

Abstract
Background
In the context of advanced immunosuppression, M. tuberculosis is known to cause detectable mycobacteremia. However, little is known about the intra-patient mycobacterial microevolution and the direction of seeding between the sputum and blood compartments.
Methods
From a diagnostic study of HIV-infected TB patients, 51 pairs of concurrent blood and sputum M. tuberculosis isolates from the same patient were available. In a previous analysis, we identified a subset with genotypic concordance, based on spoligotyping and 24 locus MIRU-VNTR. These paired isolates with identical genotypes were analyzed by whole genome sequencing and phylogenetic analysis.
Results
Of the 25 concordant pairs (49 % of the 51 paired isolates), 15 (60 %) remained viable for extraction of high quality DNA for whole genome sequencing. Two patient pairs were excluded due to poor quality sequence reads. The median CD4 cell count was 32 (IQR; 16–101)/mm3 and ten (77 %) patients were on ART. No drug resistance mutations were identified in any of the sequences analyzed. Three (23.1 %) of 13 patients had SNPs separating paired isolates from blood and sputum compartments, indicating evidence of microevolution.
Using a phylogenetic approach to identify the ancestral compartment, in two (15 %) patients the blood isolate was ancestral to the sputum isolate, in one (8 %) it was the opposite, and ten (77 %) of the pairs were identical.
Conclusions
Among HIV-infected patients with poor cellular immunity, infection with multiple strains of M. tuberculosis was found in half of the patients. In those patients with identical strains, whole genome sequencing indicated that M. tuberculosis intra-patient microevolution does occur in a few patients, yet did not reveal a consistent direction of spread between sputum and blood. This suggests that these compartments are highly connected and potentially seed each other repeatedly.
Electronic supplementary material
The online version of this article (doi:10.1186/s12879-016-1737-2) contains supplementary material, which is available to authorized users.
Keywords: Intra-patient, Microevolution, Ancestral, Concurrent sputum and blood M. tuberculosis
Background
The recent advances in molecular analytical methods have increased our understanding of the possible heterogeneity of infection with Mycobacterium tuberculosis [1]. Several perspectives around this complexity in relation to HIV-infection have been documented [2]. However, little is known on the intra-patient mycobacterial diversity and direction of seeding between the sputum and blood compartments.
Clonal variants can be detected using Variable Number of Tandem Repeats (VNTR) [3, 4] or Restriction Fragment Length polymorphism (RFLP) of the IS6110-typing genetic elements [5, 6]. The subtle genetic rearrangements caused by microevolution in IS6110 [7] are known to interrupt genes or modulate the expression of adjacent genes. These can affect interpretation of molecular epidemiological tests [8–10], whereas if this happens in the VNTR regions, such changes can modify the transcription of neighboring genes [11, 12]. These changes may have a role in the infectivity of the bacteria [13] and their survival within the host [14–17]. In a published report, it was suggested that such microevolution affected cavity formation, hence increased transmissibility of the emerging clonal variants [17].
The advent of whole genome sequencing (WGS) has led to the identification of several limitations of traditional molecular epidemiological methods in ascertaining microevolution occurring outside the classical targeted genetic elements [18, 19]. Micro-evolutionary changes may further be modified against a background of impaired immunity as a result of AIDS. Whether HIV/AIDS is the main cause of a systematic heterogeneity of a within-host population of M. tuberculosis as a result of advanced immune suppression [20] or as a result of pathogen microevolution, remains a challenge. A recent study involving four patients found as high a genetic diversity within as between patients [21].
In the present study, we considered a large cohort of HIV-positive patients who had concurrent pulmonary and blood M. tuberculosis strains and were categorized as identical, up to one spacer and/or locus difference, using the conventional methods of spoligotyping (spacer oligonucleotide typing) and Mycobacterial Interspersed Repetitive Units (MIRU)-VNTR 24 loci. We applied whole genome sequencing (WGS) to study microevolution among these strains by documenting differences in distribution of single-nucleotide polymorphisms (SNPs) between strains isolated from sputum and blood. We also aimed at ascertaining the ancestral M. tuberculosis strain between sputum and blood in each patient.
Methods
Study participants
From a previous study of 51 HIV-infected TB patients who had concurrent blood and sputum M. tuberculosis isolates at enrolment, we selected patients found to have identical genotypes from our previous study [22] using both spoligotyping [5] and MIRU-VNTR 24 loci methods [3]. In this strain selection process, we considered pairs (n = 25) with maximum one spacer and/or locus difference to be identical MTB-genotypes.
DNA sequencing
Whole genome sequencing of the DNA from M. tuberculosis isolates was performed on an Illumina HiSeq platform at Genoscreen (Lille, France) or the Beijing Genome Institute (BGI), China following the Illumina TruSeq DNA sample preparation recommendations.
SNP and indel calling for genotype and drug resistance
To confirm genotypic classification assigned using previous methods, sequences were processed through an online program, PhyResSE, which assigns lineages after calling SNP and indels that are known to be lineage specific [23, 24]. Since dynamic changes in M. tuberculosis have been found to occur during acquisition and fixation for drug resistance [25], we also used the same program to call for drug-specific SNPs and indels.
Mapping of the fastq reads and complete variant calling
For each sequence, we used the nesoni version 0.13 pipeline (https://github.com/Victorian-Bioinformatics-Consortium/nesoni) to remove Illumina adaptors sequences and low quality bases from reads using a minimum read quality of 10 and length of 45. We employed the nesoni bowtie tool for read alignment using the most recent common ancestor of the M. tuberculosis complex (MTBc; H37rv_NC_018143.1) as referenced [26, 27]. To look for differences between the reads and the reference genome, we used nesoni consensus to process the mapped reads for the SNP calling process. Quality mapping thresholds included removing reads that mapped to more than 1 position, minimum coverage of 10, minimum mapping quality of the SNP of 20 and minimum read coverage of 66 %.
A tabular list of all SNPs and indels per isolate was created using nesoni nway (Additional file 1), from which a SNP alignment was created using custom python scripts.
Phylogenetic analysis of sequence data
To infer intraspecific phylogenies with the expected small distances, we constructed a maximum likelihood tree using Randomized Axelerated Maximum Likelihood (RAxML) version 8.2 [28], based upon the SNP alignment and employing a generalized time-reversible (GTR) CAT model with Stamatakis ascertainment bias correction [28]. We calculated 100 bootstrap replicates for support of the tree nodes. We also created a neighbor joining distance matrix based upon the SNP alignment using the Molecular Evolutionary Genetics Analysis (MEGA) version 5.2.2 [29].
Results
Of the 25 patients with identical M. tuberculosis genotypes, 15 (60.0 %) had viable mycobacterial bacilli on subculture and enough DNA for whole genome sequencing. Two isolates had sequence reads of poor quality and were eliminated from the analysis. We analyzed data for 13 (86.7 %) of the patients with good quality sequence reads from both blood and sputum (Fig. 1).
Fig. 1.
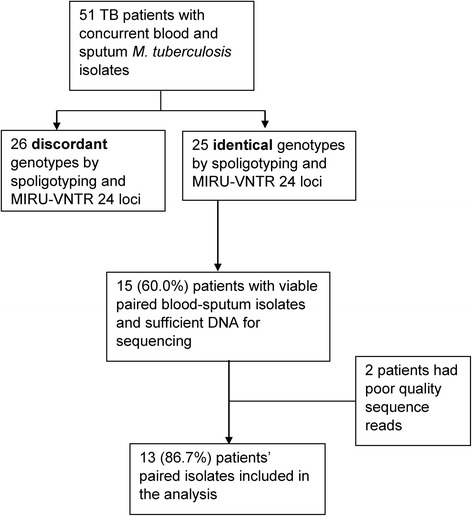
Flow chart showing the participant with concurrent sputum and blood M. tuberculosis sequences. MIRU = Mycobacterial Interspersed Repetitive Units, VNTR = Variable Number of Tandem Repeats
The 13 patients included 8 (61.5 %) women and had a median age of 32 (interquartile range; IQR, 28–37) years. The median CD4 cell count was 32 (IQR; 16–101)/mm3; 10 (76.9 %) were taking ART and only one patient was previously treated for tuberculosis. Lineage assignments based on SNP detection found five (38.5 %) patients had M tuberculosis lineage three (L3; Delhi/CAS) whereas eight (61.5 %) had lineage four (L4; Haarlem and LAM11_ZWE each 12.5 %, LAM3 and S convergent and T2; each 37.5 %) (Fig. 2). These results were in line with those found by spoligotype in the parent study [22]. The SpolDB4 unassigned T2 were found to have SNPs specific to T2- Uganda including mutations in the gyrA gene at position T80A. No drug resistance mutations were identified in any of the sequences analyzed (Fig. 2).
Fig. 2.

Participants’ with concurrent sputum and blood M tuberculosis isolates and DNA sequences. Ω = lineage based on spolDB4 online database, sub-lineage ** = spolDB4 unassigned with T2 SNPs, MIRU-VNTR = Mycobacterial Interspersed Repetitive Unit-Variable-Number Tandem Repeat, MIRU-VNTR* = Mixed allele at that locus, † = All drug susceptible and with no resistance mutations, Squared pairs = single locus variants, Rectangle in MIRU table = single locus variants (SLV)
SNP calling and neighbor joining tree for concurrent sputum and blood M tuberculosis isolates
Of the 13 patients, three (23.1 %) had SNPs (indicating evidence of microevolution) detected when comparing their concurrent sputum and blood M. tuberculosis isolates. One SNP was seen in a patient’s pair that was considered a clonal variant, single locus variants (SLV) by MIRU-VNTR 24 loci (Fig. 2) and the two were from identical pairs between pulmonary and blood compartments. The identified SNPs, their corresponding H37RV genome coordinates and the gene function as stated in Tuberculist online database [30] are indicated (Additional file 2).
The RAxML analysis using SNPs did not show a difference in branch lengths and thus could not determine the ancestral strain between most of blood vs. sputum pairs. We therefore performed phylogenetic analysis to identify the ancestral compartments through a distance matrix derived NJ tree generated using MEGA. In a total of two (15.4 %) patients the blood isolate was ancestral to the sputum isolate, in one (7.7 %) it was the opposite, and ten (76.9 %) of the pairs were identical (Fig. 3).
Fig. 3.

Neighbor Joining SNP distance matrix based tree for potential ancestral strain comparing sputum and blood M. tuberculosis strains. Created using MEGA 5.2.2; Visualized and colored using Fig Tree 1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/); Black = Ancestral strain, Green = Identical strains, p = patient, Red = descendant strains
Discussion
The advent and extended use of WGS strategies have increased our understanding of the transmission, epidemiological and molecular dynamics of the M. tuberculosis pathogen [1, 18, 31]. Recently, WGS analysis has been mainly applied to identify the number of SNPs to document the M. tuberculosis micro-evolutionary events between and within patients [21, 32]. Intra-patient M. tuberculosis microevolution has been found to be similar to the inter-patient microevolution and has been suggested to impact on the expected strain diversity within a transmission chain [21].
In the current study, we applied WGS to isolates of M. tuberculosis, which were identical by conventional typing methods, from HIV-infected patients with poor cellular immunity, in half of whom infection with multiple strains of M. tuberculosis was found. Our study documented three (23.1 %) patients with SNPs (indicating evidence of microevolution) when comparing their concurrent sputum and blood M. tuberculosis isolates. Although high intra-patient variability may be expected during the process of resistance acquisition [25, 33], all isolates in our study from both compartments were drug susceptible with no resistance conferring mutations. Categorizing these isolates as identical by both spoligotyping and MIRU-VNTR 24 loci typing methods, yet with different SNPs, underscores the power of whole genome sequencing in ascertaining microevolution occurring outside the classical targeted genetic elements of M. tuberculosis compared to traditional molecular epidemiological methods [18, 19]. Small changes have been implicated to influence bacterial phenotypes, such as strain infectivity [13] and within-host pathogen survival [14–17].Moreover, clonal MDR-variants of concurrent pulmonary and disseminated tuberculosis strains have been documented [34] which need to be recognized for appropriate therapy to be initiated. More complex intra-patient microevolution of MDR-MTBC strains under treatment has been documented through WGS analysis [35].
Studies have suggested dissemination of pulmonary tuberculosis is due to impaired immunity including compartmentalization [2, 20, 36, 37] and/or reinfection [38]. Some studies have hypothesized pulmonary infection as a spill-over of the lymphatic or haematogenous dissemination of tuberculosis [39–41]. However, few studies have approached these hypotheses using concurrent clinical M. tuberculosis isolates. Through a neighbor joining SNP distance matrix based tree, the present study found M. tuberculosis cross-seeding between pulmonary and blood compartments using clinical M. tuberculosis isolates. This may be due to the high connectedness of these compartments that may lead to repeated seeding in-between these compartments under extensive immunosuppression. Blood as the origin of tuberculosis disease, contrary to the dogma, may be supported by the fact that M. tuberculosis can persist in several sites and cell types that might constitute reservoirs that can reactivate infection producing extrapulmonary tuberculosis with or without lung involvement [42]. Indeed in the main study, 12/182 (6.6 %) of the tuberculosis patients had MTB cultured from blood with two negative sputum cultures [43]. The strains with no clear direction of seeding between sputum and blood compartments could have been influenced by advanced HIV/AIDS immune suppression resulting to increased early dissemination [20].
Our study has some limitations; complete genome sequences can currently only be obtained from cultured isolates, which may have introduced a bias, as mixed infections may have been missed if culture favored one genotype [44]. However, where blood and sputum MTB strains were identical, this strongly suggests blood and sputum acted as one compartment. Conversely, these findings validate sequencing techniques and suggest that in vitro culture did not add significant bias. Furthermore, we cannot exclude that microevolution occurred in vitro and also could not compare the observed micro-evolutionary changes between different levels of impaired immunity. However, the current study suggests that M. tuberculosis micro-evolutionary events can occur over a short time scale during disease progression.
Additionally, although the small sample size of our study may have reduced the power of our conclusions, it is worth noting that the isolation of paired strains from blood and sputum is notoriously difficult. This is due to the fact that mycobacteremia only occurs in patients with advanced immunosuppression, who fortunately are less prevalent since the wide roll-out of antiretroviral therapy. Moreover, since the most widely used automated liquid culture system today, the BD MGIT 960, is not designed for mycobacterial blood cultures, we expect that our sample size will unlikely be surpassed by future studies.
Conclusions
In conclusion, among HIV-infected patients with poor cellular immunity, infection with multiple strains of M. tuberculosis was found in half of the patients. In the patients with identical strains, whole genome sequencing showed minimal M. tuberculosis intra-patient microevolution and did not reveal a consistent direction of spread between sputum and blood, suggesting that these compartments are highly connected and potentially seed each other repeatedly. However, SNP analysis of the whole genome sequencing results indicates that micro-evolutionary events can occur even over a short time scale during disease progression, and may be observed even in a small samples size. Future studies are needed to enrich our understanding of the role of microevolution in tuberculosis disease presentation and progression. The almost binary distinction between infections with different strains versus the apomictic identical strains that populate blood and sputum compartments warrants further investigation. We recommend a larger set of sputum/blood pairs to support the interpretation that microevolution occurs and that reseeding continually occurs between compartments among severely immunocompromised HIV-infected individuals. This would also allow for an assessment of the clinical parameters that may be associated, for example, with viral suppression status due to, ART.
Abbreviations
DNA, deoxy nucleic acid; GTR, generalized time reversible; HIV, human immunodeficiency virus; MDR-TB, multi-drug resistant tuberculosis; MEGA, molecular evolutionary genetics analysis; MIRU, mycobacterial interspersed repetitive unit; MTB, mycobacterium tuberculosis; RAxML, randomized axelerated maximum likelihood; RFLP, restriction fragment length polymorphism; SLV, single locus variant; SNP, single nucleotide polymorphisim; VNTR, variable number of tandem repeats; WGS, whole genome sequencing
Acknowledgments
The authors also wish to thank the Infectious Disease Institute Makerere University for co-ordination support as well as the Mycobacteriology (BSL-3) laboratory at the department of medical microbiology Makerere University for initial specimen analysis. Institute of Tropical Medicine Antwerp, Belgium for laboratory support given to WS when performing sub-culture and molecular assays as well as during data analysis and manuscript writing.
Funding
This work was supported by the Erasmus Mundus Joint Doctorate Program of the European Union through a training grant to WS and the European Research Council-INTERRUPTB starting grant (nr.311725) to BdJ.
The findings and conclusions in this article are those of the authors and do not necessarily represent the views of the institutions or the funding body.
Availability of data and materials
The fastQ raw reads have been deposited in sequence read archive (SRA) at http://www.ebi.ac.uk/ena/data/view/PRJEB10577. All other data supporting the findings are contained within the manuscript.
Authors’ contributions
WS, BdJ, FGC and MLJ conceived the study idea, CJM, WS performed sequence analyses, all authors participated in the manuscript writing and approval.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
This was a nested study within a study that was approved by the Joint Clinical Research Centre Institutional Review Board (IRB) and the Uganda National Council of Science and Technology (UNCST HS850). Each participant gave written informed consent for participation in the main study, including a second written informed consent for the samples and isolates to be used in future studies. Additional IRB approval was obtained from the Institute of Tropical Medicine (ITM), Antwerp, Belgium for the present analyses.
Additional files
A tabular list of all SNPs and indels per M. tuberculosis isolate, sputum and blood, created using nesoni nway program. Key: Highlighted = SNPs between concurrent sputum and blood M. tuberculosis isolates, p = patient. (XLS 15043 kb)
SNPs and SNP functions identified by comparing concurrent sputum and blood M. tuberculosis strains from HIV-infected individuals. (DOC 30 kb)
Contributor Information
Willy Ssengooba, Email: willyssengooba@gmail.com.
Bouke C. de Jong, Email: bdejong@itg.be
Moses L. Joloba, Email: moses.joloba@case.edu
Frank G. Cobelens, Email: f.cobelens@aighd.org
Conor J. Meehan, Email: cmeehan@itg.be
References
- 1.Niemann S, Supply P. Diversity and Evolution of Mycobacterium tuberculosis: Moving to Whole-Genome-Based Approaches. Cold Spring Harb Perspect Med. 2014;4:a021188. doi: 10.1101/cshperspect.a021188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tuon FF, Tonacio AC, Gryschek RC. Concomitant pleural and disseminated tuberculosis in AIDS: immune response or HIV infection compartmentalization? Acta Trop. 2007;104(2-3):79–83. doi: 10.1016/j.actatropica.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 3.Supply P, Allix C, Lesjean S, Cardoso-Oelemann M, Rusch-Gerdes S, Willery E, Savine E, de Haas P, van Deutekom H, Roring S, et al. Proposal for standardization of optimized mycobacterial interspersed repetitive unit-variable-number tandem repeat typing of Mycobacterium tuberculosis. J Clin Microbiol. 2006;44(12):4498–510. doi: 10.1128/JCM.01392-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garcia de Viedma D, Alonso Rodriguez N, Andres S, Ruiz Serrano MJ, Bouza E. Characterization of clonal complexity in tuberculosis by mycobacterial interspersed repetitive unit-variable-number tandem repeat typing. J Clin Microbiol. 2005;43(11):5660–4. doi: 10.1128/JCM.43.11.5660-5664.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kamerbeek J, Schouls L, Kolk A, van Agterveld M, van Soolingen D, Kuijper S, Bunschoten A, Molhuizen H, Shaw R, Goyal M, et al. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J Clin Microbiol. 1997;35(4):907–14. doi: 10.1128/jcm.35.4.907-914.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Embden JD, Cave MD, Crawford JT, Dale JW, Eisenach KD, Gicquel B, Hermans P, Martin C, McAdam R, Shinnick TM, et al. Strain identification of Mycobacterium tuberculosis by DNA fingerprinting: recommendations for a standardized methodology. J Clin Microbiol. 1993;31(2):406–9. doi: 10.1128/jcm.31.2.406-409.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Al-Hajoj SA, Akkerman O, Parwati I, Al-Gamdi S, Rahim Z, van Soolingen D, van Ingen J, Supply P, van der Zanden AG. Microevolution of Mycobacterium tuberculosis in a tuberculosis patient. J Clin Microbiol. 2010;48(10):3813–6. doi: 10.1128/JCM.00556-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perez-Lago L, Navarro Y, Herranz M, Bouza E, Garcia-de-Viedma D. Differences in gene expression between clonal variants of Mycobacterium tuberculosis emerging as a result of microevolution. Int J Med Microbiol. 2013;303(8):674–7. doi: 10.1016/j.ijmm.2013.09.010. [DOI] [PubMed] [Google Scholar]
- 9.Warren RM, Streicher EM, Sampson SL, van der Spuy GD, Richardson M, Nguyen D, Behr MA, Victor TC, van Helden PD. Microevolution of the direct repeat region of Mycobacterium tuberculosis: implications for interpretation of spoligotyping data. J Clin Microbiol. 2002;40(12):4457–65. doi: 10.1128/JCM.40.12.4457-4465.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McEvoy CR, Falmer AA, Gey van Pittius NC, Victor TC, van Helden PD, Warren RM. The role of IS6110 in the evolution of Mycobacterium tuberculosis. Tuberculosis. 2007;87(5):393–404. doi: 10.1016/j.tube.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 11.Tantivitayakul P, Panapruksachat S, Billamas P, Palittapongarnpim P. Variable number of tandem repeat sequences act as regulatory elements in Mycobacterium tuberculosis. Tuberculosis. 2010;90(5):311–8. doi: 10.1016/j.tube.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 12.Akhtar P, Singh S, Bifani P, Kaur S, Srivastava BS, Srivastava R. Variable-number tandem repeat 3690 polymorphism in Indian clinical isolates of Mycobacterium tuberculosis and its influence on transcription. J Med Microbiol. 2009;58(Pt 6):798–805. doi: 10.1099/jmm.0.002550-0. [DOI] [PubMed] [Google Scholar]
- 13.Garcia de Viedma D, Lorenzo G, Cardona PJ, Rodriguez NA, Gordillo S, Serrano MJ, Bouza E. Association between the infectivity of Mycobacterium tuberculosis strains and their efficiency for extrarespiratory infection. J Infect Dis. 2005;192(12):2059–65. doi: 10.1086/498245. [DOI] [PubMed] [Google Scholar]
- 14.Kato-Maeda M, Bifani PJ, Kreiswirth BN, Small PM. The nature and consequence of genetic variability within Mycobacterium tuberculosis. J Clin Invest. 2001;107(5):533–7. doi: 10.1172/JCI11426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Srilohasin P, Chaiprasert A, Tokunaga K, Nishida N, Prammananan T, Smittipat N, Mahasirimongkol S, Chaiyasirinroje B, Yanai H, Palittapongarnpim P. Genetic Diversity and Dynamic Distribution of Mycobacterium tuberculosis Isolates Causing Pulmonary and Extrapulmonary Tuberculosis in Thailand. J Clin Microbiol. 2014;52(12):4267–74. doi: 10.1128/JCM.01467-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brites D, Gagneux S. Old and new selective pressures on Mycobacterium tuberculosis. Infect Genet Evol. 2012;12(4):678–85. doi: 10.1016/j.meegid.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Navarro Y, Perez-Lago L, Sislema F, Herranz M, de Juan L, Bouza E, Garcia-de-Viedma D. Unmasking subtle differences in the infectivity of microevolved Mycobacterium tuberculosis variants coinfecting the same patient. Int J Med Microbiol. 2013;303(8):693–6. doi: 10.1016/j.ijmm.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 18.Walker TM, Ip CL, Harrell RH, Evans JT, Kapatai G, Dedicoat MJ, Eyre DW, Wilson DJ, Hawkey PM, Crook DW, et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect Dis. 2013;13(2):137–46. doi: 10.1016/S1473-3099(12)70277-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gardy JL, Johnston JC, Ho Sui SJ, Cook VJ, Shah L, Brodkin E, Rempel S, Moore R, Zhao Y, Holt R, et al. Whole-genome sequencing and social-network analysis of a tuberculosis outbreak. N Engl J Med. 2011;364(8):730–9. doi: 10.1056/NEJMoa1003176. [DOI] [PubMed] [Google Scholar]
- 20.Shin SS, Modongo C, Ncube R, Sepako E, Klausner JD, Zetola NM. Advanced Immune Suppression is Associated With Increased Prevalence of Mixed-Strain Mycobacterium tuberculosis Infections Among Persons at High Risk for Drug-Resistant Tuberculosis in Botswana. J Infect Dis. 2014;211(3):347–51. doi: 10.1093/infdis/jiu421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perez-Lago L, Comas I, Navarro Y, Gonzalez-Candelas F, Herranz M, Bouza E, Garcia-de-Viedma D. Whole genome sequencing analysis of intrapatient microevolution in Mycobacterium tuberculosis: potential impact on the inference of tuberculosis transmission. J Infect Dis. 2014;209(1):98–108. doi: 10.1093/infdis/jit439. [DOI] [PubMed] [Google Scholar]
- 22.Ssengooba W, Cobelens FG, Nakiyingi L, Mboowa G, Armstrong DT, Manabe YC, Joloba ML, de Jong BC. High Genotypic Discordance of Concurrent Mycobacterium tuberculosis Isolates from Sputum and Blood of HIV-Infected Individuals. PLoS One. 2015;10(7):e0132581. doi: 10.1371/journal.pone.0132581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feuerriegel S, Schleusener V, Beckert P, Kohl TA, Miotto P, Cirillo DM, Cabibbe AM, Niemann S, Fellenberg K. PhyResSE: a Web Tool Delineating Mycobacterium tuberculosis Antibiotic Resistance and Lineage from Whole-Genome Sequencing Data. J Clin Microbiol. 2015;53(6):1908–14. doi: 10.1128/JCM.00025-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feuerriegel S, Koser CU, Niemann S. Phylogenetic polymorphisms in antibiotic resistance genes of the Mycobacterium tuberculosis complex. J Antimicrob Chemother. 2014;69(5):1205–10. doi: 10.1093/jac/dkt535. [DOI] [PubMed] [Google Scholar]
- 25.Sun G, Luo T, Yang C, Dong X, Li J, Zhu Y, Zheng H, Tian W, Wang S, Barry CE, 3rd, et al. Dynamic population changes in Mycobacterium tuberculosis during acquisition and fixation of drug resistance in patients. J Infect Dis. 2012;206(11):1724–33. doi: 10.1093/infdis/jis601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Comas I, Coscolla M, Luo T, Borrell S, Holt KE, Kato-Maeda M, Parkhill J, Malla B, Berg S, Thwaites G, et al. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat Genet. 2013;45(10):1176–82. doi: 10.1038/ng.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Comas I, Chakravartti J, Small PM, Galagan J, Niemann S, Kremer K, Ernst JD, Gagneux S. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat Genet. 2010;42(6):498–503. doi: 10.1038/ng.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–3. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–9. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lew JM, Kapopoulou A, Jones LM, Cole ST. TubercuList--10 years after. Tuberculosis. 2011;91(1):1–7. doi: 10.1016/j.tube.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 31.Walker TM, Kohl TA, Omar SV, Hedge J, Del Ojo EC, Bradley P, Iqbal Z, Feuerriegel S, Niehaus KE, Wilson DJ, et al. Whole-genome sequencing for prediction of Mycobacterium tuberculosis drug susceptibility and resistance: a retrospective cohort study. Lancet Infect Dis. 2015;15:1193–202. doi: 10.1016/S1473-3099(15)00062-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perez-Lago L, Navarro Y, Herranz M, Bouza E, Garcia-de-Viedma D. Genetic features shared by Mycobacterium tuberculosis strains involved in microevolution events. Infect Genet Evol. 2013;16:326–9. doi: 10.1016/j.meegid.2013.02.016. [DOI] [PubMed] [Google Scholar]
- 33.Comas I, Borrell S, Roetzer A, Rose G, Malla B, Kato-Maeda M, Galagan J, Niemann S, Gagneux S. Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat Genet. 2012;44(1):106–10. doi: 10.1038/ng.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andrade MK, Machado SM, Leite ML, Saad MH. Phenotypic and genotypic variant of MDR-Mycobacterium tuberculosis multiple isolates in the same tuberculosis episode, Rio de Janeiro, Brazil. Braz J Med Biol Res. 2009;42(5):433–7. doi: 10.1590/S0100-879X2009000500006. [DOI] [PubMed] [Google Scholar]
- 35.Merker M, Kohl TA, Roetzer A, Truebe L, Richter E, Rusch-Gerdes S, Fattorini L, Oggioni MR, Cox H, Varaine F, et al. Whole genome sequencing reveals complex evolution patterns of multidrug-resistant Mycobacterium tuberculosis Beijing strains in patients. PLoS One. 2013;8(12):e82551. doi: 10.1371/journal.pone.0082551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.von Reyn CF, Kimambo S, Mtei L, Arbeit RD, Maro I, Bakari M, Matee M, Lahey T, Adams LV, Black W, et al. Disseminated tuberculosis in human immunodeficiency virus infection: ineffective immunity, polyclonal disease and high mortality. Int J Tuberc Lung Dis. 2011;15(8):1087–92. doi: 10.5588/ijtld.10.0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leeds IL, Magee MJ, Kurbatova EV, del Rio C, Blumberg HM, Leonard MK, Kraft CS. Site of extrapulmonary tuberculosis is associated with HIV infection. Clin Infect Dis. 2012;55(1):75–81. doi: 10.1093/cid/cis303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adams LV, Kreiswirth BN, Arbeit RD, Soini H, Mtei L, Matee M, Bakari M, Lahey T, Wieland-Alter W, Shashkina E, et al. Molecular epidemiology of HIV-associated tuberculosis in Dar es Salaam, Tanzania: strain predominance, clustering, and polyclonal disease. J Clin Microbiol. 2012;50(8):2645–50. doi: 10.1128/JCM.00624-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Behr MA, Waters WR. Is tuberculosis a lymphatic disease with a pulmonary portal? Lancet Infect Dis. 2014;14(3):250–5. doi: 10.1016/S1473-3099(13)70253-6. [DOI] [PubMed] [Google Scholar]
- 40.Basaraba RJ, Smith EE, Shanley CA, Orme IM. Pulmonary lymphatics are primary sites of Mycobacterium tuberculosis infection in guinea pigs infected by aerosol. Infect Immun. 2006;74(9):5397–401. doi: 10.1128/IAI.00332-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McMurray DN. Hematogenous reseeding of the lung in low-dose, aerosol-infected guinea pigs: unique features of the host-pathogen interface in secondary tubercles. Tuberculosis. 2003;83(1-3):131–4. doi: 10.1016/S1472-9792(02)00079-3. [DOI] [PubMed] [Google Scholar]
- 42.Barrios-Payan J, Saqui-Salces M, Jeyanathan M, Alcantara-Vazquez A, Castanon-Arreola M, Rook G, Hernandez-Pando R. Extrapulmonary locations of mycobacterium tuberculosis DNA during latent infection. J Infect Dis. 2012;206(8):1194–205. doi: 10.1093/infdis/jis381. [DOI] [PubMed] [Google Scholar]
- 43.Nakiyingi L, Moodley VM, Manabe YC, Nicol MP, Holshouser M, Armstrong DT, Zemanay W, Sikhondze W, Mbabazi O, Nonyane BA, et al. Diagnostic accuracy of a rapid urine lipoarabinomannan test for tuberculosis in HIV-infected adults. J Acquir Immune Defic Syndr. 2014;66:270–9. doi: 10.1097/QAI.0000000000000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martin A, Herranz M, Ruiz Serrano MJ, Bouza E, Garcia de Viedma D. The clonal composition of Mycobacterium tuberculosis in clinical specimens could be modified by culture. Tuberculosis. 2010;90(3):201–7. doi: 10.1016/j.tube.2010.03.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The fastQ raw reads have been deposited in sequence read archive (SRA) at http://www.ebi.ac.uk/ena/data/view/PRJEB10577. All other data supporting the findings are contained within the manuscript.