

Key Clinical Message
3q26.33‐3q27.2 microdeletion can be classified as a clinical entity characterized by intrauterine growth retardation, feeding problems in infancy, short stature, intellectual disability, hypotonia, dysmorphic facial features (medially sparse eyebrows, narrow horizontal palpebral fissures, epicanthal folds, flat nasal bridge and tip, short philtrum, and downturned corners of mouth), and teeth and feet abnormalities.
Keywords: 3q26.33‐3q27.2 microdeletion, 3q27.3 microdeletion, blepharophimosis, intellectual disability, Ohdo syndrome
Introduction
Chromosomal microdeletion/microduplication syndromes are frequently associated with intellectual disability (ID), multiple congenital anomalies, and/or autism spectrum disorders. It has been possible to routinely identify the microdeletion/microduplication syndromes by chromosomal microarray analysis (CMA) over the last years 1. Mandrile et al. 2 recently described three unrelated patients carrying an overlapping 3q26.33‐3q27.2 microdeletion who share a common clinical phenotype. Subsequently, two additional cases of microdeletion were published 3, 4. It has been proposed that the 3q26.33‐3q27.2 microdeletion may represent a novel condition caused by the haploinsufficiency of dosage‐sensitive genes. Also, it was suggested that it could be a new clinically recognizable syndrome, as reported patients share common features such as neonatal hypotonia, severe feeding problems, specific facial features, abnormal dentition, recurrent upper airways infections, developmental delay, and severe growth impairment of prenatal onset 2.
We report a case with 3q26.33‐3q28 microdeletion with blepharophimosis and ID which was presented at the European Society of Human Genetics meeting in 2002 as Ohdo syndrome 5. We also review all previously reported cases and compare them to our case.
Case Report
The patient is a 16‐year‐old girl. She is the first child of healthy, nonconsanguineous Estonian parents. The family history is unremarkable, and she has a younger sister who is healthy. The pregnancy was complicated with fetal ultrasound finding of intrauterine growth restriction at 29 weeks of gestation. Amniocentesis done subsequently showed normal female karyotype (46,XX). Delivery was at 35 weeks of gestation and all growth parameters were below the third centile: weight was 1220 g (−3.0 SD), length 40 cm (−3.0 SD), and occipitofrontal circumference (OFC) 28 cm (−3.0 SD). Estonian age‐ and gender‐specific growth curves were used to evaluate and correct growth parameters at birth and later. On initial examination, she was noted to have facial dysmorphism as well as supravalvular aortic and pulmonary stenosis. Williams syndrome was suspected, but FISH analysis for 7q11.23 microdeletion was normal.
Weak suck and gastroesophageal reflex were noted from the first week of life which resulted in severe feeding difficulties and poor weight gain. She was fed high‐caloric food via nasogastral tube and later via gastrostomy. However, her poor weight gain persisted. Her psychomotor development was delayed. At 9 months the development corresponded to the age of 5–7 months according to Griffiths scale. She started to walk at 3 years of age.
At 4.5 years of age, her height was 91 cm (−3.0 SD), weight 11.5 kg (−3.0 SD), and OFC 46.5 cm (−3.0 SD). She had facial dysmorphism with blepharophimosis, ptosis, wide nasal bridge, depressed nasal tip, smooth philtrum, narrow mouth with downturned corners, micrognathia, and low‐set dysplastic ears (Fig. 1A and B). Her teeth were peg‐like, crowded, and with dysplastic enamel. Clinodactyly of the fifth finger was also observed. She had muscular hypotonia and mild ID. Brain MRI showed brain atrophy in frontal lobe. Considering all clinical features, Ohdo syndrome was diagnosed.
Figure 1.

(A) The facial view and (B) profile of our patient at 4.5 years of age, note a peculiar face with blepharophimosis, ptosis, a broad nasal bridge, a flat nasal tip, a flat philtrum, a small mouth, downturned corners of mouth, micrognathia, low‐set dysplastic ears, and peg‐like, irregular teeth; (C) the facial view at the age of 16 years.
At 13 years of age, the Psychoeducational Profile (PEP) test showed that her development corresponded to the age of 4 years 5 months up to 4 years 9 months. Her developmental profile was quite stable and the strongest abilities were perception, eye‐mouth cooperation, and verbal abilities. Eye examination revealed mild hyperopia (both eyes +0.5 D) and astigmatism.
At the age of 16 years, her height is 157 cm (−1.5 SD), weight 46.8 kg (−1.5 SD), and OFC 52 cm (−2 SD). Her facial features show blepharophimosis, ptosis, depressed nasal tip, smooth philtrum, narrow mouth, and low‐set dysplastic ears, and prognathism has developed as a new feature (Fig. 1C). In addition, she has thoracic kyphosis and mild left club foot. At early school age, sensorineural hearing loss was diagnosed and she has been using hearing aid. Her speech was dysarthric. She is clumsy. Neurological examination shows increased muscle tonus (legs more than arms) and increased deep tendon reflexes. She has a habit to bite her nails when anxious and has frequent tics of eyelids. She studies in the school for mildly intellectually disabled children. Hyperopia has progressed (right eye +2.25 D and left +2.0 D).
Molecular Analyses
DNA was extracted from a peripheral blood sample by a standard salting out procedure. Chromosomal microarray analysis using Illumina HumanCytoSNP‐12 array (Illumina Inc., San Diego, CA, USA) was performed according to the manufacturer's protocol. Genotypes were called and the data were analyzed using GenomeStudio v2011.1 software (Illumina Inc., San Diego, CA, USA). Chromosomal microarray analysis revealed an 8.4‐Mb deletion in the long arm of chromosome 3 (arr [hg19] 3q26.33q28(182,674,821–191,025,402)x1) (Fig. 2).
Figure 2.
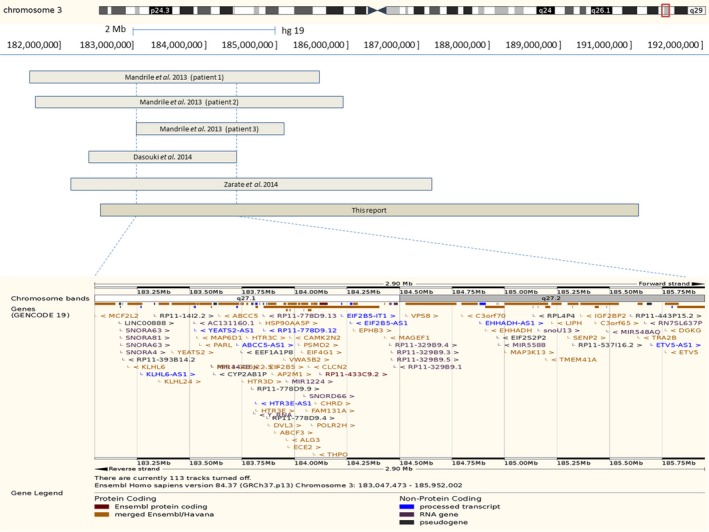
Schematic representation of the SRO of 3q26.33‐3q27.2 microdeletion syndrome in all described patients and our case by the Human Genome Browser hg19 assembly. Coordinates for the deletion described by Dasouki et al. 3 were converted from hg18 to hg19 [chr3:182,470,516–184,469,308]. SRO is 1.4 Mb in size and contains 39 protein coding genes according to the Ensembl database (http://www.ensembl.org/Homo_sapiens/).
The diagnostic whole exome sequencing analysis was simultaneously performed by Genome Diagnostics Nijmegen (http://www.genomediagnosticsnijmegen.nl/) and covered by the Estonian Health Insurance Fund. First, no pathogenic mutations were reported, but secondary analysis led to an additional report of a suspected chromosomal deletion in chromosome 3 independent of CMA.
Discussion
We report on a patient carrying an 8.4 Mb 3q26.33‐3q28 microdeletion providing new insights into recently described microdeletion syndromes in distal 3q region. Up to now, six cases including the present one of overlapping 3q26.33‐3q27.2 microdeletions have been published 2, 3, 4. All described cases have presented with pre‐ and postnatal growth retardation, ID, hypotonia, dysmorphic facial features, and various teeth abnormalities (Table 1). Feeding problems in infancy, microcephaly, behavioral problems, speech delay, hearing loss, recurrent infections, feet abnormalities, and pubertal delay and/or genital abnormalities were seen in two thirds of the patients. Most frequently described facial features were flat facial profile, medially sparse eyebrows, epicanthal folds, flat nasal bridge and tip, short philtrum, and downturned corners of mouth. Considering the wide range of clinical features, the microdeletion syndrome may be clinically recognizable. The smallest region of overlap (SRO) for 3q26.33‐3q27.2 microdeletion syndrome is estimated to be 1.4 Mb in size (3:183,047,473–185,952,002) and encompasses chromosomal bands 3q27.1‐3q27.2 (Fig. 2). Therefore, the syndrome could be renamed according to a smaller 3q27.1‐3q27.2 deletion.
Table 1.
The size of deletion, karyotype and clinical features of five previously published cases with the 3q36.33‐3q27.2 microdeletion and our case is given in detail in this table
| Patient 1 2 | Patient 2 2 | Patient 3 2 | 3 | 4 | Our case | Summary | |
|---|---|---|---|---|---|---|---|
| Size of deletion | 4.14 Mb | 4.28 Mb | 2.09 Mb | 1.99 Mb | 5 Mb | 8.4 Mb | |
| Karyotype | arr [hg19] 3q26.33q.27.2 (181,648,378–185,786,898)x1 | arr [hg19] 3q26.33q.27.2 (181,692,255–185,969,168)x1 | arr [hg19] 3q27.1q.27.2 (183,047,473–185,140,522)x1 | arr [hg18] 3q26.33q27.1 (183,953,210–185,952,002)x1, Xp22.33Xq28 (0–154,913,754)x2 | arr [hg19] 3q26.33q.27.5 (182,189,525–187,212,935)x1 | arr [hg19] 3q26.33q28 (182,674,821–191,025,402)x1 | |
| Age at last examination | 6 years | 17 years | 12 years | 9.5 years | 16 years | 16 years | |
| Intrauterine growth retardation | + | + | + | + | + | + | 6/6 |
| Feeding problems | + | + | + | − | + | + | 5/6 |
| Short stature | + (−3.68 SD) | + (−4 SD) | + (−2 SD) | + (≪3rd centile) | ± (−1.35 SD) | ± (−1.5 SD) | 6/6 |
| Microcephaly | + (−4.4 SD) | − | − | + (48 cm, 7y) | + (−5.93 SD) | + (−2 SD) | 4/6 |
| Flat facial profile | + | + | + | + | − | + | 5/6 |
| Medially sparse eyebrows | + | + | + | + | + | + | 6/6 |
| Epicanthal folds | + | + | + | − | − | + | 4/6 |
| Blepharophimosis/short palpebral fissures | − | − | + Narrow horizontal opening | − Puffy eyelids | + | + | 3/6 |
| Flat nasal bridge and tip | + | + | + | − | − | + | 4/6 |
| Anteverted nares | + | − | − | − | − | − | 1/6 |
| Short philtrum | + | + | + (flat) | − (flat) | + | − | 4/6 |
| Downturned corners of mouth | + | + | − | − | + | + | 4/6 |
| Eye abnormalities | Myopia/astigmatism | Bilateral keratoconus | Myopia/astigmatism | − | − | − | 3/6 |
| Teeth abnormalities | Incomplete dentition | Primary dentition at 15 years of age | Pointed, wide spared, fused | Dental crowding, delayed teeth eruption | Poor dentition, delayed teeth eruption | Peg‐like, irregular and with dysplastic enamel | 6/6 |
| Ears abnormalities | Preauricular pit | − | Mildly simple and thickened | − | − | Low‐set dysplastic | 3/6 |
| Hearing loss | − | Mild conductive hearing loss | Decreased hearing secondary to fluid | − | Moderate to severe sensorineural hearing loss | Sensorineural hearing loss) | 4/6 |
| Thin skin | + | + | − | − | + | − | 3/6 |
| Hands abnormalities | Clinodactyly of F4 | − | Mildly tapered fingers | − | Clinodactyly of F5 | − | 3/6 |
| Feet abnormalities | Pes planus | Pes planus | Mild pes planus | − | Pes planus | Club foot | 5/6 |
| Skeletal | Mild kyphosis, mild pectus carinatum | − | Hypermobility of the hips | − | − | Thoracal kyphosis | 3/6 |
| Heart anomaly | Patent ductus arteriosus) | − | − | − | − | Supravalvular aortic and pulmonary stenosis) | 2/6 |
| Genitalia/puberty | Retractable left testicle | Undescended testis, micropenis, delayed puberty | Hypoplasia labia minora and pubic pad | − | − | Delayed puberty | 4/6 |
| Delayed milestones | + | + | + | + | + | + | 6/6 |
| Speech delay | Not acquired | − | − | Delay | Delay | Dysarthric | 4/6 |
| Intellectual disability | Severe | Severe | Borderline | + | + | Mild | 6/6 |
| Behavioral abnormalities | − | Hyperactivity | Attention deficit disorder | Asperger syndrome | − | Tics and nail biting | 4/6 |
| Recurrent infections | + | + | + | + | − | − | 4/6 |
| Hypotonia | + | + | + | + | + | + | 6/6 |
| Other abnormalities | Thrombocytopenia | − | Thrombocytopenia | Thrombocytopenia, neutropenia | Tonic‐clonic seizures | − |
Our patient was initially clinically diagnosed with Ohdo syndrome 5 mainly due to striking blepharophimosis (Fig. 1), and also considering the co‐occurrence of teeth abnormalities, heart defect, and ID. Ohdo et al. 6 first described a syndrome of blepharophimosis, ptosis, hypoplastic teeth, heart defect, and mental handicap, to which his name is currently attached (MIM 249620). Since then, several mostly sporadic cases have been reported greatly widening the clinical phenotype 7, 8, 9, 10, 11. Later, Verloes et al. 12 proposed to classify blepharophimosis–ID syndromes into five distinct subgroups. For three of them, the etiology has been clarified as deletions in 3p and mutations in KAT6B (the Say‐Barber/Biesecker/Young‐Simpson type) 13 or MED12 (X‐linked Maat–Kievit–Brunner type) 14, respectively. The exact etiology of the original Ohdo type 6 and Verloes type has not been established yet. Based on the clinical findings in our patient and the presence of short palpebral fissures or narrow horizontal palpebral fissures in two previously reported 3q26.33‐3q27.2 microdeletion cases, we hypothesize that additional possible candidate gene(s) for blepharophimosis–ID syndrome can be located in the established SRO in 3q27.1‐3q27.2 2, 4.
The SRO encompasses 39 protein coding genes, including several known autosomal‐recessive disease genes, such as EIF2B5 (leukoencephalopathy with vanishing white matter), ALG3 (congenital disorder of glycosylation type Id), and CLCN2 (leukoencephalopathy with ataxia) (Fig. 2). As our patient had no white matter abnormalities on brain MRI scan and metabolic testing showed normal results, we did not suspect those genes to be responsible for the phenotype.
Out of the genes located within the SRO, CHRD (chordin, MIM 603475) is of great interest regarding the clinical phenotype. Chordin dorsalizes early vertebrate embryonic tissues by binding to bone morphogenetic proteins and sequestering them in latent complexes 15. It has been shown that the fully penetrant chordin‐null mouse phenotype includes dysmorphic ears, absence of the thymus, persistent truncus arteriosus, abnormal aortic arch artery structure, and cleft palate, which is virtually identical to that observed in Tbx1‐null homozygotes and human individuals with 22q11 deletion syndrome 16. However, penetrance of the chordin phenotype is highly dependent on genetic background and the expression of other genes, including Tbx1. We suggest that the CHRD gene could be a good candidate for explaining dysmorphic facial phenotype and cardiac anomalies in some patients with 3q26.33‐3q27.2 microdeletion syndrome.
It has been suggested that haploinsufficiency of THPO (thrombopoietin, MIM 600044) gene causes mild thrombocytopenia and haploinsufficiency of LAMP3 (lysosome‐associated membrane protein 3) may cause immune deficiency, respiratory distress, and dental anomalies 2, 3. Indeed, at least half of the 3q26.33‐3q27.2 microdeletion cases have thrombocytopenia (Table 1), and mutations in THPO gene causing improved translational efficiency have been shown to be responsible for hereditary autosomal‐dominant thrombocythemia 17, 18. However, LAMP3 gene is not located in the SRO and therefore, is probably not causative for the common phenotype as was suggested before.
All described 3q26.33‐3q27.2 microdeletion patients have presented with intrauterine growth retardation and short stature. PARL gene (presenilin‐associated rhomboid‐like protein, MIM 607858) located in 3q27.1 chromosomal region could be responsible for growth delay. The Parl knockout mice displayed growth retardation, cachexia, and severe atrophy of muscle, spleen, and thymus 19.
The microdeletion in our patient is larger compared to previously described patients with 3q26.33‐3q27.2 microdeletion syndrome and in addition overlaps also with another recently described microdeletion syndrome – 3q27.3, which is associated with dysmorphic features, marfanoid habitus, ID, and psychosis with mood disorder 20. Two SROs have been defined for 3q27.3 microdeletion syndrome 20. The first one included several candidate genes associated with psychotic episodes and mood disorders as well as recognizable facial dysmorphism (slender face, deep‐set eyes, high nasal bridge, hooked nose above a short philtrum, thin upper lip surrounding a small mouth, and prognathism). The second SRO in the 3q27.3q28 has been hypothesized to link with marfanoid habitus (scoliosis, long and thin habitus with leanness, arachnodactyly, and pectus excavatum) and severe ID, which could be explained by the deletion of the AHSG gene. The 3q26.33‐3q28 microdeletion in our patient includes both SROs described by Thevenson et al. 20. However, our patient has no marfanoid habitus and recognizable facial dysmorphism specific for 3q27.3 microdeletion. She presented only kyphosis and prognathism in older age. She has not had psychiatric manifestations yet besides mild ID, a habit to bite her nails, and frequent tics of eyelids.
In conclusion, the 3q26.33‐3q28 microdeletion in our patient overlaps with two recently published cases of microdeletion syndromes, 3q26.33‐3q27.2 and 3q27.3, with more similarities to the former. 3q26.33‐3q27.2 microdeletion can be classified as a clinical entity characterized by intrauterine growth retardation, feeding problems in infancy, short stature, ID, hypotonia, dysmorphic facial features (flat facial profile, medially sparse eyebrows, narrow horizontal palpebral fissures, epicanthal folds, flat nasal bridge and tip, short philtrum, and downturned corners of mouth), and teeth and feet abnormalities. Further functional studies on possible candidate genes inside the deleted regions are needed to clarify the pathogenesis of both microdeletion syndromes.
Conflict of Interest
None declared.
Acknowledgments
We thank this family for their kind cooperation. This work was supported by grant PUT355 from the Estonian Science Foundation.
References
- 1. Miller, D. T. , Adam M. P., Aradhya S., Biesecker L. G., Brothman A. R., Carter N. P., et al. 2010. Consensus statement: chromosomal microarray is a first‐tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 86:749–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mandrile, G. , Dubois A., Hoffman J. D., Uliana V., Di Maria E., Malacarne M., et al. 2013. 3q26.33‐3q27.2 microdeletion: a new microdeletion syndrome? Eur. J. Med. Genet. 56:216–221. [DOI] [PubMed] [Google Scholar]
- 3. Dasouki, M. , Roberts J., Santiago A., Saadi I., and Hovanes K.. 2014. Confirmation and further delineation of the 3q26.33‐3q27.2 microdeletion syndrome. Eur. J. Med. Genet. 57:76–80. [DOI] [PubMed] [Google Scholar]
- 4. Zarate, Y. A. , Bell C., and Schaefer B.. 2013. Description of another case of 3q26.33‐3q27.2 microdeletion supports a recognizable phenotype. Eur. J. Med. Genet. 56:624–625. [DOI] [PubMed] [Google Scholar]
- 5. Ounap, K. , Žordania R., Laidre P., and Nõmmela R.. 2002. Ohdo blepharophimosis syndrome: report of two new unrelated cases and review of literature. European Human Genetics Conference 2002. Strasbourg, France: Eur. J. Hum. Genet. 10(Suppl. 1):121. [Google Scholar]
- 6. Ohdo, S. , Madokoro H., Sonoda T., and Hayakawa K.. 1986. Mental retardation associated with congenital heart disease, blepharophimosis, blepharoptosis, and hypoplastic teeth. J. Med. Genet. 23:242–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Biesecker, L. G. 1991. The Ohdo blepharophimosis syndrome: a third case. J. Med. Genet. 28:131–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Clayton‐Smith, J. , Krajewska‐Walasek M., Fryer A., and Donnai D.. 1994. Ohdo‐like blepharophimosis syndrome with distinctive facies, neonatal hypotonia, mental retardation and hypoplastic teeth. Clin. Dysmorphol. 3:115–120. [PubMed] [Google Scholar]
- 9. Maat‐Kievit, A. , Brunner H. G., and Maaswinkel‐Mooij P.. 1993. Two additional cases of the Ohdo blepharophimosis syndrome. Am. J. Med. Genet. 47:901–906. [DOI] [PubMed] [Google Scholar]
- 10. Maat‐Kievit, J. A. , Milla P. J., Collins J. E., Baraitser M., and Winter R. M.. 1994. A case with blepharophimosis resembling Ohdo syndrome. Clin. Dysmorphol. 3:125–127. [PubMed] [Google Scholar]
- 11. White, S. M. , Ades L. C., Amor D., Liebelt J., Bankier A., Baker E., et al. 2003. Two further cases of Ohdo syndrome delineate the phenotypic variability of the condition. Clin. Dysmorphol. 12:109–113. [DOI] [PubMed] [Google Scholar]
- 12. Verloes, A. , Bremond‐Gignac D., Isidor B., David A., Baumann C., Leroy M. A., et al. 2006. Blepharophimosis‐mental retardation (BMR) syndromes: a proposed clinical classification of the so‐called Ohdo syndrome, and delineation of two new BMR syndromes, one X‐linked and one autosomal recessive. Am. J. Med. Genet. A 140:1285–1296. [DOI] [PubMed] [Google Scholar]
- 13. Clayton‐Smith, J. , O'Sullivan J., Daly S., Bhaskar S., Day R., Anderson B., et al. 2011. Whole‐exome‐sequencing identifies mutations in histone acetyltransferase gene KAT6B in individuals with the Say‐Barber‐Biesecker variant of Ohdo syndrome. Am. J. Hum. Genet. 89:675–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vulto‐van Silfhout, A. T. , de Vries B. B., van Bon B. W., Hoischen A., Ruiterkamp‐Versteeg M., Gilissen C., et al. 2013. Mutations in MED12 cause X‐linked Ohdo syndrome. Am. J. Hum. Genet. 92:401–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Scott, I. C. , Blitz I. L., Pappano W. N., Imamura Y., Clark T. G., Steiglitz B. M., et al. 1999. Mammalian BMP‐1/Tolloid‐related metalloproteinases, including novel family member mammalian Tolloid‐like 2, have differential enzymatic activities and distributions of expression relevant to patterning and skeletogenesis. Dev. Biol. 213:283–300. [DOI] [PubMed] [Google Scholar]
- 16. Choi, M. , and Klingensmith J.. 2009. Chordin is a modifier of tbx1 for the craniofacial malformations of 22q11 deletion syndrome phenotypes in mouse. PLoS Genet. 5:e1000395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ghilardi, N. , Wiestner A., Kikuchi M., Ohsaka A., and Skoda R. C.. 1999. Hereditary thrombocythaemia in a Japanese family is caused by a novel point mutation in the thrombopoietin gene. Br. J. Haematol. 107:310–316. [DOI] [PubMed] [Google Scholar]
- 18. Wiestner, A. , Schlemper R. J., van der Maas A. P., and Skoda R. C.. 1998. An activating splice donor mutation in the thrombopoietin gene causes hereditary thrombocythaemia. Nat. Genet. 18:49–52. [DOI] [PubMed] [Google Scholar]
- 19. Cipolat, S. , Rudka T., Hartmann D., Costa V., Serneels L., Craessaerts K., et al. 2006. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1‐dependent cristae remodeling. Cell 126:163–175. [DOI] [PubMed] [Google Scholar]
- 20. Thevenon, J. , Callier P., Poquet H., Bache I., Menten B., Malan V., et al. 2014. 3q27.3 microdeletional syndrome: a recognisable clinical entity associating dysmorphic features, marfanoid habitus, intellectual disability and psychosis with mood disorder. J. Med. Genet. 51:21–27. [DOI] [PubMed] [Google Scholar]