain of parkin (15–17).
How these two phosphorylation events act together to convert parkin from an inactive to a catalytically active state is not fully understood. Alone, the two events are not sufficient to induce translocation of parkin to mitochondria, but they are required for its translocation and are sufficient to release the latent E3 ubiquitin ligase activity of parkin in vivo (15, 16).
A leading hypothesis for parkin activation is that Ser65-phosphorylated ubiquitin (hereafter referred to as “phosphoubiquitin”) may activate parkin in an allosteric manner (15, 18, 19). Indeed, it has been demonstrated in vitro that the presence of phosphoubiquitin in a ubiquitylation assay solution is sufficient to activate parkin without the need for phosphorylation of the parkin UBL domain (18).
The aim of this study was therefore to investigate whether ubiquitin can exert a direct influence on the multidomain structure of parkin, which ultimately leads to release of its latent ubiquitin ligase activity.
Results
Ubiquitin Binding to Parkin
Direct allosteric activation of parkin by ubiquitin would require a physical interaction of the two molecules in solution. Indeed, we detected such a non-covalent association by NMR spectroscopy. Addition of an equimolar amount of unlabeled parkin to 15N-labeled ubiquitin markedly changed the HSQC spectrum of ubiquitin (Fig. 2A). Several resonances of ubiquitin, such as Ile36, shifted in a fast exchange-like manner upon the addition of parkin, while displaying a small degree of line broadening. Conversely, other resonances, such as Val70, were severely broadened and were no longer detectable, suggesting that ubiquitin binds to parkin by a specific interaction. Such non-covalent binding of ubiquitin to an E3 ligase is not typical because catalytic processing of ubiquitin exclusively involves its covalent transfer via cysteine residues (20). Thus, we inferred that binding of ubiquitin may have a specific role in parkin function. The complete loss of Leu8 and Val70 signals suggested that parkin binds to the canonical hydrophobic patch on ubiquitin (Fig. 2B) (20). Moreover, loss of the amide cross-peaks of Arg72, Leu73, and Arg74 of ubiquitin indicated that parkin also interacts with the flexible C terminus of ubiquitin in solution.
FIGURE 2.

Physical interaction of parkin with ubiquitin. A, transverse relaxation-optimized spectroscopy-HSQC spectrum of 100 μm 15N-labeled ubiquitin before (black) and after (red) addition of 1 mol eq of unlabeled full-length parkin, showing selective line broadening after the addition of parkin. B, residue-specific NMR signal loss in ubiquitin upon the addition of parkin. Green bars, residues not observed in the reference spectrum of free ubiquitin; red bars, residues for which near-complete signal loss occurred upon the addition of parkin. Red line, average intensity ratio. C, structural model of ubiquitin binding to parkin based on PDB code 5CAW (19). Residues (one-letter code) with significant line broadening in the NMR experiment are colored red.
Direct determination of the ubiquitin-binding site on parkin by NMR spectroscopy was obstructed by inefficient folding of parkin in isotope-labeled minimal media. While this study was in progress, a crystal structure of phosphoubiquitin bound to the parkin core was reported (19). Superimposing ubiquitin on that crystal structure brought residues showing severe line-broadening in our NMR experiments close to the surface of parkin, thereby explaining the line-broadening as interaction of ubiquitin's hydrophobic patch and C terminus with residues on the surface of parkin (Fig. 2C).
Accordingly, these data argue that parkin and ubiquitin specifically interact in solution. Notably, ubiquitin can interact with parkin even without phosphorylation of Ser65. Because the intracellular concentration of ubiquitin is on the order of ∼85 μm (21), which is much higher than that of other proteins, including parkin and phosphoubiquitin (15), our data suggest that cytosolic (inactive) parkin may be often bound to unmodified ubiquitin in vivo. Nevertheless, it has been reported that only phosphoubiquitin or phosphomimetic (S65D) ubiquitin activates parkin in cells (15). On the basis of these considerations, we hypothesized that, although unmodified ubiquitin can bind to parkin, binding of phosphoubiquitin might exert a specific effect that facilitates the release of latent E3 activity. The nature of such an effect is not evident from the currently available static structural data alone because, in the recent crystal structure of phosphoubiquitin-bound parkin, the catalytic cysteine residue is still buried inside the protein and thereby unable to participate in the ubiquitylation cascade (Fig. 1, C and D) (19). Thus, we assumed that phosphoubiquitin binding to parkin must play a more subtle role in the activation of parkin.
Thermodynamic Evaluation of Ubiquitin Binding to Full-length Parkin
To evaluate the binding of ubiquitin and parkin in solution in more detail, we employed isothermal titration calorimetry (ITC), which confirmed the binding of ubiquitin to parkin. Intriguingly, addition of ubiquitin to parkin was found to be an endothermic reaction with a dissociation constant of ∼70 μm (Fig. 3A). Because it is not ubiquitin per se, but rather phosphoubiquitin that is thought to activate parkin, we predicted that the affinity of phosphoubiquitin for parkin would be stronger than that of unmodified ubiquitin.
FIGURE 3.

Ubiquitin binding to parkin is entropy-driven. A and B, isothermal titration calorimetry thermogram for ubiquitin (A) and phosphomimetic ubiquitin (B) binding to full-length parkin, showing an endothermic reaction. Upper panels show raw data; lower panels show the integrated heat values. C, dissociation constants determined from the ITC experiments. Unmodified ubiquitin and phosphomimetic ubiquitin bind to full-length parkin with similar avidity (p = 0.16; Student's t test). D, entropy drives the binding of both ubiquitin and phosphomimetic ubiquitin to parkin. The binding process is disfavored enthalpically, but this is counteracted by a large favorable entropic term. Error bars indicate the S.E. in C and D of three independent experiments (n = 3).
To examine this possibility, we employed the S65D ubiquitin mutant, which has been successfully used as a mimic for phosphorylated ubiquitin (15, 16, 22). ITC experiments with phosphomimetic ubiquitin resulted in a dissociation constant of ∼35 μm; however, statistical analysis precluded the conclusion that phosphomimetic ubiquitin bound to full-length parkin more avidly than wild-type ubiquitin (Fig. 3, B and C). Interestingly, the entropic contribution to the binding reaction was enhanced in the case of phosphomimetic ubiquitin. These results suggest that parkin has a specific, entropy-driven binding mode for phosphorylated ubiquitin.
Ubiquitin Binding to the Parkin Core
Direct interpretation of the thermodynamic driving force behind the binding is complex, because changes in both solvent and protein conformational entropy must be taken into account. Moreover, the presence of the UBL domain and the ∼70-residue linker between the UBL and the parkin core (Fig. 1A) further complicated the interpretation. Therefore, to analyze the effect of ubiquitin binding on the catalytic core of parkin specifically, we deleted the N-terminal UBL domain and its adjacent linker in parkin and measured binding of the parkin core (hereafter parkin ΔUBL) to ubiquitin using ITC.
Parkin ΔUBL showed increased affinity for unmodified ubiquitin (Fig. 4A; Kd ∼18 μm) as compared with full-length parkin (Fig. 3A; Kd ∼70 μm). Notably, deletion of the UBL domain did not change the endothermic nature of the reaction, indicating that the entropic driving force behind the reaction is independent of the UBL domain and the adjacent linker.
FIGURE 4.

Binding of ubiquitin, phosphomimetic ubiquitin, and parkin UBL to the parkin core (parkin ΔUBL). A–C, isothermal titration calorimetry thermograms for ubiquitin (A), phosphomimetic ubiquitin (B), and parkin UBL (C) binding to parkin ΔUBL. Upper panels, raw data; lower panels, integrated heat values. D, dissociation constants determined from the ITC experiments. Phosphomimetic ubiquitin binds to the parkin core more avidly than wild-type ubiquitin (p < 0.001; Student's t test). E, thermodynamic parameters for the binding of ubiquitin, phosphomimetic ubiquitin, and the parkin UBL to parkin ΔUBL. Error bars indicate the S.E. in D and E of three independent experiments (n = 3).
Because parkin bound unmodified and phosphomimetic ubiquitin with comparable affinity, we next determined whether this behavior would be preserved in the absence of the UBL domain of parkin. ITC experiments showed that phosphomimetic ubiquitin bound to parkin ΔUBL with a Kd of ∼1.5 μm (Fig. 4, B and D). Thus, parkin ΔUBL showed a 10-fold preference for phosphomimetic ubiquitin over unmodified ubiquitin. Again, the reaction was entropy-driven, and the entropic contribution was larger for phosphomimetic ubiquitin than for unmodified ubiquitin (Fig. 4E).
Because binding of (phospho-) ubiquitin to the parkin core is entropy-driven, we considered the role of the UBL domain in full-length parkin from a thermodynamic point of view. Using ITC, we found that the parkin UBL domain bound parkin ΔUBL with a Kd of ∼16 μm (Fig. 4C). Accordingly, parkin ΔUBL showed essentially the same affinity toward unmodified ubiquitin and its UBL domain in trans but bound phosphomimetic ubiquitin ∼10-fold more tightly (Fig. 4E). Unexpectedly, we found that binding of the UBL domain to parkin ΔUBL is an exothermic reaction. Thus, although ubiquitin and the parkin UBL bind to parkin ΔUBL with essentially the same affinity, the driving force behind the reaction is fundamentally reversed. Taken together, these observations indicate that the UBL domain and ubiquitin exert thermodynamically distinct influences on the parkin core, implying that they have different functional roles.
Global Structural Rearrangements in Parkin
Next, we asked whether ubiquitin binding to parkin caused conformational changes in parkin upon complex formation in solution. To this end, we exploited the fact that ubiquitin has no tryptophan residues, whereas parkin has seven that are clustered in the vicinity of its catalytic center and the important REP element (Fig. 5A). Thus, the tryptophan emission spectrum of parkin specifically reflects the environment in the vicinity of the catalytic core of parkin.
FIGURE 5.

Structural changes in the parkin core upon ubiquitin binding. A, distribution of tryptophan residues in rat parkin. Pink sphere, catalytic cysteine. B, changes in the tryptophan emission spectrum of full-length parkin upon binding to ubiquitin. C, red shift of the baricentric mean of the fluorescence emission spectrum as a function of the molar ratio of ubiquitin to parkin. D, changes in the tryptophan emission spectrum of parkin ΔUBL upon binding to ubiquitin. a.u., arbitrary unit. E, red shift of the baricentric mean of the fluorescence emission spectrum as a function of the molar ratio of ubiquitin to parkin ΔUBL.
Addition of ubiquitin to full-length parkin resulted in a small but measurable red shift of its emission spectrum, suggesting that on average the tryptophan residues of parkin become increasingly solvent-exposed upon ubiquitin binding (Fig. 5B). The phosphomimetic form of ubiquitin had a larger effect on the emission spectrum of parkin, which may indicate a slightly stronger affinity of phosphomimetic ubiquitin for parkin (Fig. 5, B and C). Equivalent experiments with parkin ΔUBL confirmed that (phosphomimetic) ubiquitin induced structural changes in the core RING domains of parkin (Fig. 5D). Once again, phosphomimetic ubiquitin was a more effective agent as compared with ubiquitin (Fig. 5, D and E). Taken together, these results imply that binding of phosphoubiquitin to parkin induces subtle structural changes in the core domains of parkin (Fig. 5E).
Changes in the Conformational Dynamics of Ubiquitin upon Complex Formation
The comparably small red shift of the fluorescence emission spectra upon complex formation indicated that (phospho-) ubiquitin binding-induced conformational changes in parkin must be rather modest. Indeed, the static structural differences between free parkin (7–9) and its complex form with phosphoubiquitin (19) seem to be limited. We thus considered the possibility that ubiquitin affects the structural dynamics, rather than the static structure of parkin, to modulate parkin function. To obtain detailed insight into the conformational dynamics of the free and phosphoubiquitin-bound parkin core (parkin ΔUBL), we performed molecular dynamics (MD) simulations with parkin ΔUBL, (phospho-) ubiquitin, and the parkin-phosphoubiquitin complex in explicit water.
First, we obtained MD trajectories for ubiquitin and phosphoubiquitin. To enable a statistical analysis of the output of our simulations, we repeated each calculation 10 times, starting from the same structure, but with different randomized initial velocities. In the case of phosphoubiquitin, two distinct conformations have been reported; the structure of the major state of phosphoubiquitin in solution was used for our simulations because this is the conformation of phosphoubiquitin that binds to parkin (18, 19).
On the nanosecond time scale, ubiquitin and phosphoubiquitin exhibited similarly rigid overall motion with residual flexibility in the loop regions (residues 7–10 and 46–48), as well as the well known high flexibility in the C-terminal tail (Fig. 6, A and B). Second, we obtained trajectories of the parkin ΔUBL-phosphoubiquitin complex. In the complex, the backbone fluctuations of phosphoubiquitin were essentially identical to that of the free form in the regions of secondary structure. However, the backbone motion of phosphoubiquitin was even more suppressed in the loop regions (residues 7–10 and 46–48) and the C-terminal tail (Fig. 6, A and B, red trace). This suggests that phosphoubiquitin gives up some of its residual dynamic character upon formation of a complex with parkin.
FIGURE 6.

Changes in conformational fluctuations in ubiquitin upon binding to the parkin core. A, time traces of the r.m.s.d. calculated for Cα atoms after rotational and translational fitting to the Cα coordinates of the first time frame during molecular dynamics simulations of ubiquitin (black), phosphoubiquitin (blue), and phosphoubiquitin in the parkin-phosphoubiquitin complex (red). All simulations were performed 10 times with different randomized initial velocities. Average r.m.s.d. values are shown. The shaded area represents the S.E. of the respective r.m.s.d. B, root mean square fluctuation (RMSF) of Cα atoms of ubiquitin (black), phosphoubiquitin (blue), and phosphoubiquitin in the parkin-phosphoubiquitin complex (red) averaged over the course of the simulations. The average RMSF value of 10 simulations, each of 50 ns, is shown. Error bars indicate the S.E.
Conformational Dynamics of the Parkin Core
Next, we obtained MD trajectories of the free parkin core (parkin ΔUBL). The core domains of parkin adopted a stable conformation with a total Cα r.m.s.d. value of ∼4–5 Å (Fig. 7A). Among these domains, the IBR domain showed the highest RMSF values, indicating the high mobility of this domain with respect to the other domains of parkin. RING1 was rigid, with average RMSF values ranging from ∼1.5 to 2 Å (Fig. 7, B and C). This result is reasonable considering that RING1 engages in non-covalent interactions with all other domains of parkin (Fig. 1B). Interestingly, the catalytic cysteine residue Cys431 was observed at a minimum of backbone flexibility (Fig. 7, B and C). Likewise, the key auto-inhibitory residue Trp403, alanine mutation of which results in activated parkin (7), was observed in another local minimum of backbone flexibility (Fig. 7B). Thus, auto-inhibition of parkin works not only by the static auto-inhibitory interactions, as described by recent x-ray crystallographic reports (7–9), but also by the intrinsically rigid motion of the protein backbone among residues flanking key auto-regulatory sites.
FIGURE 7.

Conformational fluctuations in the parkin core before and after binding to phosphoubiquitin. A, time traces of the r.m.s.d. calculated for Cα atoms after rotational and translational fitting to the Cα coordinates of the first time frame during molecular dynamics simulations of parkin ΔUBL (black) and the parkin ΔUBL-phosphoubiquitin complex (red). All simulations were performed 10 times with different randomized initial velocities. Average r.m.s.d. values are shown. The shaded area represents the S.E. of the respective r.m.s.d. B, RMSF of Cα atoms of parkin ΔUBL (black) and the parkin ΔUBL-phosphoubiquitin complex (red) averaged over the time interval (5 ns; 50 ns). The average RMSF value of 10 simulations, each of 50 ns, is shown. Error bars indicate the S.E. Residue numbers refer to parkin from R. norvegicus. and the alignment was performed by Clustal Omega (53). The respective domains are indicated. C, conformational fluctuations in the free parkin core (B) as visualized on the structure of the first time frame. Residues are color-coded as follows: red, RMSF > RMSFaverage + 1σ; yellow, RMSF > RMSFaverage; blue, RMSF < RMSFaverage − 1σ; gray, all other residues. Magenta sphere, catalytic cysteine. D, changes in conformational fluctuations in the parkin core upon binding to phosphoubiquitin. The ratio of the RMSF values of B is visualized on the structure of free parkin. The phosphoubiquitin-binding site is indicated. Color code: blue, ρi > ρaverage + 1σ; light blue, ρi > ρaverage; gray, all other residues; ρi, RMSF (B) ratio for residue i of the free to the phosphoubiquitin-bound form of the parkin core. Magenta sphere, catalytic cysteine.
Changes in the Conformational Dynamics of Parkin upon Complex Formation
Many residues of parkin showed differences in conformational dynamics between the free and phosphoubiquitin-bound forms, as indicated by the respective RMSF values (Fig. 7B). Interestingly, phosphoubiquitin binding to RING1 changed the backbone fluctuations in parkin, and this effect was not confined to the immediate vicinity of the binding site. Rather, the global conformational dynamics in the whole parkin molecule were redistributed after complex formation (Fig. 7, B and D).
Phosphoubiquitin binding had no significant effect on the RING2 domain, which showed very similar dynamics between the free and phosphoubiquitin-bound form (Fig. 7, B and D). This is reasonable, considering that the RING2 domain is located away from the ubiquitin-binding site. The conformational fluctuations in all other domains of parkin were significantly affected by phosphoubiquitin binding. The RING1 domain engaged in non-covalent interactions with phosphoubiquitin, rendering it even more rigid as indicated by average RMSF values of ∼1 Å (Fig. 7, B and D). Several residues in the RING0 domain also showed increased rigidity, whereas regions more remote to the ubiquitin-binding site seemed to be less affected (Fig. 7, B and D). Importantly, the highly dynamic IBR domain showed increased rigidity after complex formation.
Taken together, these data suggest that the conformational fluctuations in the parkin core are redistributed by phosphoubiquitin binding leading to significantly enhanced rigidity in the RING1-IBR element, while exerting no apparent influence on RING2 and large parts of the RING0 domain.
Discussion
Thermodynamic Change upon Ubiquitin Binding
We have shown that both ubiquitin and phosphomimetic ubiquitin can bind to full-length parkin and the isolated parkin core (parkin ΔUBL) in solution (Figs. 2–4). Binding of (phosphomimetic) ubiquitin to parkin is enthalpically disfavored, but it is driven by an increase in the total entropy of the system (Figs. 3 and 4). Because the total entropy change of binding (−TΔStotal) can be dominated by changes in protein conformational entropy (−TΔSconf) (23–28), we considered changes in protein dynamics upon complex formation. However, MD analysis indicated that both phosphoubiquitin and the parkin core become less dynamic after complex formation. Accordingly, the entropy-driven nature of ubiquitin binding to parkin must be explained by increases in solvent entropy.
The entropic nature of ubiquitin binding to parkin can be explained by the hydrophobic effect; the phosphoubiquitin-binding site on the RING1 domain of parkin contains several hydrophobic residues whose exposed side chains engage in interactions with phosphoubiquitin (Ala152, Val226, Val303, Ala321, Phe325, Ile326, Ile343, Leu344, and Ile345 in PDB code 5CAW in Fig. 8A) (19). In the absence of phosphoubiquitin, these hydrophobic side chains cannot participate in hydrogen bonding with the solvent, leading to the formation of highly ordered water molecules caging the binding site. Phosphoubiquitin binding displaces many of these highly ordered water molecules, resulting in less ordered water (higher solvent entropy), which drives the reaction.
FIGURE 8.
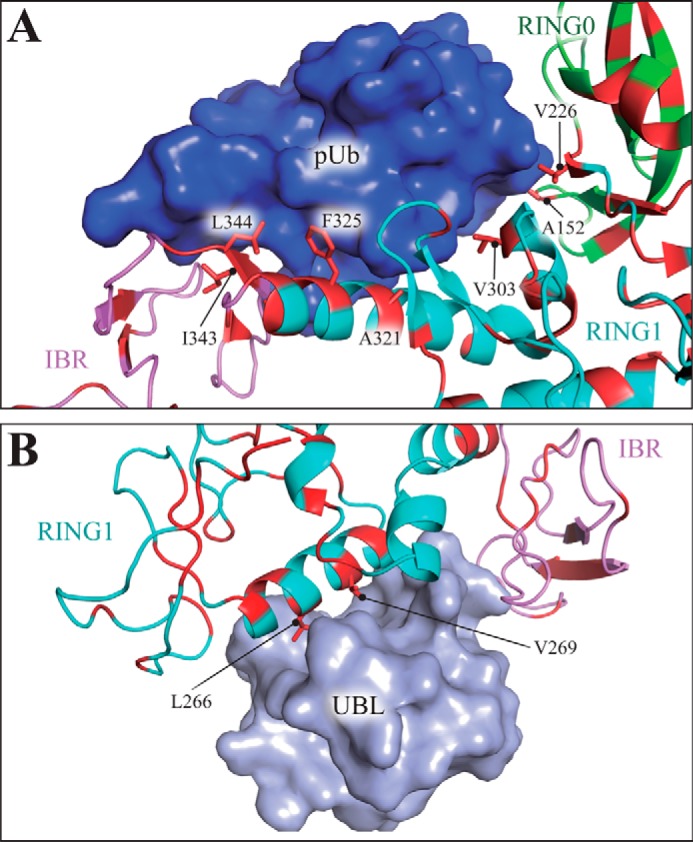
Distinct properties of the UBL- and phosphoubiquitin-binding sites on parkin. A, binding site of phosphoubiquitin on parkin contains several hydrophobic residues (red), many of which engage in interactions with phosphoubiquitin, thereby requiring solvent exposure before complex formation. B, conversely, only two hydrophobic side chains of the parkin core engage in interactions with the UBL domain of parkin. The structures are drawn from PDB code 5CAV (A) (19) and 4K95 (B) (7). Color code: RING0, green; RING1, cyan; IBR, violet; phosphoubiquitin, dark blue; UBL, light blue; residues with exposed hydrophobic side chains (Phe, Val, Leu, Ile, Trp, Met, and Ala) are shown in red.
Conversely, only two hydrophobic side chains (Leu266 and Val269 in PDB code 4K95, see Fig. 8B (7)) are exposed to the solvent in the case of the UBL-binding site. Thus, the solvent at this site is unlikely to form highly ordered water clusters, and UBL binding does not result in a significant entropy gain. These considerations are in good agreement with our observation that binding of the UBL domain to parkin is driven by enthalpy (Fig. 4E).
Effect of AR-JP-associated Mutations on Parkin
Pathogenic mutations can affect parkin by a variety of mechanisms (Fig. 9). Some of the mutations appear to perturb the stability of structural elements of parkin or directly alter the enzymatic active site. Importantly, two pathogenic mutations L283P and G284R are located in a phosphoubiquitin-binding element of the RING1 domain, and the AR-JP mutation A240R interferes with E2 binding (7, 19). Other Parkinson disease-associated mutations are located at domain-domain interfaces. This highlights that both intra- and intermolecular interactions have to be taken into account to understand parkin dysregulation in AR-JP.
FIGURE 9.

AR-JP mutations may unbalance the conformational dynamics of parkin by a variety of mechanisms. Pathogenic mutations linked to autosomal recessive juvenile parkinsonism (3, 30, 54–62) are mapped onto the crystal structure of parkin (spheres). Several mutants appear to affect the structure of parkin by interfering with Zn2+ binding (blue) or the correct formation of secondary structure elements (magenta). The mutants G430D and C431F directly affect the active site (yellow). Other PD mutants may alter domain-domain interactions (green). Ala240 (orange) is critical for binding to the E2, and Leu283 and Gly284 (red) participate in phosphoubiquitin binding (19). Many of the remaining PD mutants are less well understood (gray) but may modulate the overall conformational dynamics of the enzyme. The structure of parkin was drawn from PDB code 4K95 (7).
Intriguingly, the role of a large number of pathogenic mutations remains largely unexplained (Fig. 9, gray). Because autoinhibition of parkin activity is critical for proper regulation of its activity, this group of point mutations may exert a subtle influence on the conformational dynamics of the autoinhibited enzyme (Fig. 7), resulting in a slight imbalance of its autoregulatory mechanism. However, a detailed thermodynamic and kinetic analysis will be required to elucidate the exact impact of these point mutations on parkin activity.
Role of PINK1 in Parkin Activation
On the basis of our data and recently published reports, it is possible to construct a model of parkin activation.
First, the mitochondrial protein kinase PINK1 phosphorylates ubiquitin at residue Ser65 (12–14). Although unmodified ubiquitin can bind to parkin (Figs. 2–5), phosphorylation by PINK1 increases the affinity of ubiquitin for the parkin core (Fig. 4D). Importantly, the absence of the parkin UBL domain enhances ubiquitin binding to parkin (Figs. 3D and 4D), suggesting that ubiquitin and the parkin UBL domain compete for binding to parkin, even though they bind at distinct sites (19, 29). Phosphoubiquitin binding displaces (19, 29) the auto-inhibitory (30) UBL domain from the parkin core, leading to solvent exposure of Ser65 of the UBL domain, which is buried in the structure of free parkin.
This exposure renders Ser65 accessible to PINK1, which then phosphorylates the UBL domain of parkin. This phosphorylation event prevents the UBL from rebinding back onto the parkin core (19, 29). Thus, PINK1 plays a dual regulatory role in this pathway by (i) enhancing the affinity of ubiquitin for the parkin core (Fig. 4D) and (ii) decreasing the affinity of the UBL domain for the parkin core. Here, PINK1 intriguingly uses the same mechanism, phosphorylation of Ser65, but on different molecules (ubiquitin and the UBL domain of parkin) with opposite effects (respectively, enhanced and decreased affinity for the parkin core) to shift the UBL/ubiquitin competition equilibrium toward a phosphoubiquitin-bound and UBL-released state of parkin, comprising an elegant positive feedback mechanism (Fig. 10A).
FIGURE 10.

Model of parkin activation by phosphoubiquitin. A, left, in the initial state, parkin adopts an auto-inhibited conformation with the UBL domain bound to the parkin core (7). PINK1 phosphorylates Ser65 of ubiquitin, which competes with the UBL for binding to the parkin core. Release (19, 29) of the UBL domain from the parkin core to the solvent enables PINK1 to phosphorylate Ser65 of the UBL, which prevents the UBL from rebinding back on the parkin core. Accordingly, the action of PINK1 drives the UBL/phosphoubiquitin competition equilibrium toward a phosphoubiquitin-bound/UBL-exposed state of parkin (right, schematic). B, structure of an active RBR/E2∼Ub transfer complex of the RBR-type E3 ubiquitin ligase HOIP (31). The linear ubiquitin chain-determining domain is omitted for clarity. An allosteric ubiquitin molecule (Uballo) orients the IBR and RING1 domains with respect to one other. C, as compared with the inactive conformation of parkin (A), the RING2 domain in active HOIP (B) is rotated and translated, bringing it in close proximity to the RING1-IBR element. Such a large scale structural change is permitted by the long linker between the IBR and RING2 domains, but it requires release of the REP element from RING1. This displacement of the REP element is probably driven by competitive binding of E2∼Ub to the E2-binding site on RING1 (blue arrow). D, comparison of the inactive and active states of the RING2 domain of parkin. Left, in the inactive conformation, the catalytic cysteine is occluded by the RING0 domain. Right, structural model of the RING2 domain of parkin in the active state as generated by SWISS-MODEL (63) on the basis of the active state of HOIP (B). The sequence identity of the RING2 domain between HOIP and parkin, as judged by SWISS-MODEL, is 43%. Apart from formation of a new α-helix, the overall structure of the RING2 domain is not markedly changed, indicating that simple rotation and translation of the RING2 domain with respect to the RING1-IBR element is sufficient to bring the catalytic cysteine within close proximity of the activated Gly76 of the incoming ubiquitin (Ubact), leading to formation of an active parkin-E2∼Ub transfer complex. Domains other than RING0 and RING2 are omitted for clarity. Structures are drawn from PDB codes 4K95, 5CAW, and 5EDV, respectively.
Structural Rearrangements in the Parkin Core
We observed structural changes in the core RING domains of parkin by monitoring the fluorescence red shift that accompanies the solvent exposure of tryptophan residues upon complex formation. The absolute magnitude of red shift is comparably small, indicating that the tryptophan residues do not become completely exposed to the solvent as would be the case in protein unfolding (λmax ≃ 355 nm). Rather, the observed red shift may suggest that, upon phosphoubiquitin binding, subtle rearrangements in the conformational ensemble of parkin take place in solution.
Conformational rearrangements on a much larger scale are required to convert the conformation of auto-inhibited parkin into a fully activated form, because superposition of the E2∼Ub conjugate onto the E2-binding site in any of the known crystal structures of parkin results in models in which the active sites of the E2 and E3 enzymes are placed ∼50 Å apart from each other (7–9, 19). Thus, currently available crystal structures of parkin cannot explain the transthiolation reaction.
Role of Ubiquitin in Parkin Activation
Phosphoubiquitin binding to the parkin core results in modest local changes to the static structure of the parkin core such as the straightening of an α-helix in the RING1 domain, although most other regions seem to be rather unaffected (Fig. 10A, right) (19). By contrast, our MD analysis indicated that the structural rigidity of parkin is increased in the phosphoubiquitin-bound state, and importantly, this effect is most pronounced in the RING1-IBR element (Fig. 7, B and D).
Comparison of parkin to a recently published structure of an active RBR/E2∼Ub transfer complex of HOIP suggests a possible mechanism for the conversion of parkin to an activated state. In that structure (31), the RING1-IBR element makes non-covalent contacts with an allosteric ubiquitin molecule, which according to the authors was not expected to be found in the electron density, but it proved absolutely necessary for crystal formation (Fig. 10B). Thus, we infer that ubiquitin exerts a stabilizing effect on the RING1-IBR element in the crystal of the HOIP RBR/E2∼Ub, in analogy to the stabilizing effect that phosphoubiquitin exerts on the RING1-IBR element in parkin (Fig. 7D).
Fixation of RING1 and the dynamic (Fig. 7C) IBR domain relative to one other (Fig. 7D) would permit the RING2 domain to undergo diffusion with respect to the phosphoubiquitin/RING1-IBR arrangement, facilitated by the linker between the IBR and RING2 domains (Fig. 10C). Importantly, the final conformation of the RING2 domain is unlikely to show marked structural changes (Fig. 10D), suggesting that simple rotational and translational diffusion of the RING2 domain toward the phosphoubiquitin-fixed RING1-IBR element is sufficient to bring the catalytic cysteine within close proximity of the C-terminal glycine residue (Gly76) of the activated ubiquitin, thereby yielding an active state of parkin.
Competition of E2∼Ub and REP for Binding to Parkin RING1
The mechanism that we propose requires release of the REP element from RING1 to allow large scale movement of the RING2 domain with respect to RING1-IBR. On the one hand, the REP element occludes the E2-binding site in free and phosphoubiquitin-bound parkin, a feature that is often interpreted as preventing E2 binding (Fig. 1B). On the other hand, previous NMR experiments have demonstrated (7) that in solution the E2 enzyme UbcH7 is able to bind to the parkin core even in the presence of the REP element. Moreover, the REP mutation W403A further enhances binding of the E2 to parkin by weakening interactions between REP and RING1 (7). Thus, we conclude that an incoming E2 enzyme can competitively displace the REP element in solution. Importantly, the active HOIP RBR-E2∼Ub complex shows that not only the E2 enzyme itself but also the activated ubiquitin present on E2 engages in non-covalent interactions with the E3 enzyme (Fig. 10B) (31). This indicates that E2∼Ub may be more effective than a free E2 enzyme at binding to and thereby displacing the REP element from parkin. Indeed, it has been previously reported that RING-type E3 enzymes can show a 50-fold preference for ubiquitin-conjugated E2 over the free E2 enzyme (32).
Conclusion
Our study has shown that phosphoubiquitin is an activating agonist of parkin that functions by an intriguing dual mechanism of (i) competition with the antagonistic UBL domain for parkin core binding and (ii) stabilization of the RING1-IBR element, which prepares transition of parkin into an active state upon E2∼Ub binding.
Experimental Procedures
Proteins
Ubiquitin was expressed and purified as described previously (33). Parkin from Rattus norvegicus was expressed from a pGEX-6P1 expression vector that was obtained from Addgene with kind permission from Dr. Kalle Gehring (7). Full-length parkin (residues 1–465) and its fragment lacking the N-terminal UBL domain (parkin ΔUBL; residues 141–465) were expressed and purified as reported previously (7). In brief, BL21(DE3) Escherichia coli cells harboring the parkin expression vector were grown to an A600 of 0.5 at 37 °C. The temperature was decreased to 16 °C, and expression was induced by the addition of 25 μm isopropyl 1-thio-β-d-galactopyranoside overnight. GST-parkin was purified by glutathione-Sepharose 4FF (GE Healthcare) column chromatography. After cleavage of the GST tag by PreScission protease, parkin was further purified by size-exclusion chromatography on a HiLoad 16/60 Superdex 75pg column (GE Healthcare). The parkin UBL domain (residues 1–76) was expressed and purified as described previously (30).
NMR Spectroscopy
NMR spectra were acquired on an Avance II 700 MHz spectrometer equipped with a 5-mm 15N/13C/1H z-gradient triple resonance cryoprobe (Bruker). The free induction decays were apodized, zero-filled, and Fourier-transformed using NMRPipe (34). Resolution in the indirect dimension was improved by linear prediction. Spectra were analyzed in CCPN (35). Ubiquitin was measured at a concentration of 100 μm at 310 K, and its heteronuclear single quantum coherence (HSQC) spectrum was acquired as transverse relaxation-optimized spectroscopy-type experiment. Figures of spectra were created by the Python packages Nmrglue and Matplotlib (36, 37).
Isothermal Titration Calorimetry
Isothermal titration calorimetry (ITC) experiments were conducted at 298 K on an ITC200 system (MicroCal). All protein samples were dialyzed against ITC buffer (20 mm HEPES, pH 7.5, 100 mm sodium chloride, 0.1 mm tris(2-carboxyethyl)phosphine) overnight and thoroughly degassed before each experiment. Samples were incubated at room temperature (298 K) before each experiment. The concentration of the syringe protein (ubiquitin, phosphomimetic ubiquitin, or parkin UBL) ranged from 0.8 to 1.5 mm, whereas the cell contained protein (parkin or parkin ΔUBL) at 87–100 μm. The syringe protein was injected into the cell at 3-min intervals. The resulting data were processed using Origin 7 (MicroCal Software, Inc.). Errors in the derived data represent the standard error of the mean of three independent experiments.
Molecular Dynamics
All molecular dynamics simulations were performed by GROMACS 5.0.6 (38–43) in explicit solvent using the AMBER99SB-ILDN force field (43) and the following crystal structures: Pediculus humanus parkin ΔUBL (residues 143–461; PDB code 5CAW (19)) in complex with phosphoubiquitin (residues 1–75); R. norvegicus parkin ΔUBL (residues 140–465; PDB code 4K7D (7)); human ubiquitin (residues 1–75; PDB code 1UBQ (44)); Ser65-phosphorylated human ubiquitin (major conformation; residues 1–75; PDB code 4WZP (18)). Force field parameters for phosphoserine were obtained from Ref. 45. Residues missing in the crystal structures due to disorder were rebuilt using the ModLoop interface to Modeler (46). The respective protein was solvated in a triclinic box with SPC/E water. In structures involving phosphoubiquitin, the system charge was neutralized by addition of two Na+ ions. Zn2+ ions of the RING domains of parkin were distance restrained to the respective cysteine Sγ atoms (47, 48). Energy minimization was performed by the steepest-descent algorithm with a maximum force Fmax of 1000 kJ mol−1 nm−1. Equilibration of protein and solvent was conducted in two steps. First, the system was simulated under the canonical (NVT) ensemble for 100 ps with position restraints on all protein atoms. Second, 100 ps of position-restrained NPT simulation was performed to stabilize the density of the system.. The Parrinello-Rahman algorithm (49) was applied for pressure coupling (1 bar, 2.0-ps coupling constant), and the temperature was coupled using the modified Berendsen (40) thermostat (310 K, 0.1 ps coupling constant). The integration time step was set to 2 fs. Bonds were constrained by the LINCS algorithm (42). The parameters for fast particle mesh Ewald electrostatics (50) were as follows: Lennard-Jones cutoff distance, 0.8 nm; Coulomb cutoff distance, 0.8 nm; Fast Fourier Transform grid spacing, 0.08 nm; interpolation order, cubic. For each simulation, 10 independent calculations were performed starting from the same structure with different initial velocities randomly assigned from the Maxwell-Boltzmann distribution at 310 K. All simulations were carried out on Linux workstations running Fedora 21 with CUDA-compatible NVIDIA graphics processing units. Analysis was done by routines integrated in GROMACS, and figures were created using the Python package Matplotlib (37). r.m.s.d. and RMSF values were calculated with reference to the structure of first time frame of the simulation by rotational and translational fitting of the Cα coordinates. RMSF values were calculated for time frames in the range of 5–50 ns.
Fluorescence Spectroscopy
Fluorescence was measured using a FluoroMax4 fluorescence spectrometer (HORIBA). Tryptophan fluorescence was selectively excited at 300 nm, and emission spectra were acquired over wavelengths of 310–400 nm with the slit width set to 5 nm at 298 K. Ubiquitin has no tryptophan residues and contributes only baseline noise to the spectra. In parkin, tryptophan residues cluster in the catalytic center with the single exception of the linker residue Trp97, which is not resolved in any of the reported crystal structures. Before the experiments, proteins were dialyzed into 20 mm HEPES, pH 7.5, 100 mm sodium chloride, 0.1 mm tris(2-carboxyethyl)phosphine. Parkin was diluted to a final protein concentration of 7 μm and measured at a volume of 500 μl. Each acquisition was repeated 10 times, and the averaged spectrum was subjected to analysis. The spectral contribution of the buffer was subtracted from the acquired spectra. The red shift of the fluorescence spectrum upon ligand addition was evaluated by calculating the baricentric mean of the spectrum. The baricentric mean was calculated as shown in Equation 1,
 |
in which F(λ) is the tryptophan fluorescence emission intensity at λ nm, and the sum is over the entire spectrum (310–400 nm). Fluorescence red shifts are reported as differences from the initial baricentric mean: Δλbcm = λi − λ0, in which λi is the baricentric mean of the spectrum after the ith addition of ligand, and λ0 is the baricentric mean of the fluorescence spectrum before the addition of ligand. A red shift of the tryptophan emission spectrum was interpreted as an increased solvent exposure of tryptophan residues (51, 52).
Author Contributions
E. W. and D. M. conducted all experiments. K. S. and M. S. designed and supervised the study. All authors discussed the results and wrote the manuscript.
Acknowledgments
We express our gratitude to Dr. Noriyuki Matsuda and Dr. Yutaka Ito for helpful discussions. We thank Dr. Kousuke Inomata for providing the expression vector for phosphomimetic ubiquitin (S65D). We further express our gratitude to Dr. Kalle Gehring for the expression vector for R. norvegicus parkin.
Note Added in Proof
After our manuscript was accepted, we became aware of results previously reported by Kumar et al. (64). Their results partially overlap with the conclusions of this study but were not discussed in the original version of the manuscript. The authors sincerely apologize for this oversight.
This work was supported by Japan Society for the Promotion of Science KAKENHI Grant 15H04342. The authors declare that they have no conflicts of interest with the contents of this article.
- AR-JP
- autosomal recessive juvenile parkinsonism
- HSQC
- heteronuclear single quantum coherence
- HOIP
- HOIL-1L interacting protein
- IBR
- in-between RING
- ITC
- isothermal titration calorimetry
- MD
- molecular dynamics
- RING
- really interesting new gene
- r.m.s.d.
- root mean square deviation
- RMSF
- root mean square fluctuation
- RBR
- RING-in-between-RING
- UBL
- ubiquitin-like
- Ub
- ubiquitin
- PDB
- Protein Data Bank
- REP
- repressor element.
References
- 1. Kitada T., Asakawa S., Hattori N., Matsumine H., Yamamura Y., Minoshima S., Yokochi M., Mizuno Y., and Shimizu N. (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608 [DOI] [PubMed] [Google Scholar]
- 2. Shulman J. M., De Jager P. L., and Feany M. B. (2011) Parkinson's disease: genetics and pathogenesis. Annu. Rev. Pathol. 6, 193–222 [DOI] [PubMed] [Google Scholar]
- 3. Lücking C. B., Dürr A., Bonifati V., Vaughan J., De Michele G., Gasser T., Harhangi B. S., Meco G., Denèfle P., Wood N. W., Agid Y., Brice A., French Parkinson's Disease Genetics Study Group, and European Consortium on Genetic Susceptibility in Parkinson's Disease (2000) Association between early-onset Parkinson's disease and mutations in the parkin gene. N. Engl. J. Med. 342, 1560–1567 [DOI] [PubMed] [Google Scholar]
- 4. Wenzel D. M., Lissounov A., Brzovic P. S., and Klevit R. E. (2011) UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature 474, 105–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Matsuda N., Sato S., Shiba K., Okatsu K., Saisho K., Gautier C. A., Sou Y. S., Saiki S., Kawajiri S., Sato F., Kimura M., Komatsu M., Hattori N., and Tanaka K. (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 189, 211–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Narendra D. P., Jin S. M., Tanaka A., Suen D. F., Gautier C. A., Shen J., Cookson M. R., and Youle R. J. (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8, e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Trempe J. F., Sauvé V., Grenier K., Seirafi M., Tang M. Y., Ménade M., Al-Abdul-Wahid S., Krett J., Wong K., Kozlov G., Nagar B., Fon E. A., and Gehring K. (2013) Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 340, 1451–1455 [DOI] [PubMed] [Google Scholar]
- 8. Wauer T., and Komander D. (2013) Structure of the human Parkin ligase domain in an autoinhibited state. EMBO J. 32, 2099–2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Riley B. E., Lougheed J. C., Callaway K., Velasquez M., Brecht E., Nguyen L., Shaler T., Walker D., Yang Y., Regnstrom K., Diep L., Zhang Z., Chiou S., Bova M., Artis D. R., et al. (2013) Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nat. Commun. 4, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Smit J. J., Monteferrario D., Noordermeer S. M., van Dijk W. J., van der Reijden B. A., and Sixma T. K. (2012) The E3 ligase HOIP specifies linear ubiquitin chain assembly through its RING-IBR-RING domain and the unique LDD extension. EMBO J. 31, 3833–3844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Duda D. M., Olszewski J. L., Schuermann J. P., Kurinov I., Miller D. J., Nourse A., Alpi A. F., and Schulman B. A. (2013) Structure of HHARI, a RING-IBR-RING ubiquitin ligase: autoinhibition of an Ariadne-family E3 and insights into ligation mechanism. Structure 21, 1030–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Iguchi M., Kujuro Y., Okatsu K., Koyano F., Kosako H., Kimura M., Suzuki N., Uchiyama S., Tanaka K., and Matsuda N. (2013) Parkin-catalyzed ubiquitin-ester transfer is triggered by PINK1-dependent phosphorylation. J. Biol. Chem. 288, 22019–22032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Clark I. E., Dodson M. W., Jiang C., Cao J. H., Huh J. R., Seol J. H., Yoo S. J., Hay B. A., and Guo M. (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441, 1162–1166 [DOI] [PubMed] [Google Scholar]
- 14. Kazlauskaite A., Kelly V., Johnson C., Baillie C., Hastie C. J., Peggie M., Macartney T., Woodroof H. I., Alessi D. R., Pedrioli P. G., and Muqit M. M. (2014) Phosphorylation of Parkin at serine65 is essential for activation: elaboration of a Miro1 substrate-based assay of Parkin E3 ligase activity. Open Biol. 4, 130213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Koyano F., Okatsu K., Kosako H., Tamura Y., Go E., Kimura M., Kimura Y., Tsuchiya H., Yoshihara H., Hirokawa T., Endo T., Fon E. A., Trempe J. F., Saeki Y., Tanaka K., and Matsuda N. (2014) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510, 162–166 [DOI] [PubMed] [Google Scholar]
- 16. Kane L. A., Lazarou M., Fogel A. I., Li Y., Yamano K., Sarraf S. A., Banerjee S., and Youle R. J. (2014) PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 205, 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kazlauskaite A., Kondapalli C., Gourlay R., Campbell D. G., Ritorto M. S., Hofmann K., Alessi D. R., Knebel A., Trost M., and Muqit M. M. (2014) Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J. 460, 127–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wauer T., Swatek K. N., Wagstaff J. L., Gladkova C., Pruneda J. N., Michel M. A., Gersch M., Johnson C. M., Freund S. M., and Komander D. (2015) Ubiquitin Ser65 phosphorylation affects ubiquitin structure, chain assembly and hydrolysis. EMBO J. 34, 307–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wauer T., Simicek M., Schubert A., and Komander D. (2015) Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature 524, 370–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Komander D., and Rape M. (2012) The ubiquitin code. Annu. Rev. Biochem. 81, 203–229 [DOI] [PubMed] [Google Scholar]
- 21. Kaiser S. E., Riley B. E., Shaler T. A., Trevino R. S., Becker C. H., Schulman H., and Kopito R. R. (2011) Protein standard absolute quantification (PSAQ) method for the measurement of cellular ubiquitin pools. Nat. Methods 8, 691–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Okatsu K., Koyano F., Kimura M., Kosako H., Saeki Y., Tanaka K., and Matsuda N. (2015) Phosphorylated ubiquitin chain is the genuine Parkin receptor. J. Cell Biol. 209, 111–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tzeng S. R., and Kalodimos C. G. (2013) Allosteric inhibition through suppression of transient conformational states. Nat. Chem. Biol. 9, 462–465 [DOI] [PubMed] [Google Scholar]
- 24. Tzeng S. R., and Kalodimos C. G. (2012) Protein activity regulation by conformational entropy. Nature 488, 236–240 [DOI] [PubMed] [Google Scholar]
- 25. Tzeng S. R., and Kalodimos C. G. (2009) Dynamic activation of an allosteric regulatory protein. Nature 462, 368–372 [DOI] [PubMed] [Google Scholar]
- 26. Frederick K. K., Marlow M. S., Valentine K. G., and Wand A. J. (2007) Conformational entropy in molecular recognition by proteins. Nature 448, 325–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Marlow M. S., Dogan J., Frederick K. K., Valentine K. G., and Wand A. J. (2010) The role of conformational entropy in molecular recognition by calmodulin. Nat. Chem. Biol. 6, 352–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wand A. J. (2001) Dynamic activation of protein function: a view emerging from NMR spectroscopy. Nat. Struct. Biol. 8, 926–931 [DOI] [PubMed] [Google Scholar]
- 29. Sauvé V., Lilov A., Seirafi M., Vranas M., Rasool S., Kozlov G., Sprules T., Wang J., Trempe J. F., and Gehring K. (2015) A Ubl/ubiquitin switch in the activation of Parkin. EMBO J. 34, 2492–2505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chaugule V. K., Burchell L., Barber K. R., Sidhu A., Leslie S. J., Shaw G. S., and Walden H. (2011) Autoregulation of Parkin activity through its ubiquitin-like domain. EMBO J. 30, 2853–2867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lechtenberg B. C., Rajput A., Sanishvili R., Dobaczewska M. K., Ware C. F., Mace P. D., and Riedl S. J. (2016) Structure of a HOIP/E2∼ubiquitin complex reveals RBR E3 ligase mechanism and regulation. Nature 529, 546–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Spratt D. E., Wu K., Kovacev J., Pan Z.-Q., and Shaw G. S. (2012) Selective recruitment of an E2∼ ubiquitin complex by an E3 ubiquitin ligase. J. Biol. Chem. 287, 17374–17385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tenno T., Fujiwara K., Tochio H., Iwai K., Morita E. H., Hayashi H., Murata S., Hiroaki H., Sato M., Tanaka K., and Shirakawa M. (2004) Structural basis for distinct roles of Lys63- and Lys48-linked polyubiquitin chains. Genes Cells 9, 865–875 [DOI] [PubMed] [Google Scholar]
- 34. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 35. Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., and Laue E. D. (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696 [DOI] [PubMed] [Google Scholar]
- 36. Helmus J. J., and Jaroniec C. P. (2013) Nmrglue: an open source Python package for the analysis of multidimensional NMR data. J. Biomol. NMR 55, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hunter J. D. (2007) Matplotlib: A 2D graphics environment. Computing In Science & Engineering 9, 90–95 [Google Scholar]
- 38. Lindahl E., Hess B., and Van Der Spoel D. (2001) GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 7, 306–317 [Google Scholar]
- 39. Berendsen H. J., van der Spoel D., and van Drunen R. (1995) GROMACS: A message-passing parallel molecular dynamics implementation. Comp. Phys. Commun. 91, 43–56 [Google Scholar]
- 40. Bussi G., Donadio D., and Parrinello M. (2007) Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 [DOI] [PubMed] [Google Scholar]
- 41. Miyamoto S., and Kollman P. A. (1992) SETTLE: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 13, 952–962 [Google Scholar]
- 42. Hess B., Bekker H., Berendsen H. J., and Fraaije J. G. (1997) LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 [Google Scholar]
- 43. Lindorff-Larsen K., Piana S., Palmo K., Maragakis P., Klepeis J. L., Dror R. O., and Shaw D. E. (2010) Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 78, 1950–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vijay-Kumar S., Bugg C. E., and Cook W. J. (1987) Structure of ubiquitin refined at 1.8 A resolution. J. Mol. Biol. 194, 531–544 [DOI] [PubMed] [Google Scholar]
- 45. Craft J. W. Jr., and Legge G. B. (2005) An AMBER/DYANA/MOLMOL phosphorylated amino acid library set and incorporation into NMR structure calculations. J. Biomol. NMR 33, 15–24 [DOI] [PubMed] [Google Scholar]
- 46. Fiser A., and Sali A. (2003) ModLoop: automated modeling of loops in protein structures. Bioinformatics 19, 2500–2501 [DOI] [PubMed] [Google Scholar]
- 47. Tropp J. (1980) Dipolar relaxation and nuclear Overhauser effects in nonrigid molecules: the effect of fluctuating internuclear distances. J. Chem. Phys. 72, 6035–6043 [Google Scholar]
- 48. Torda A. E., Scheek R. M., and Van Gunsteren W. F. (1989) Time-dependent distance restraints in molecular dynamics simulations. Chem. Phys. Lett. 157, 289–294 [Google Scholar]
- 49. Parrinello M., and Rahman A. (1981) Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 [Google Scholar]
- 50. Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., and Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 [Google Scholar]
- 51. Eftink M. R. (1991) Fluorescence techniques for studying protein structure. Methods Biochem. Anal. 35, 127–205 [DOI] [PubMed] [Google Scholar]
- 52. Burstein E. A., Abornev S. M., and Reshetnyak Y. K. (2001) Decomposition of protein tryptophan fluorescence spectra into log-normal components. I. Decomposition algorithms. Biophys. J. 81, 1699–1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Thompson J. D., Gibson T. J., and Higgins D. G. (2002) Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinformatics, 2–3 [DOI] [PubMed] [Google Scholar]
- 54. Muñoz E., Tolosa E., Pastor P., Martí M. J., Valldeoriola F., Campdelacreu J., and Oliva R. (2002) Relative high frequency of the c. 255delA parkin gene mutation in Spanish patients with autosomal recessive parkinsonism. J. Neurol. Neurosurg. Psychiatry 73, 582–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Oliveira S. A., Scott W. K., Martin E. R., Nance M. A., Watts R. L., Hubble J. P., Koller W. C., Pahwa R., Stern M. B., and Hiner B. C., Ondo W. G., Allen F. H. Jr., Scott B. L., Goetz C. G., Small G. W., et al. (2003) Parkin mutations and susceptibility alleles in late-onset Parkinson's disease. Ann. Neurol. 53, 624–629 [DOI] [PubMed] [Google Scholar]
- 56. Shimura H., Hattori N., Kubo Si, Mizuno Y., Asakawa S., Minoshima S., Shimizu N., Iwai K., Chiba T., Tanaka K., and Suzuki T. (2000) Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 25, 302–305 [DOI] [PubMed] [Google Scholar]
- 57. Hedrich K., Marder K., Harris J., Kann M., Lynch T., Meija-Santana H., Pramstaller P. P., Schwinger E., Bressman S. B., Fahn S., and Klein C. (2002) Evaluation of 50 probands with early-onset Parkinson's disease for Parkin mutations. Neurology 58, 1239–1246 [DOI] [PubMed] [Google Scholar]
- 58. Periquet M., Lücking C. B., Vaughan J. R., Bonifati V., Dürr A., De Michele G., Horstink M., Farrer M., Illarioshkin S. N., Pollak P., Borg M., Brefel-Courbon C., Denefle P., Meco G., Gasser T., et al. (2001) Origin of the mutations in the parkin gene in Europe: exon rearrangements are independent recurrent events, whereas point mutations may result from Founder effects. Am. J. Hum. Genet. 68, 617–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Macedo M. G., Verbaan D., Fang Y., van Rooden S. M., Visser M., Anar B., Uras A., Groen J. L., Rizzu P., van Hilten J. J., and Heutink P. (2009) Genotypic and phenotypic characteristics of Dutch patients with early onset Parkinson's disease. Mov. Disord. 24, 196–203 [DOI] [PubMed] [Google Scholar]
- 60. Beasley S. A., Hristova V. A., and Shaw G. S. (2007) Structure of the Parkin in-between-ring domain provides insights for E3-ligase dysfunction in autosomal recessive Parkinson's disease. Proc. Natl. Acad. Sci. U.S.A. 104, 3095–3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Huynh D. P., Scoles D. R., Nguyen D., and Pulst S. M. (2003) The autosomal recessive juvenile Parkinson disease gene product, parkin, interacts with and ubiquitinates synaptotagmin XI. Hum. Mol. Genet. 12, 2587–2597 [DOI] [PubMed] [Google Scholar]
- 62. Chen D., Gao F., Li B., Wang H., Xu Y., Zhu C., and Wang G. (2010) Parkin mono-ubiquitinates Bcl-2 and regulates autophagy. J. Biol. Chem. 285, 38214–38223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schwede T., Kopp J., Guex N., and Peitsch M. C. (2003) SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 31, 3381–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kumar A., Aguirre J. D., Condos T. E., Martinez-Torres R. J., Chaugule V. K., Toth R., Sundaramoorthy R., Mercier P., Knebel A., Spratt D. E., Barber K. R., Shaw G. S., and Walden H. (2015) Disruption of the autoinhibited state primes the E3 ligase parkin for activation and catalysis. EMBO J. 34, 2506–2521 [DOI] [PMC free article] [PubMed] [Google Scholar]