

Abstract
Objective:
We investigated the association between 52 risk variants identified through genome-wide association studies and disease severity in multiple sclerosis (MS).
Methods:
Ten unique MS case data sets were analyzed. The Multiple Sclerosis Severity Score (MSSS) was calculated using the Expanded Disability Status Scale at study entry and disease duration. MSSS was considered as a continuous variable and as 2 dichotomous variables (median and extreme ends; MSSS of ≤5 vs >5 and MSSS of <2.5 vs ≥7.5, respectively). Single nucleotide polymorphisms (SNPs) were examined individually and as both combined weighted genetic risk score (wGRS) and unweighted genetic risk score (GRS) for association with disease severity. Random-effects meta-analyses were conducted and adjusted for cohort, sex, age at onset, and HLA-DRB1*15:01.
Results:
A total of 7,125 MS cases were analyzed. The wGRS and GRS were not strongly associated with disease severity after accounting for cohort, sex, age at onset, and HLA-DRB1*15:01. After restricting analyses to cases with disease duration ≥10 years, associations were null (p value ≥0.05). No SNP was associated with disease severity after adjusting for multiple testing.
Conclusions:
The largest meta-analysis of established MS genetic risk variants and disease severity, to date, was performed. Results suggest that the investigated MS genetic risk variants are not associated with MSSS, even after controlling for potential confounders. Further research in large cohorts is needed to identify genetic determinants of disease severity using sensitive clinical and MRI measures, which are critical to understanding disease mechanisms and guiding development of effective treatments.
Multiple sclerosis (MS) is a severe autoimmune inflammatory disease of the central nervous system. Neurologic damage in MS is caused by irreversible demyelination of axons and lesion formation. Although early disease may manifest as attacks with full recovery, over time MS is extremely debilitating for the majority of patients. Only 37% of individuals with mild MS are employed, and within 15 years of diagnosis, 50%–60% of patients will require assistance with walking, posing tremendous economic and societal burden.1,2
Evidence suggests that both genetic and environmental components contribute to the risk of MS. The strongest genetic risk factor is within the human leukocyte antigen (HLA)-DRB1 locus, specifically the 15:01 allele,3 and studies support the presence of additional independent susceptibility alleles within the major histocompatibility complex (MHC) Class I and Class II regions.4 However, these genes have not been convincingly associated with progression.3,5 Environmental risk factors for MS include infection with Epstein-Barr virus, tobacco smoke exposure, low serum levels of vitamin D, and childhood/adolescent obesity.6,7 With the exception of tobacco smoke and low vitamin D,8,9 environmental influences on MS disease severity are unknown.
Through international collaboration, genome-wide association studies (GWAS) followed by replication have identified a large number of non-MHC MS risk variants.10,11 We hypothesized that MS risk variants might also influence disease severity. We investigated the association of the Multiple Sclerosis Severity Score (MSSS) with both a weighted genetic risk score (wGRS) and unweighted genetic risk score (GRS) comprising 52 established risk variant alleles, and each susceptibility variant alone in 7,125 MS cases from 10 independent cohorts.
METHODS
Study populations.
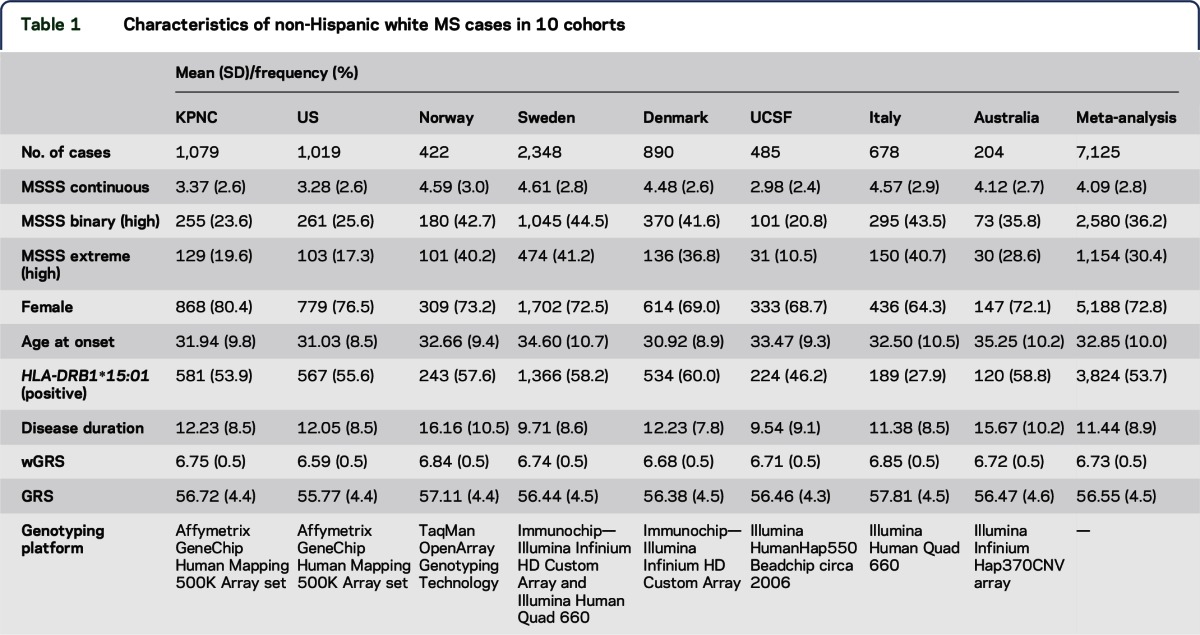
Ten independent and well-characterized MS case data sets were analyzed (table 1). The analysis included 1,079 white non-Hispanic MS patients recruited from Kaiser Permanente Medical Care Plan in the Northern California Region (KPNC).12 The following additional cases were included: 1,019 MS white non-Hispanic patients recruited from 2 other clinical sites in the United States (US1 and US2)5; 422 MS patients recruited through a population-based study in Oslo, Norway (Norway)13; 2,348 MS patients recruited through a population-based study in Sweden (Sweden)14; 890 MS patients from a cohort in Denmark (Denmark)11; 485 white non-Hispanic MS patients from a University of California San Francisco (UCSF)15 cohort; 678 MS patients from 2 cohorts recruited in Italy (Italy1 and Italy2)10,16; and 204 patients from a Tasmanian cohort study (Australia).17,18 Each case included in the meta-analysis fulfilled disease criteria for MS.19 Classical HLA-DRB1*15:01 typing was used in the KPNC, Sweden, and UCSF studies. Validated tagging single nucleotide polymorphisms (SNPs) for DRB1*15:01 were used in the US1 and US2, Italy1 and Italy2, and Australia (rs9271366), Norway (rs9270986), and Denmark (rs3135388) studies. Each of these tagging SNPs was tested against the classical high-resolution HLA-DRB1 typing performed in the KPNC data set; the correlations (r2) were 0.99, 0.91, and 0.95, respectively.
Table 1.
Characteristics of non-Hispanic white MS cases in 10 cohorts
Standard protocol approvals, registrations, and patient consents.
Each study protocol was approved by the appropriate Institutional Review Board of the participating academic institution. All participants provided written informed consent.
Genotyping and imputation.
In addition to HLA-DRB1*15:01 (described above), a group of 52 independent non-MHC GWAS SNPs were chosen for the present study. They were selected because they were previously identified and further replicated through GWAS, and they demonstrated larger magnitudes of effect on disease risk. All participants were genotyped using separate platforms: Affymetrix platform using the GeneChip Human Mapping 500K Array set (KPNC and United States), TaqMan OpenArray Genotyping Technology (Norway), Illumina Infinium HD Custom Array and Illumina Human Quad 660 (Sweden, Denmark, and Italy), and Illumina HumanHap550 Beadchip ©2006 (UCSF). All cohorts, except United States and Australia, contained genotyping information for all 52 variants. In the United States, tagging SNPs were used as proxies for 2 missing candidate SNPs: rs6693456 tagged rs11581062 (r2 = 0.95), and rs8106574 tagged rs1077667 (r2 = 0.63), based on publicly available data. In the Australia data set, 15 SNPs were missing: rs1323292, rs7522462, rs17174870, rs10201872, rs669607, rs12212193, rs17066096, rs13192841, rs354033, rs1520333, rs10466829, rs2119704, rs7200786, rs13333054, and rs2425752. Genetic data within 1-MB regions around all missing SNPs were available for each patient; therefore, imputation was possible against the 1000 Genomes reference. After imputation, missingness was lower than 2% in all cohorts. However, to recover all missing genotypes, the risk allele frequency from each cohort was used to estimate the missing genotypes. The estimation for each individual was made using a multinomial distribution generated from 1,000 random samples from each respective cohort. The probability for each of the 3 possible genotypes was generated. When a single genotype was missing for an individual, one sample was drawn randomly from the generated distribution from that cohort. This was done for a total of 146 individuals (KPNC = 92, Norway = 19, Sweden = 22, Denmark = 9, UCSF = 15, and Italy = 8) and a total of 48 SNPs (KPNC = 44, Norway = 15, Sweden = 4, Denmark = 2, UCSF = 10, and Italy = 8).
Weighted and unweighted genetic risk scores.
wGRS was calculated for each MS patient using the discovery odds ratios (ORs) as the weight for each of 52 non-MHC risk alleles derived from 9,772 MS cases and 17,376 controls,10 as previously described. In brief, the number of risk alleles for each SNP was multiplied by the weight for that variant, and then the sum across all 52 variants was calculated (table 1).10,20 GRS was calculated as the sum of risk allele copies for each SNP without weighting (table 1). Both wGRS and GRS were analyzed as continuous variables.
Multiple Sclerosis Severity Score.
MSSS is a probabilistic algorithm that uses the Expanded Disability Severity Scale (EDSS) to calculate disease severity and duration of disease, which was defined as time between first symptom and EDSS assessment.21 The MSSS for each patient in the current study was assigned using Global MSSS reference data derived from a large independent cohort of 9,892 patients with EDSS and disease duration ranging from 1 to >30 years. EDSS scores from a pooled reference patient cohort were ranked as previously described,21 and the average of the lowest and highest ranks for each possible EDSS value was calculated, taking into account also, scores reported for 2 years before and after each designated time point. These averages were normalized to account for the number of available assessments for each year (disease duration) and multiplied by 10. MSSS was analyzed as a continuous variable and as 2 dichotomous variables, as previously described.5 In brief, a binary MSSS variable was based on the median MSSS value, defined as MSSS ≤5 vs >5, with a smaller score indicating more benign or “mild” disease. The second variable was based on extreme ends of the MSSS distribution, defined as MSSS <2.5 (benign) vs ≥7.5 (severe) (table 1, figure 1).
Figure 1. Density plot of MSSS by cohort.

The density plot represents the frequency distribution of MSSS in each of the 10 cohorts and the total 7,125 individuals with MS. KPNC = Kaiser Permanente Medical Care Plan in the Northern California Region; MS = multiple sclerosis; MSSS = Multiple Sclerosis Severity Score. UCSF = University of California at San Francisco.
Statistical analysis.
All 10 data sets were included in a random-effects meta-analysis. Random-effects meta-analysis allows for heterogeneity across studies because of inherent differences and/or differential biases among each cohort, unlike fixed-effects models, which assume a single common effect underlies each study. A random-effects meta-analysis is generally more conservative, generating wider confidence intervals (CIs) and larger p values. Weighted and unweighted GRS, and all 52 non-MHC risk variants, were tested with the 3 MSSS outcomes in the meta-analysis.
In addition, analyses restricted to cases with a preestablished disease duration greater than or equal to 10 years were conducted to increase the likelihood that the MSSS measurement was stable. Both adjusted linear and logistic regression models were used to estimate adjusted beta values (β), ORs, and 95% CIs. The meta-analysis was adjusted for sex, age at onset, and HLA-DRB1*15:01. All analyses were conducted in STATA v13.1 (StataCorp, College Station, TX). The present study had sufficient power for all analyses (table e-1 at Neurology.org/ng).
RESULTS
A total of 7,125 individuals were included in the meta-analysis of the 3 MSSS outcomes (table 1). The overall distributions of MSSS demonstrated similarities across individual cohorts (figure 1) and were comparable to those in other cohorts reported previously21; however, there were some notable differences. Cohorts from Nordic countries and Italy had a larger proportion of cases with greater disease severity; disease duration, on average, was shorter in the Swedish and US (UCSF) cohorts (table 1). The sex distribution was also very similar across cohorts, with a 3:1 female to male ratio; KPNC had the highest proportion of females (80.4%), and Italy had the lowest proportion of females (64.3%) (table 1). Age at onset was similar among all cohorts and normally distributed, ranging from 30.9 years of age in Denmark to 35.3 years of age in Australia (table 1, figure 2). On average, disease duration since first symptom was 11.4 years across all cohorts and normally distributed, ranging from 9.5 years in UCSF to 16.2 years in Norway (table 1, figure e-1). Last, the distribution of HLA-DRB1*15:01 was typical of established genetic patterns in the literature (table 1): MS participants of Northern European descent were more likely to be HLA-DRB1*15:01 positive than participants in the Italy cohort.22 The non-MHC wGRS and GRS were similar across cohorts (figures e-2 and e-3).
Figure 2. Density plot of age at onset by cohort.

The density plot represents the frequency distribution of age at onset in each of the 10 cohorts and the total 7,125 individuals with MS. KPNC = Kaiser Permanente Medical Care Plan in the Northern California Region; MS = multiple sclerosis; MSSS = Multiple Sclerosis Severity Score. UCSF = University of California at San Francisco.
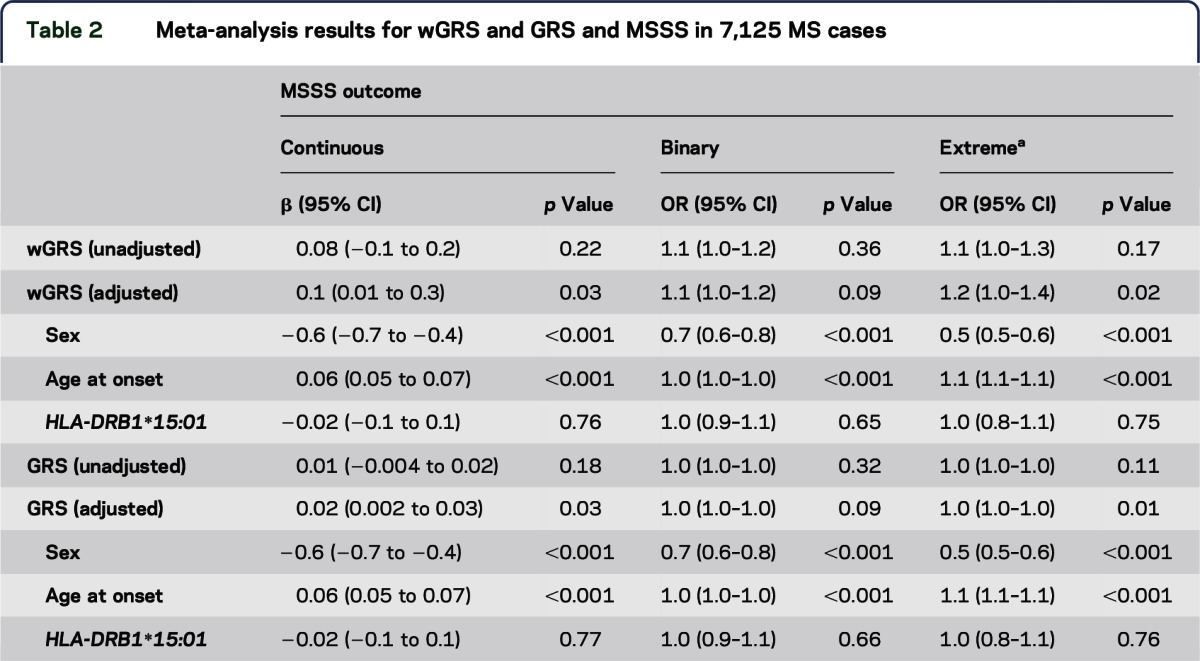
Meta-analyses accounting for the random effect of each cohort were used. We observed some evidence for association with wGRS and GRS for the MSSS outcomes (table 2). However, after restricting the data set to individuals who had a disease duration of 10 years or more, no significant associations remained (table e-2). Sex, age at onset, and HLA-DRB1*15:01 status were all analyzed as fixed effects. Sex and age at onset were consistently associated with MSSS in all 3 models, even after restricting to individuals with 10 years or more of disease (all p values <0.001) (table 2 and table e-2). Male sex and a later age at onset were both associated with more severe disease. HLA-DRB1*15:01 was not associated with MSSS in any of the models.
Table 2.
Meta-analysis results for wGRS and GRS and MSSS in 7,125 MS cases
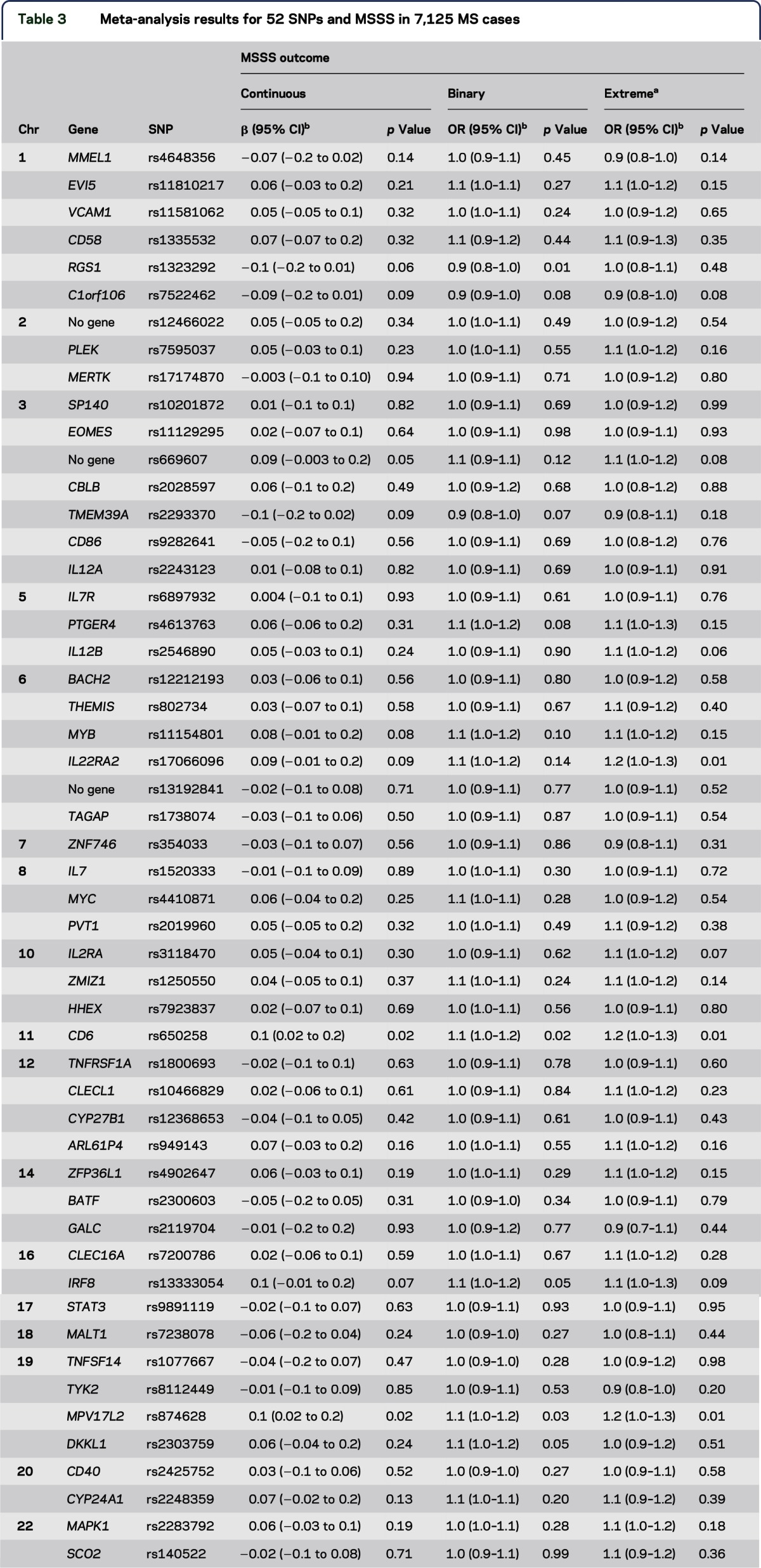
The 52 non-MHC risk variants were also tested individually in random-effects meta-analytic models adjusting for sex, age at onset, and HLA-DRB1*15:01 across all MS cases (full cohort), and in MS cases with disease duration of 10 years or more (restricted cohort) (table 3, table e-3). In the full cohort, 2 variants showed evidence of association for all 3 MSSS outcomes: rs874628 and rs650258, within MPV17L2 and 44-kb upstream of CD6, respectively. Similar results were observed in the restricted cohort (data not shown). All associations were in the same direction as the wGRS and GRS. However, after accounting for multiple testing, no single variant remained significant. When analyses were stratified by DRB1*15:01-positive and -negative carrier status in cases, results were similar (data not shown). We also restricted the above analyses to combined US, European, and Australian cohorts separately and did not observe any cohort-specific associations between MSSS and the wGRS or GRS (data not shown).
Table 3.
Meta-analysis results for 52 SNPs and MSSS in 7,125 MS cases
DISCUSSION
The identification of disease-modifying genes in MS is critical to further our understanding of disease pathogenesis, given the phenotype heterogeneity observed in patients for clinical manifestations, temporal course, and disease severity. This important topic has been previously reviewed, and several challenges have been described.23 Minimal progress has been made in the past decade owing mostly to the need for large, well-designed studies to detect presumably modest genetic effects, similar to what has been observed for GWAS, and clinical outcome data for large numbers of patients that accurately capture measures likely to be modified by genetic variation. Here, a meta-analysis of association between 52 established non-MHC MS genetic risk factors with larger effect sizes demonstrated in GWAS and disease severity was performed. Thus far, this is the largest combined MS patient cohort used to evaluate the relationship between established risk variants and disease severity. Ten independent and well-characterized data sets comprising 7,125 individuals with clinically definite MS were studied. The hypothesis that one or more known MS genetic risk variants are also associated with disease severity characterized by MSSS was comprehensively tested. Genetic factors that influence disease susceptibility tested here were not shown convincingly to affect disease severity.
The individuals included in this analysis are representative of the international MS population with regard to sex distribution, average age at onset, and proportion of HLA-DRB1*15:01 allele carriers. Male sex and older age at onset were associated with more severe disease, which is consistent with other reports.24 While HLA-DRB1*15:01 is an established risk factor for MS, it was not associated with MS disease severity here, as in previous studies that examined MSSS3,5 and brain atrophy.25
Available MS family data show that familial factors do not substantially influence eventual disease severity. However, they may increase the probability of a progressive clinical course, either from onset or after a phase of relapsing remitting disease, and evidence suggests that genetic influences are involved.26 Findings from studies of other neurodegenerative diseases, such as Alzheimer disease and Parkinson disease, have linked disease progression with specific genetic markers, also raising the possibility that MS progression might have a genetic component,27,28 given evidence for both neurodegeneration and inflammation in MS. However, to date, there has been limited success identifying non-MHC genetic associations with disease severity or other clinical phenotypes in MS. One study showed that a small subset (n = 16) of risk variants investigated in the present study was not associated with clinical and MRI outcomes in 179 recently diagnosed MS patients.29 However, the cumulative burden of non-MHC risk variants (n = 110) contributed modestly to the number of relapses in 842 MS patients.30 Furthermore, some variants studied (n = 17) were associated with attack severity, recovery, or frequency in 503 recently diagnosed MS cases.31 Relapse data were not available for the present study. These observations require further investigation and much larger samples sizes for confirmation. The most compelling evidence for genetic associations with cerebrospinal fluid antibody levels, both immunoglobulin G index and oligoclonal band positive status, was reported recently for MHC and immunoglobulin heavy-chain region genes in 6,950 patients.32 Both antibodies are markers of more severe disease in MS. The results, in conjunction with the present study findings, underscore the need for new studies to identify or exclude genetic contributions to disease severity in MS.
There is evidence in animal models related to MS to suggest that risk alleles have an effect on progression. The experimental autoimmune encephalomyelitis (EAE) model, which mirrors an inflammatory autoimmune disease of the central nervous system in rats and mice, has offered numerous experimental insights into MS. When genetically dissected into high-resolution quantitative trait loci, Eae25 and Eae29 have been shown to influence both susceptibility and progression.33,34 In addition, differential expression of an interleukin 2 (IL2) repressor in the gene ZEB1 results in EAE severity changes.35 Similarly, congenic rats with Eae18b locus have been shown to develop milder disease, with decreased demyelination and reduced recruitment of inflammatory cells to the brain.36 Evidence for multiple linked quantitative trait loci within the Tmevd2/Eae interval controlling disease severity in mice has also been reported.37 However, these findings have not been replicated in humans.
The present study had many strengths. First, the MSSS has been favored over EDSS to capture disease severity in MS because it incorporates disease duration to account for time.21 This approach builds on earlier work38 and is advantageous for large studies where EDSS measurements may have only been recorded at a single time point. We used data from a large, independent cohort reference (Global MSSS) to assign MSSS for each case in the present study. Sparse data were therefore avoided for cells in the reference table and potentially much larger variances, and missing data, in general; both could result from small sample sizes if our cohorts, which vary greatly in size, had been characterized instead by local MSSS.21 Furthermore, the large sample size used to create the Global MSSS made it possible to reduce the effect of stochastic fluctuations over time. Specifically, EDSS scores in the surrounding 2 years for each disease duration year are accounted for in the ranking; very large data sets are required to do this. It is important to note that the Global MSSS reference data set is at least ∼5 times larger than the largest individual cohort in the present study. The power of Global MSSS for genetic studies based on the large pooled reference data has also been previously demonstrated.21
Additional strengths included application of a statistical model for association testing that accounted for random (cohort) effects, and we adjusted for potential confounders such as age and sex. Moreover, use of MSSS extremes analysis reduced the possibility of phenotype misclassification because individuals with benign disease were compared with individuals with severe disease, and individuals in the middle of the MSSS spectrum who are most likely to be misclassified were excluded. While restricting analyses to extreme categories of MSSS reduced the overall number of individuals in our data set, given our overall large sample size, close to 4,000 individuals were still available for association testing.
Analyses were also restricted to individuals with 10 or more years of disease duration since symptom onset, to help ensure stability of the MSSS. At least 1 year of duration is required before the EDSS can be used to calculate the MSSS with the published algorithm.21 Median time to requiring unilateral assistance ranges from 15 to 30 years in MS, based on the present estimates.24 Conservatively, the present study used disease duration near the median value for the overall data set (table 1), but before the early end of the transition to requiring unilateral assistance (or an EDSS of 6). When analyses were restricted by disease duration (≥5, ≥10, ≥15, and ≥20 years), wGRS, GRS, and individuals SNPs (data not shown) were not associated with any of the 3 MSSS outcomes (figures e-4 and e-5).
There were also several limitations in this study. The EDSS used to calculate the MSSS is heavily weighted by physical measurements of disability. While MSSS is a widely accepted measure of disability for MS research, there are concerns about its sensitivity, given that different combinations of disease duration and EDSS can result in similar measures of the MSSS. In addition, it is possible that population-specific EDSS rankings could differ from those derived using the Global MSSS. A genetic association might therefore only be uncovered when a population is examined independently. Even larger studies will be required to determine whether studies of disability based on locally derived values for MSSS reveal population-specific genetic effects. Furthermore, because subclinical disease can occur many years before clinical symptom onset, the measure of disease duration required to derive the MSSS may not be accurate. Recent work shows that age could potentially be used for ranking EDSS in future studies when disease duration is unknown, because the exact age of symptom onset may not be certain.39 Longitudinal data on progression were not available for cases in the present study. There is also potential selection bias if individuals with mild disease are more willing to participate in research studies than those with severe disease. However, more than 1,100 participants were categorized in the highest or “most severe” MSSS phenotype in the present study (table 1), which provided a sufficient representation of cases with severe disease. Finally, our negative findings may have resulted from population or individual factors that could be not assessed in the present study. Additional genetic variants (untested), environmental exposures, or medication histories were not available and may distinguish MSSS differences in the presence or absence of the established genetic risk variants that were investigated.
The current data set includes 52 non-MHC MS genetic risk variants,10 and a larger list of the most recently identified non-MHC MS risk variants through GWAS and follow-up studies is now available.11 The 52 non-MHC MS risk variants used here were stronger genetic markers of MS susceptibility; the average OR based on GWAS for our 52 non-MHC variants is 1.19 vs average OR for new 48 non-MHC variants, which is 1.09.10,11 GWAS of disease severity, to date, have not yet yielded new candidates at genome-wide significance5,10; and while disease risk and severity variants appear to differ in MS, this still remains a question. We have comprehensively pursued testing of the strongest established disease risk variants identified, thus far, for evidence of influence on MS severity. Larger studies of cases with additional genotype data for newly discovered GWAS variants are warranted. Finally, whole-genome data were not available to formally adjust for potential effects of population stratification. However, this was not a case-control study, each cohort was treated separately in the analysis, and consistent nonsignificant estimates for each of the 10 independent cohorts were observed. Our findings in this large, well-powered study are unlikely to result from population stratification.
Results derived from investigation of a large number of recently established GWAS variants in 7,125 MS cases suggest that the genetics underlying MS susceptibility and disease severity, as measured by MSSS, do not substantially overlap. Sensitive measures of severity and progression are needed, and for larger prospective cohort studies of incident cases. Comprehensive quantitative trait data derived from high-resolution brain MRIs can be used as dependent variables for a GWAS, as recently demonstrated with 284 MS patients,40 or for whole-genome sequencing studies. Longitudinal studies will be informative for genetic investigations. However, such studies will be necessarily balanced by a lack of statistical power to detect modest genetic effects unless very large numbers of individuals can be assembled and followed for long periods of time.
Supplementary Material
GLOSSARY
- CI
confidence interval
- EAE
experimental autoimmune encephalomyelitis
- EDSS
Expanded Disability Severity Scale
- GRS
unweighted genetic risk score
- GWAS
genome-wide association studies
- KPNC
Kaiser Permanente Medical Care Plan in the Northern California Region
- MHC
major histocompatibility complex
- MS
multiple sclerosis
- MSSS
Multiple Sclerosis Severity Score
- OR
odds ratio
- SNP
single nucleotide polymorphism
- UCSF
University of California at San Francisco
- wGRS
weighted genetic risk score
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR AFFILIATIONS
From the Division of Epidemiology (M.F.G., X.S., M.A.G., L.F.B), Genetic Epidemiology and Genomics Laboratory, School of Public Health, University of California, Berkeley; Department of Epidemiology and Biostatistics, School of Medicine (F.B.S.B.), Case Western Reserve University, Cleveland, OH; Department of Clinical Neuroscience (I.K., A.H., J.H, T.O.), Karolinska Institutet, Stockholm, Sweden; Department of Neurology (H.F.H., E.G.C., S.D.B.), Oslo University Hospital; Institute of Clinical Medicine (H.F.H., S.D.B.), University of Oslo, Norway; Kaiser Permanente Division of Research (L.S., C.S., L.F.B.), Oakland, CA; Sonoma West Medical Center (A.B.), Sebastopol, CA; Institute of Environmental Medicine (L.A.), Karolinska Institutet, Stockholm, Sweden; Centre for Occupational and Environmental Medicine (L.A.), Stockholm County Council, Sweden; Program in Translational NeuroPsychiatric Genomics (N.A.P., P.L.D.J.), Institute for the Neurosciences, Department of Neurology, Division of Genetics (N.A.P., P.L.D.J.), Department of Medicine, Brigham & Women's Hospital, Harvard Medical School (N.A.P., P.L.D.J.), Boston, MA; Broad Institute of Harvard and Massachusetts Institute of Technology (N.A.P., P.L.D.J.), Cambridge; Danish Multiple Sclerosis Center (A.B.O., H.B.S., F.S., P.S.S.), Department of Neurology, Rigshospitalet, University of Copenhagen, Denmark; Department of Neurology (R.G., S.J.C., B.A.C.C., J.R.O., S.L.H.), University of California, San Francisco; Department of Health Sciences (S.D.A.), Interdisciplinary Research Center of Autoimmune Diseases IRCAD (S.D.A., M.A.L.), University of Eastern Piedmont, Novara, Italy; SCDU Neurology (M.A.L.), AOU “Maggiore della Carità,” Novara, Italy; Laboratory of Genetics of Neurological Complex Disorders (F.M.B., M.S.), Institute of Experimental Neurology (INSPE), Division of Neuroscience, San Raffaele Scientific Institute, Milan, Italy; Department of Neurology (F.M.B.), Institute of Experimental Neurology (INSPE), Division of Neuroscience, San Raffaele Scientific Institute, Milan, Italy; and Menzies Institute for Biomedical Research (I.v.d.M., B.V.T., Y.Z.), University of Tasmania, Hobart, Australia.
AUTHOR CONTRIBUTIONS
Dr. George: conception and design, analysis and interpretation of the data, drafting of the manuscript, and final approval of the version to be published. Dr. Briggs: conception and design, analysis and interpretation of the data, reviewing and critically revising the manuscript, and final approval of the version to be published. Ms. Shao and Ms. Gianfrancesco: analysis and interpretation of the data, reviewing and critically revising the manuscript, final approval of the version to be published. Dr. Kockum and Dr. Harbo: acquisition of the data, clinical evaluation of patients, collection of samples, evaluation of manuscript, and final approval of the version to be published. Dr. Celius: clinical evaluation of patients, collection of samples, evaluation of manuscript, and final approval of the version to be published. Dr. Bos, Dr. Hedström, and Dr. Shen: data preparation, evaluation of manuscript, and final approval of the version to be published. Dr. Bernstein: clinical evaluation of patients, evaluation of manuscript, and final approval of the version to be published. Dr. Alfredsson, Dr. Hillert, Dr. Olsson, Dr. Patsopoulos, Dr. De Jager, Dr. Oturai, Dr. Søndergaard, Dr. Sellebjerg, and Dr. Sorensen: acquisition of the data, collection of samples, evaluation of manuscript, and final approval of the version to be published. Ms. Gomez, Ms. Caillier, Dr. Cree, and Dr. Hauser: clinical evaluation of patients, evaluation of manuscript, and final approval of the version to be published. Dr. Oksenberg: acquisition of the data, collection of samples, evaluation of manuscript, and final approval of the version to be published. Dr. D'Alfonso: acquisition of the data, clinical evaluation of patients, collection of samples, evaluation of manuscript, and final approval of the version to be published. Dr. Leone: acquisition of the data, collection of samples, evaluation of manuscript, and final approval of the version to be published. Dr. Martinelli Boneschi: acquisition of the data, clinical evaluation of patients, collection of samples, evaluation of manuscript, and final approval of the version to be published. Dr. Sorosina: acquisition of the data, collection of samples, evaluation of manuscript, and final approval of the version to be published. Dr. van der Mei and Dr. Taylor: acquisition of the data, clinical evaluation of patients, collection of samples, evaluation of manuscript, and final approval of the version to be published. Mr. Zhou: data preparation, evaluation of manuscript, and final approval of the version to be published. Dr. Shaefer: conception and design, acquisition of the data, reviewing and critically revising the manuscript, and final approval of the version to be published. Dr. Barcellos: conception and design, acquisition of the data, analysis and interpretation of the data, drafting and writing of the manuscript, and final approval of the version to be published.
STUDY FUNDING
This work was supported by the NIH/National Institute of Neurological Disorders and Stroke R01NS049510, NIH/National Institute of Allergy and Infectious Diseases R01AI076544, and NIH/National Institute of Environmental Health Sciences R01 ES017080; Foundation for Multiple Sclerosis (grants “Progetto Speciale Immunochip,” 2011/R/14, Fondazione cariplo [2010-0728]); and NIH/National Institute of Neurological Disorders and Stroke R01NS026799 and NIH/National Institute of Neurological Disorders and Stroke R01NS049477.
DISCLOSURE
Dr. George, Dr. Briggs, and Ms. Shao report no disclosures. Ms. Gianfrancesco has received research support from NIH/National Institute of Neurological Disorders and Stroke. Dr. Kockum has received speaker honoraria from Merc Serono; has received research support from NHR Fonden and Karolinska University Hospital; and holds stock in Novo Nordisk and several Swedish companies (all stock unrelated to this study). Dr. Harbo has received research support and/or travel funding from Biogen Idec, Sanofi-Aventis, TEVA, and Novartis and has received research support from the Norwegian Research Council, Norwegian Health Authorities South-East, the University of Oslo, the Oslo Multiple Sclerosis Society, the Odd Fellow MS Society (Norway), and Novartis. Dr. Celius has received unrestricted research grants from Novartis and Biogen Idec and payment for advice and lectures from Almirall, Biogen Idec, Genzyme, Sanofi, TEVA, Merck-Serono, and Novartis. Dr. Bos, Dr. Hedström, and Dr. Shen report no disclosures. Dr. Bernstein receives research funding from Eli Lilly and Roach (for Alzheimer studies) and has received research support from Genentech, Biogen, Lundbeck, Accera, and AZ Therapeutics. Dr. Alfredsson has received speaker honoraria from TEVA and Biogen Idec and has received research support from the following government entities: the Swedish Medical Research Council; the Swedish Council for Working Life, Health and Welfare; AFA Insurance; and the Swedish Brain Foundation. Dr Hillert has received honoraria for serving on advisory boards for BiogenIdec, Sanofi-Genzyme, and Novartis; has received speaker fees from BiogenIdec, Merck-Serono, Bayer-Schering, TEVA, Novartis, and Sanofi-Genzyme; has received research support from BiogenIdec, Merck-Serono, TEVA, Novartis, Sanofi-Genzyme, Bayer-Schering, the Swedish Research Council, the Karolinska Institute, and the Swedish Brain Foundation; and has served on the editorial board of the Journal of Neuroimmunology. Dr. Olsson has served on the scientific advisory boards of/received speaker honoraria from Biogen Idec, Merck, Novartis, TEVA, Sanofi-Aventis, Genzyme, Almirall, and Actelion; has received research support from Biogen Idec, Bayer Schering, Genzyme, Merck, Sanofi-Aventis, Novartis, AstraZeneca, the Swedish Research Council, the Knut and Alice Wallenbergs Foundation, the AFA Foundation, and the Swedish Brain Foundation; and has served on the editorial board of Current Opinion in Immunology. Dr. Patsopoulos has served as a consultant for Merck and has received research support from Intel Corporation, the National Multiple Sclerosis Society, and the Harvard NeuroDiscovery Center. Dr. De Jager has served on the scientific advisory boards of TEVA Neuroscience and Sanofi/Genzyme; has received travel funding/speaker honoraria from Biogen Idec, Source Healthcare Analytics Pfizer Inc, and TEVA; has served on editorial boards of the Journal of Neuroimmunology, Neuroepigenetics, and Multiple Sclerosis; and has received research support from Biogen Idec, Vertex, Sanofi/Genzyme, and the National MS Society. Dr. Oturai has served on the scientific advisory boards for Novartis, Sanofi/Genzyme, and Biogen Idec; has received speaker honoraria from Biogen Idec, Merck Serono, TEVA, Sanofi/Genzyme, and Novartis; and has received research support from Biogen Idec, Novartis, the Danish Council for Strategic Research, Brdr. Rønje Holding, the Danish Multiple Sclerosis Society, the Warwara Larsen Foundation, and the Johnsen Foundation. Dr. Søndergaard has received travel funding/speaker honoraria from Biogen Idec, Genzyme, and TEVA and has received research support from Sclerose Foreningen, MS Networks and Immunogenetics, and A.P. Møller Fonden-Fonden til lægevidenskabens fremme. Dr. Sellebjerg has served on scientific advisory boards, been on the steering committees of clinical trials, served as a consultant, received support for congress participation, received speaker honoraria, or received research support for his laboratory from Bayer Schering, Biogen Idec, Genzyme, Merck Serono, Novo Nordisk, Novartis, Sanofi-Aventis, Schering Plough, TEVA, the Danish Strategic Research Council, the Danish Multiple Sclerosis Society, and the Lounkær Foundation and has served on the editorial board of Multiple Sclerosis and Related Disorders. Dr. Sorensen has served on scientific advisory boards for/received speaker honoraria from Biogen Idec, Merck Serono, Novartis, Genmab, TEVA, GlaxoSmithKline, Bayer Schering, Sanofi-Aventis, and Genzyme; has received research support from Biogen Idec, Bayer Schering, Merck Serono, TEVA, Sanofi-Aventis, Novartis, RoFAR, Roche, Genzyme, the Danish Multiple Sclerosis Society, the Danish Medical Research Council, and the European Union Sixth Framework Programme: Life sciences, Genomics and Biotechnology for Health; and has served on the editorial boards of the European Journal of Neurology, Therapeutic Advances in Neurological Disorders, and Multiple Sclerosis Journal. Ms. Gomez and Ms. Caillier report no disclosures. Dr. Cree has been a consultant for Abbvie, Biogen Idec, EMD Serono, Genzyme/Sanofi-Aventis, MedImmune, Shire, Novartis, and TEVA Neurosciences; has received research support from Acorda, Avanir, Biogen Idec, EMD Serono, Hoffman La Roche, MedImmune, Novartis, Receptos, and TEVA; and has served on the editorial board of Annals of Neurology. Dr. Oksenberg has served on the editorial board of Genes and Immunity; has been an employee of the University of California at San Francisco; and has received research support from NIH/National Institute of Neurological Disorders and Stroke and the National Multiple Sclerosis Society. Dr. Hauser has served on the scientific advisory boards of Bionure, Annexon, Symbiotix, Neurona, and Molecular Stethoscope; has received publishing royalties from McGraw-Hill; has been an employee of the University of California, San Francisco; has received research support from NIH, the National Multiple Sclerosis Society, the Conrad Hilton Foundation; and received stock options from Bionure, Annexon, and Symbiotix (all transferred to his institution). Dr. D'Alfonso has served on the editorial board of Genes and Immunity and has received research support from the Italian Ministry of Education, University and Research, the Italian Foundation for Multiple Sclerosis, and Fondazione Cariplo. Dr. Leone has served on the editorial board of the European Journal of Neurology and has received research support from the Italian Ministry of Health. Dr. Martinelli Boneschi has received support for congress participation or speaker honoraria from TEVA, Biogen Idec, Merck-Serono, Sanofi-Aventis and Genzyme and has received research support from Merck Serono, TEVA, Biogen Idec, Genzyme, GW-Pharma, the Italian Foundation of Multiple Sclerosis, the Italian Ministry of Health, the Fondazione Cariplo, and the European Union Seventh Framework Programme: Research and Innovation. Dr. Sorosina reports no disclosures. Dr. van der Mei has received funding for travel and speaker honoraria from Biogen Idec and has received research support from the National Health and Medical Research Council of Australia and Multiple Sclerosis Research Australia. Dr. Taylor has served on the scientific advisory boards of Biogen Idec, Australia Novartis Gilenya, and Genzyme International; has received funding for travel and speaker honoraria from Bayer Schering Pharma, MS Society of Western Australia, CSL Australia Pty. Ltd., Biogen Idec, and Novartis; has served on the editorial boards of International MS Journal and the Journal of Neurology Neurosurgery and Psychiatry; and has received research support from National Health and Medical Research Council Australia, MS Research Australia, National Health and Medical Research Council, HRC New Zealand, National MS Society of USA, the Australian Research Council, and the MS society of Tasmania. Mr. Zhou reports no disclosures. Dr. Schaefer has received research support from GlaxoSmithKline, National Institute of Environmental Health Sciences, National Institute of Mental Health, National Institute of Allergy and Infectious Diseases, National Institute on Aging, and the National Multiple Sclerosis Society. Dr. Barcellos has received research support from NIH/National Institute of Environmental Health Sciences, NIH/National Institute of Neurological Disorders and Stroke, US Environmental Protection Agency, NIH/National Cancer Institute, NIH/National Institute of Arthritis and Musculoskeletal and Skin Diseases, the National Multiple Sclerosis Society, the Peder Sather Grant for Collaborative Research, and the Robert Wood Johnson Health and Society Program Pilot Grant. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Tedeholm H, Skoog B, Lisovskaja V, Runmarker B, Nerman O, Andersen O. The outcome spectrum of multiple sclerosis: disability, mortality, and a cluster of predictors from onset. J Neurol 2015;262:1148–1163. [DOI] [PubMed] [Google Scholar]
- 2.Naci H, Fleurence R, Birt J, Duhig A. Economic burden of multiple sclerosis: a systematic review of the literature. Pharmacoeconomics 2010;28:363–379. [DOI] [PubMed] [Google Scholar]
- 3.Barcellos LF, Sawcer S, Ramsay PP, et al. Heterogeneity at the HLA-DRB1 locus and risk for multiple sclerosis. Hum Mol Genet 2006;15:2813–2824. [DOI] [PubMed] [Google Scholar]
- 4.Patsopoulos NA, Barcellos LF, Hintzen RQ, et al. Fine-mapping the genetic association of the major histocompatibility complex in multiple sclerosis: HLA and non-HLA effects. PLoS Genet 2013;9:e1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.The International Multiple Sclerosis Genetics Consortium. Genome-wide association study of severity in multiple sclerosis. Genes Immun 2011;12:615–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ascherio A. Environmental factors in multiple sclerosis. Expert Rev Neurother 2013;13(12 suppl):3–9. [DOI] [PubMed] [Google Scholar]
- 7.Munger KL, Chitnis T, Ascherio A. Body size and risk of MS in two cohorts of US women. Neurology 2009;73:1543–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wingerchuk DM. Smoking: effects on multiple sclerosis susceptibility and disease progression. Ther Adv Neurol Disord 2012;5:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ascherio A, Munger KL, Simon KC. Vitamin D and multiple sclerosis. Lancet Neurol 2010;9:599–612. [DOI] [PubMed] [Google Scholar]
- 10.The International Multiple Sclerosis Genetics Consortium; Wellcome Trust Case Control Consortium 2, Sawcer S, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011;476:214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beecham AH, Patsopoulos NA, Xifara DK, et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet 2013;45:1353–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Briggs FB, Acuna BS, Shen L, et al. Adverse socioeconomic position during the life course is associated with multiple sclerosis. J Epidemiol Community Health 2014;68:622–629. [DOI] [PubMed] [Google Scholar]
- 13.Smestad C, Brynedal B, Jonasdottir G, et al. The impact of HLA-A and -DRB1 on age at onset, disease course and severity in Scandinavian multiple sclerosis patients. Eur J Neurol 2007;14:835–840. [DOI] [PubMed] [Google Scholar]
- 14.Hedstrom AK, Baamhielm M, Olsson T, Alredsson L. Tobacco smoking, but not Swedish snuff use, increases the risk of multiple sclerosis. Neurology 2009;73:696–701. [DOI] [PubMed] [Google Scholar]
- 15.Okuda DT, Srinivasan R, Oksenberg JR, et al. Genotype-phenotype correlations in multiple sclerosis: HLA genes influence disease severity inferred by 1HMR spectroscopy and MRI measures. Brain 2009;132:250–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leone MA, Barizzone N, Esposito F, et al. Association of genetic markers with CSF oligoclonal bands in multiple sclerosis patients. PLoS One 2013;8:e64408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pittas F, Ponsonby AL, van der Mei IA, et al. Smoking is associated with progressive disease course and increased progression in clinical disability in a prospective cohort of people with multiple sclerosis. J Neurol 2009;256:577–585. [DOI] [PubMed] [Google Scholar]
- 18.Simpson S Jr, Pittas F, van der Mei I, Blizzard L, Ponsonby A, Taylor B. Trends in the epidemiology of multiple sclerosis in Greater Hobart, Tasmania: 1951 to 2009. J Neurol Neurosurg Psychiatry 2011;82:180–187. [DOI] [PubMed] [Google Scholar]
- 19.McDonald WI, Compston A, Edan G, et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis. Ann Neurol 2001;50:121–127. [DOI] [PubMed] [Google Scholar]
- 20.De Jager PL, Chibnik LB, Cui J, et al. Integration of genetic risk factors into a clinical algorithm for multiple sclerosis susceptibility: a weighted genetic risk score. Lancet Neurol 2009;8:1111–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roxburgh RH, Seaman SR, Masterman T, et al. Multiple Sclerosis Severity Score: using disability and disease duration to rate disease severity. Neurology 2005;64:1144–1151. [DOI] [PubMed] [Google Scholar]
- 22.Ballerini C, Guerini FR, Rombola G, et al. HLA-multiple sclerosis association in continental Italy and correlation with disease prevalence in Europe. J Neuroimmunol 2004;150:178–185. [DOI] [PubMed] [Google Scholar]
- 23.Kantarci OH, de Andrade M, Weinshenker BG. Identifying disease modifying genes in multiple sclerosis. J Neuroimmunol 2002;123:144–159. [DOI] [PubMed] [Google Scholar]
- 24.Tremlett H, Zhao Y, Rieckmann P, Hutchinson M. New perspectives in the natural history of multiple sclerosis. Neurology 2010;74:2004–2015. [DOI] [PubMed] [Google Scholar]
- 25.Van der Walt A, Stankovich J, Bahlo M, et al. Heterogeneity at the HLA-DRB1 allelic variation locus does not influence multiple sclerosis disease severity, brain atrophy or cognition. Mult Scler 2011;17:344–352. [DOI] [PubMed] [Google Scholar]
- 26.Hensiek AE, Seaman SR, Barcellos LF, et al. Familial effects on the clinical course of multiple sclerosis. Neurology 2007;68:376–383. [DOI] [PubMed] [Google Scholar]
- 27.Pankratz N, Nichols WC, Elsaesser VE, et al. Alpha-synuclein and familial Parkinson's disease. Mov Disord 2009;24:1125–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kamboh MI, Demirci FY, Wang X, et al. Genome-wide association study of Alzheimer's disease. Transl Psychiatry 2012;2:e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalincik T, Guttman CRG, Krasensky J, et al. Multiple sclerosis susceptibility loci do not alter clinical and MRI outcomes in clinically isolated syndrome. Genes Immun 2013;14:244–248. [DOI] [PubMed] [Google Scholar]
- 30.Hilven K, Patsopoulos NA, Dubois B, Goris A. Burden of risk variants correlates with phenotype of multiple sclerosis. Mult Scler 2015;21:1670–1680. [DOI] [PubMed] [Google Scholar]
- 31.Mowry EM, Carey RF, Blasco MR, et al. Multiple sclerosis susceptibility genes: associations with relapse severity and recovery. PLoS One 2013;8:e75416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goris A, Pauwels I, Gustavsen MW, et al. Genetic variants are major determinants of CSF antibody levels in multiple sclerosis. Brain 2015;138:632–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marta M, Stridh P, Becanovic K, et al. Multiple loci comprising immune-related genes regulate experimental neuroinflammation. Genes Immun 2010;11:21–36. [DOI] [PubMed] [Google Scholar]
- 34.Beyeen AD, Adzemovic MZ, Ockinger J, et al. IL-22RA2 associates with multiple sclerosis and macrophage effector mechanisms in experimental neuroinflammation. J Immunol 2010;185:6883–6890. [DOI] [PubMed] [Google Scholar]
- 35.Stridh P, Thessen Hedreul M, Beyeen AD, et al. Fine-mapping resolves Eae23 into two QTLs and implicates ZEB1 as a candidate gene regulating experimental neuroinflammation in rat. PLoS One 2010;5:e12716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adzemovic MZ, Ockinger J, Zeitelhofer M, et al. Expression of Ccl11 associates with immune response modulation and protection against neuroinflammation in rats. PLoS One 2012;7:e39794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spach KM, Case LK, Noubade R, et al. Multiple linked quantitative trait loci within the Tmevd2/Eae3 interval control the severity of experimental allergic encephalomyelitis in DBA/2J mice. Genes Immun 2010;11:649–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weinshenker BG, Wingerchuk DM, Liu Q, et al. Genetic variation in the tumor necrosis factor alpha gene and the outcome of multiple sclerosis. Neurology 1997;49:378–385. [DOI] [PubMed] [Google Scholar]
- 39.Manouchehrinia A, Helga W, Kingwell E, et al. Age related multiple sclerosis severity score: disability ranked by age. Neurology 2016;86(16 suppl):S24.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gourraud PA, Sdika M, Khankhanian P, et al. A genome-wide association study of brain lesion distribution in multiple sclerosis. Brain 2013;136:1012–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.