

Key Clinical Message
There is some question about the relationship between hereditary spherocytosis (HS) and pulmonary arterial hypertension, even associated with splenectomy. The finding of BMPR2 mutations in our patient suggests that other factors are necessary for the development of the disease, and perhaps, the incidence of pulmonary hypertension is not increased in patients with HS.
Keywords: ACVRL1, ambrisentan, BMPR2, hereditary spherocytosis, pulmonary arterial hypertension
Introduction
Hereditary spherocytosis (HS) is a hemolytic anemia that affects one in 3000 people with dominant inheritance in 75% of cases. Several mutations involved in the synthesis of different proteins of the erythrocyte membrane are the genetic basis of this disease, being ankyrin deficiency the most frequent in western countries 1. The most typical symptoms are those resulting from hemolysis, anemia, jaundice, gallstones, and splenomegaly. The blood count shows spherocytes with reduced membrane surface. There are varying degrees of severity of the disease, with around 80% mild or moderate and 20% severe or very severe. Due to decreased surface, erythrocytes are unable to pass through the spleen, and consequently, they are retained therein and separated from the circulation. Therefore, splenectomy is a common treatment in severe forms of HS and other hemolytic disorders. Splenectomy appears to be associated with a higher incidence of both arterial and venous thrombotic events and possibly with the development of pulmonary hypertension (PH) 2, 3, although in the case of HS data are controversial. Among the ~10 reported cases of PH associated with HS, around 50% were due to thromboembolism.
The major genetic risk to develop pulmonary arterial hypertension (PAH) is the presence of mutations in the bone morphogenetic protein receptor type 2 (BMPR2) gene, present at up to 80% of hereditary forms of PAH and with variable proportion in other forms of PAH 4.
To the best of our knowledge, no case with HS has been studied for mutations in BMPR2 and other genes commonly associated with PAH. We present a patient with HS with splenectomy carrying a potentially pathogenic mutation in the gene encoding BMPR2, which developed late PAH.
Case Report
The patient was a 48‐year‐old woman, never smoker or drinker of alcohol, with HS diagnosed shortly after birth and treated with splenectomy performed in the seventh month of life. She was carrying the c.845G>A (p.C282Y) mutation in heterozygosis, conferring the diagnosis of hemochromatosis, but never developed clinical manifestations of the disease. About 8 months before, she began to notice dizziness on exertion. At that time, she had normal laboratory tests with no anemia (hemoglobin 14.4 g/dL) and moderate increase in the levels of ferritin, related to hemolysis. A chest radiograph showed no abnormalities and electrocardiogram showed a pattern of right overload. The echocardiography estimated a pulmonary artery systolic pressure of 55 mmHg. Computer thoracic angiography, perfusion lung scan, and Doppler ultrasound ruled out the presence of thromboembolism, but because of the underlying disease, anticoagulation with rivaroxaban was started. In the next 3 months, symptoms worsened and several near‐syncope with moderate efforts were present. She was referred to our PH Unit. At that time, the patient was in functional class (FC) III, slightly tachypneic at rest, with oxygen saturation by pulse oximetry (SpO2) 92% breathing air room. She walked 442 meters in 6 min (6MWT), with a minimum SpO2 86%. Laboratory test showed normal hemoglobin levels, normal hepatic enzymes, and increased levels of N‐terminal probrain natriuretic peptide (NT‐proBNP), 876 pg/mL. Thoracic computed tomography (CT) showed no thrombi in the pulmonary arteries (Fig. 1A, and B). A new echocardiogram showed moderate pericardial effusion, enlarged and dysfunctional right ventricle (55 mm, TAPSE 11 mm), and tricuspid regurgitation velocity (TRV) of 4.22 m/s (Fig. 2). Cardiac catheterization showed the following values: mean pulmonary artery pressure 55 mmHg, systolic pulmonary artery pressure 97 mmHg, arterial wedge pressure 11 mmHg, pulmonary resistance 1005 dyn cm sec−5, and cardiac index 2.02 L/m per m2. The vasodilator test was negative and coronary arteries were normal.
Figure 1.
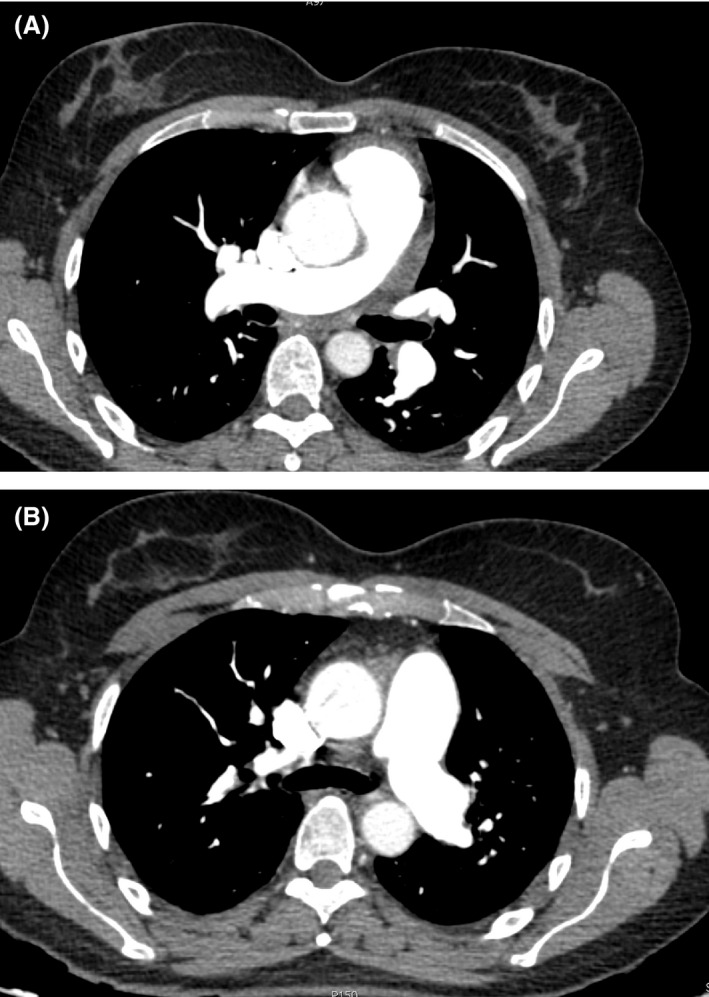
CT scan images at the time the patient was referred to the Pulmonary Hypertension Unit, which ruled out thromboembolic disease.
Figure 2.
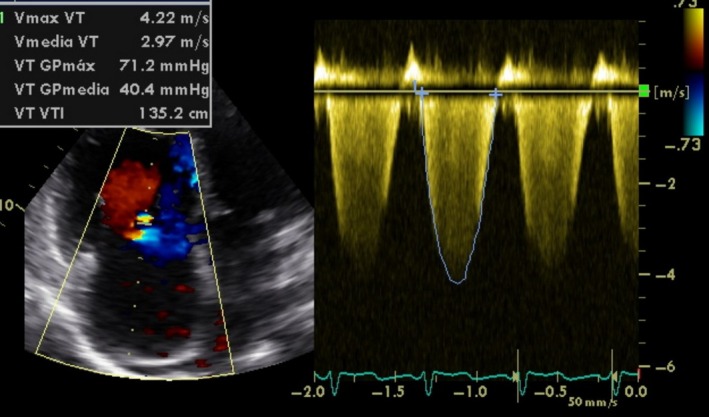
Echocardiogram showing an elevated tricuspid regurgitation velocity corresponding with an estimated systolic pulmonary pressure of 82 mmHg.
After informed consent, we conducted a genetic study that included BMPR2, ENG, ACVRL1, and KCNA5 genes. This study showed a missense mutation in BMPR2, c.742A>G (p.R248G) located in exon six, and another mutation in ACVRL1, c.760G>A (p.D254N. These mutations are described for the first time. None of them was seen in 55 healthy individuals. To predict their potential pathogenicity, we used combined computer algorithms including Polyphen‐2, Pmut, Sort Intolerant from Tolerant (SIFT), and MutationTaster 2 software. One mutation was considered pathogenic when two or more of these computer programs labeled it as pathogenic. c.742A>G was considered pathogenic by all but Polyphen‐2 software and c.760G>A by all but Pmut software. The other genes studied showed only some polymorphisms without pathogenic capacity.
Treatment was started with the endothelin‐receptor antagonist ambrisentan at a dose of 10 mg once a day, with an excellent response. After 7 months, she was in FC I with no dizziness on daily efforts and SpO2 96% at rest. She walked 495 meters in the 6MWT and NT‐proBNP decreased until 287 pg/mL. The echocardiography showed a TRV of 2.42 m/s with an estimated systolic pulmonary pressure of 35 mmHg (Fig. 3). No adverse events occurred with ambrisentan.
Figure 3.
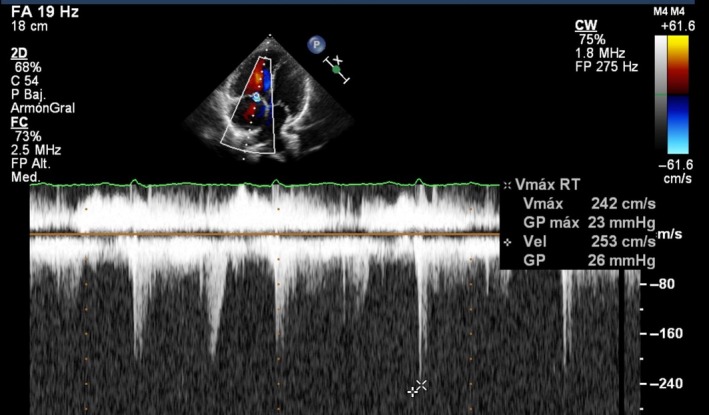
Echocardiogram 5 months after starting treatment with ambrisentan. The tricuspid regurgitation velocity is within normal limits.
Discussion
We present a rare case of HS with splenectomy who developed PAH in adulthood and whose genetic study showed a potentially pathogenic mutation in BMPR2 and ACVRL1 genes. There was no history of thromboembolic disease and imaging studies ruled out the presence of pulmonary thromboembolism. She had no family history of PH.
HS is included within the hemolytic anemias and, although its incidence is quite high, very few cases associated with PH have been published, a high percentage of them due to thromboembolic disease. There is considerable evidence of an association between splenectomy and PH in sickle cell disease and thalassemia 5, but it is more uncertain in patients with HS. A study including 26 children with HS and splenectomy did not find any risk factors for PH or evidence of elevated pulmonary artery pressure after a mean follow up of 4.5 years 6. Another study with 36 adults, 28 of which with previous splenectomy, failed to find any patient with PH 7. The TRV was not different between patients with and without splenectomy. All this findings seem to suggest that the prevalence of PAH associated with HS could be not very different from the expected to general population and other risk factors are needed to developed PAH. We had the opportunity to perform genetic testing, which showed the presence of one mutation in the BMPR2 gene and another in the ACVRL1 gene, perhaps with a key role in the development of the disease in our patient. ACVRL1 belongs to the family of transforming growth factor (TGF) beta as BMPR2, and it has been associated with PAH 8. According to existing data, we cannot know exactly the role that both HS and splenectomy had in the development of PAH. There is the possibility that they could contribute in some way, perhaps due to the production of microthrombi, although we could not demonstrate thromboembolic disease. Platelet activation, oxidative stress, and decrease in nitric oxide secondary to endothelial damage related to free plasma hemoglobin due to repeated hemolysis are potential mechanisms to develop PH at least in sickle cell disease 9, but it is unknown what role they can play in the HS with a much lower degree of hemolysis.
Our patient was a carrier of a gene associated with hemochromatosis, but did not suffer any symptoms related to it. Hemochromatosis has not been associated with PAH. We do not think that this disease had any role in the development of PAH in our patient.
Although this patient had a serious illness, with the presence of dizziness during exertion and severe hemodynamic compromise, the response to treatment with an antagonist of endothelin‐1 was excellent and, after 7 months, she was in FC I with no relevant secondary adverse events.
In summary, we present a rare case of PAH associated with HS and splenectomy without evidence of thromboembolism and, as far as we know, it is the first with genetic study showing a mutation in BMPR2 and ACVRL1 genes. According to the findings of our case, in a patient with HS without thromboembolic disease who develops PH, it should be ruled out other risk factors for the disease, perhaps including genetic testing.
Conflict of Interest
None declared.
References
- 1. An, X. , and Mohandas N.. 2008. Disorders of red cell membrane. Br. J. Haematol. 141:367–375. [DOI] [PubMed] [Google Scholar]
- 2. Hoeper, M. M. , Niedermeyer J., Hoffmeyer F., Flemming P., and Fabel H.. 1999. Pulmonary hypertension after splenectomy? Ann. Intern. Med. 130:506–509. [DOI] [PubMed] [Google Scholar]
- 3. Jaïs, X. , Ioos V., Jardim C., O., Sitbon Parent F., Hamid A., et al. 2005. Splenectomy and chronic thromboembolic pulmonary hypertension. Thorax 60:1031–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Machado, R. D. , Southgate L., Eichstaedt C. A., Aldred, M. A. Austin, E. D. Best, D. H. et al. 2015. Pulmonary arterial hypertension: a current perspective on established and emerging molecular genetic defects. Hum. Mutat. 36:113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hayes, M. M. , Vedamurthy A., George G., Dweik, R. Klings, E. S. Machado R. F. et al. 2014. Pulmonary hypertension in sickle cell disease. Ann. Am. Thorac. Soc. 1:1488–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Das, A. , Bansal D., Ahluwalia J., Das R. Rohit, M. K. Attri S. V. et al. 2014. Risk factors for thromboembolism and pulmonary artery hypertension following splenectomy in children with hereditary spherocytosis. Pediatr. Blood Cancer 61:29–33. [DOI] [PubMed] [Google Scholar]
- 7. Crary, S. E. , Ramaciotti C., and Buchanan G. R.. 2011. Prevalence of pulmonary hypertension in hereditary spherocytosis. Am. J. Hematol. 86:E73–E76. [DOI] [PubMed] [Google Scholar]
- 8. Pousada, G. , Baloira A., Vilariño C., Cifrian J. M., and Valverde D.. 2014. Novel mutations in BMPR2, ACVRL1 and KCNA5 genes and hemodynamic parameters in patients with pulmonary arterial hypertension. PLoS One 9:e100261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Potoka, K. P. , and Gladwim M. T.. 2015. Vasculopathy and pulmonary hypertension in sickle cell disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 308:L314–L324. [DOI] [PMC free article] [PubMed] [Google Scholar]