

Abstract
Heart diseases, especially coronary artery diseases (CAD), are the leading causes of morbidity and mortality in developed countries. Effective therapy is available to ensure patient survival and to prevent long term sequelae after an acute ischemic event caused by CAD, but appropriate therapy requires rapid and accurate diagnosis. Research into the pathology of CAD have demonstrated the usefulness of measuring concentrations of chemicals released from the injured cardiac muscle can aid the diagnosis of diseases caused by myocardial ischemia. Since the mid-1950s successively better biochemical markers have been described in research publications and applied for the clinical diagnosis of acute ischemic myocardial injury. Aspartate aminotransferase of the 1950s was replaced by other cytosolic enzymes such as lactate dehydrogenase, creatine kinase and their isoenzymes that exhibited better cardiac specificity. With the availability of immunoassays, other muscle proteins, that had no enzymatic activity, were also added to the diagnostic arsenal but their limited tissue specificity and sensitivity lead to suboptimal diagnostic performance. After the discovery that cardiac troponins I and T have the desired specificity, they have replaced the cytosolic enzymes in the role of diagnosing myocardial ischemia and infarction. The use of the troponins provided new knowledge that led to revision and redefinition of ischemic myocardial injury as well as the introduction of biochemicals for estimation of the probability of future ischemic myocardial events. These markers, known as cardiac risk markers, evolved from the diagnostic markers such as CK-MB or troponins, but markers of inflammation also belong to these groups of diagnostic chemicals. This review article presents a brief summary of the most significant developments in the field of biochemical markers of cardiac injury and summarizes the most recent significant recommendations regarding the use of the cardiac markers in clinical practice.
Key words: coronary artery disease, myocardial infarction, biochemical markers, cardiac markers, cardiac troponins
EPIDEMIOLOGY OF CORONARY ARTERY DISEASES
Heart disease, along with malignancies, are the top two causes of death in developed countries. In the United States approximately 25 % of all deaths occur because of cardiac diseases. This is equivalent to 610,000 deaths each year from heart disease [1]. Sixty-one percent of the deaths, or 370,000 events, are due to coronary heart disease (CHD) [1] that is caused by cholesterol plaque buildup with consequent narrowing of the coronary arteries. This is known as coronary artery disease (CAD). When this plaque ruptures it activates the coagulation cascade locally and the developing thrombus restricts or completely stops blood flow to the cardiac muscle downstream from the occlusion, causing the clinical conditions known as angina and heart attack (myocardial infarction). According to the 2013 US national statistics, the 370,000 deaths per year are the consequence of approximately 735,000 heart attacks of which about one third (1/3) are recurrent events [1]. Based on population statistics it can be calculated that approximately 310 heart attacks per hundred-thousand people older than 18 years of age and approximately 150 deaths per hundred-thousand adults can be expected each year in developed nations. The number of people with heart disease and the death rate from CHD varies between sexes, racial groups and geographic region within a country and it increases with increasing age even within the same country, but CHD and heart attack are large public health issues and consume large amounts of health care dollars. As the severity of consequences of a heart attack increases with each minute of delay in diagnosis and treatment, early diagnosis is mandatory to minimize long term sequelae of an acute coronary event.
Non-standard abbreviations (in alphabetical order)
AACC: American Association for Clinical Chemistry,
ACC: American College of Cardiology,
ACS: acute coronary syndromes,
AHA: American Heart Association,
AMI: acute myocardial infarction,
AST: aspartate transaminase,
CAD: coronary artery disease,
CABG: coronary artery bypass graft,
CHD: coronary heart disease,
CK: creatine kinase,
CK-MB: creatine kinase MB isoenzyme,
CRP: C-reactive protein,
cTnI: cardiac troponin I,
cTns: cardiac troponins,
cTnT: cardiac troponin T,
CV: coefficient of variation,
ECG, EKG: electrocardiogram,
ESC: European Society of Cardiology,
HAMA: human anti-mouse antibody,
hs: high-sensitivity,
IFCC: International Federation of Clinical Chemistry and Laboratory Medicine,
IMA: ischemia modified albumin,
LD, LDH: lactate dehydrogenase,
LD-1: lactate dehydrogenase isoenzyme 1,
LoD: limit of detection,
MI: myocardial infarction,
MoAb: monoclonal antibody,
NACB: National Academy of Clinical Biochemistry,
NQWMI: non-Q wave MI,
NSTEMI: non-ST elevation MI,
PCI: percutaneous coronary intervention,
QWMI: Q-wave MI,
SGOT serum glutamic oxaloacetic transaminase,
STEMI: ST-elevation MI,
TF-CB: Task Force on Clinical Applications of Cardiac Bio-Markers,
TnI: troponin I,
TnT: troponin T,
UA: unstable angina,
URL: upper reference limit,
WHO: World Health Organization
CORONARY HEART DISEASE AND ACUTE HEART ATTACK
For many people the first sign of having CHD is chest pain, caused by heart attack, but others may learn about their CHD from their doctor after receiving results of laboratory tests as part of their annual check up. Diet, smoking, presence of diabetes, hypertension or hyperlipidemia, among other things, can help estimate one's chance of having CHD or CAD, therefore measurement of blood pressure, cholesterol, blood sugar, body weight and body mass index as well as obtaining relevant medical history to collect information about one's diet, exercise and smoking habits can be the first steps of working up a patient for possible presence of CAD. If it appears that one is at high risk for CAD, additional tests can be performed using a variety of diagnostic procedures consisting of electrocardiogram (ECG or EKG), echocardiogram, exercise test, cardiac catheterization and coronary angiogram. During an acute chest pain event the concentration of biochemicals can be measured in one's blood to asses if the person's CAD has progressed into a heart attack or if the chest pain is due to some other disease entity than coronary artery closure. The laboratory markers used in the diagnosis and differential diagnosis of acute chest pain are collectively called cardiac markers, myocardial injury markers or biochemical markers of myocardial injury. This article will present a brief overview of the most significant cardiac markers and it will discuss the use of those markers for the diagnosis of cardiac diseases but it will not talk in details about the non-laboratory diagnostic modalities.
THE EARLY CARDIAC MARKERS
The first blood test to aid in the diagnosis of a heart attack was described in 1954 by Ladue, Wroblewski and Karmen in Science [2]. They found that the activity of the enzyme, serum glutamic oxaloacetic transaminase (SGOT), later known as aspartate transaminase (AST), increased and remained elevated for several days following a heart attack. Because AST activity could be increased in other conditions than a heart attack search was started for other, better laboratory diagnostic markers of myocardial injury. This search lead to the discovery of several other cytoplasmic enzymes that could also be used for the detection of a heart attack. Of the many candidates that were described in the literature lactate dehydrogenase (LDH or LD) and creatine kinase (CK) gained wide clinical acceptance. However, because these enzymes were also present in other tissues than the myocardium, their diagnostic specificity was limited especially in the presence of concurrent liver or skeletal muscle diseases or even strenuous exercise. Their diagnostic sensitivity also suffered from the presence of a sizeable baseline enzyme concentration in the circulation without any cardiac pathology, sometimes masking a small infarction because of the high “background”. To overcome the sensitivity and specificity limitations of LD and CK measurement, their more cardiac specific isoenzymes were introduced to clinical practice. In the mid-1960's it was discovered that the LD-1 isoenzyme of LD had better cardiac specificity than total LD activity and the LD-1 to LD-2 ratio was proposed to be a better diagnostic tool for the diagnosis of myocardial infarction (MI) than total enzyme activity measurement [3]. Unfortunately, LD-1 elevation could also be caused by minor in vivo or in vitro hemolysis [4]. LD-1 isoenzyme measurement was further limited because practical, relatively rapid analytical techniques could not be developed. Most laboratories were required to use gel electrophoresis for LD isoenzyme measurement although an immunoinhibition assay was later introduced to the medical market [5]. In spite of the many limitations of LD and LD-1 measurement it remained in clinical use until the 'troponin era' because LD-1 concentrations remained elevated for up to a week after an acute myocardial infarction (AMI) thus allowing diagnosis of a heart attack in patients who presented to the hospital after the other markers' concentrations returned to normal.
The measurement of the MB isoenzyme of creatine kinase had a better record. The CK enzyme is a dimer of two polypeptide chain, encoded by two genes and translated separately. The CK-M and CK-B monomers form the dimer of CK-MB, and because heart muscle expresses the B gene at a higher rate than other skeletal muscle the CK-MB isoenzyme exhibits better cardiac specificity than total CK. This specificity could be further enhanced by calculating the so called CK-MB ratio from separate measurements of CK-MB and total CK. CK-MB concentration measurement or CK-MB ratio reporting demonstrated sufficient specificity for clinical practice and after the CK-MB specific antibody was developed in the Ladenson lab [6] the possibility of practical, automated CK-MB measurement became reality. The availability of CK-MB immunoassays on automated chemistry analyzers and the significantly improved diagnostic specificity of CK-MB above all the other cardiac enzymes made CK-MB measurement the “gold standard” of AMI diagnosis until the troponins replaced it in this role.
DIAGNOSIS OF AMI ACCORDING TO THE WHO CRITERIA
Although no official definition of heart attack was ever developed, consensus existed for the purpose of establishing the diagnosis of MI. This consensus became to be known as the “WHO criteria” and it had been used since the early 1960's in research publications and later in clinical practice for AMI diagnosis. The “WHO criteria” were also referred to as the “WHO two out of three rule” and it was based on the presence of characteristic chest pain, elevation of cardiac enzymes such as CK, CK-MB or LD, and new abnormalities on electrocardiogram (ECG), such as a newly developed Q-wave or ST-segment elevation. The simultaneous presence of at least two of these three criteria was sufficient for the diagnosis of an acute MI. If a minimum of two criteria was not present the chest pain would be called angina. AMI, caused by the acute occlusion of a coronary artery, was believed to have started at the time the chest pain started and diagnostic cardiac enzyme elevations were timed starting from the onset of chest pain. Because old ECG abnormalities could hide a new ischemic event, biochemical diagnosis was essential to establish the diagnosis of AMI and to start therapy, therefore the ideal cardiac marker was expected to become positive, or exceed a predetermined cut-off concentration, shortly after the onset of chest pain. In addition to cardiac specificity, cardiac markers were also graded on the fact of how rapidly they would diagnose an AMI. For example, CK-MB concentration could take 4-6 hours to become positive after the onset of chest pain.
Because the early markers didn't have absolute cardiac specificity, characteristic rise and fall pattern of cardiac enzyme concentration was required to rule out false positive diagnosis of AMI. This necessitated multiple sample collections within the first 12-24 hours of hospitalization before the definitive diagnosis could be pronounced. As the concentration of the gold standard CK-MB could become negative approximately three days after an AMI, the need for the much less tissue specific LD and LD-1 measurement remained necessary.
As experience accumulated with these cardiac markers it was recognized that patients with small muscle mass may never reach diagnostic concentrations of CK-MB, and that patients with regenerative skeletal muscle disease such as Duchenne muscular dystrophy or acute skeletal muscle trauma due to recent surgery or moving vehicle accident, could have false negative CK-MB results due to the very high concentrations of the MM isoform of CK released from skeletal muscle [7], [8]. It was also recognized that the elevated CK-MB concentration in patients with regenerating muscle diseases was due to enhanced CK-B gene expression by skeletal muscle [9].
These limitations, while they did not stop the reign of CK-MB, accelerated the search for the “ideal” cardiac marker that would demonstrate absolute cardiac specificity, would not be present in blood without cardiac muscle damage and its concentration would rise rapidly after a heart attack, and it would be inexpensive to test for and the analysis could be performed on automated instruments. The search for this ideal cardiac marker turned from cytosolic enzymes to structural proteins of the myocytes. The first practical result of this search was the discovery of myoglobin as a cardiac marker. Although myoglobin did not provide any cardiac specificity it satisfied another requirement of an ideal cardiac marker [10]: It could become positive within one to two hours of the onset of chest pain so it became the best early biochemical marker of myocardial damage and later it gained a significant role as a “rule out” marker for non-cardiac chest pain patients.
THE EMERGENCE OF CARDIAC TROPONINS AS CARDIAC MARKERS
Several structural muscle proteins were evaluated as possible biochemical markers of myocardial damage, including myosin heavy and light chains, fatty acid binding proteins, natriuretic peptides, tropomyosin and members of the troponin complex. The search was directed by the idea of finding one protein that had different genes and amino acid sequences in skeletal and cardiac muscle, and developing monoclonal antibodies against the cardiac isoform to be used in an immunoassay for quantitative measurement of the marker's concentration in blood following a cardiac event. The target protein was supposed to be a small molecule as it was known by this time that smaller proteins exited damaged cells faster than larger ones after an ischemic event. Many markers showed promise initially but large-scale clinical trials that exposed serious flaws for all of them prevented their acceptance for clinical use except for cardiac troponin I and T.
The troponin complex is part of the regulatory apparatus of the myocyte. It consists of three components: the calcium binding troponin C, the inhibitory troponin I and the tropomyosin binding troponin T. The troponin complex is involved in the Ca-mediated muscle contraction that is exerted via conformational changes of the individual components [11]. Only troponin I (TnI) and troponin T (TnT) have cardiac specific genes in addition to other genes that encode for slow-twitch, fast-twitch and smooth muscle isoforms. The laboratory of Hugo Katus in Germany focused on developing an immunoassay for the measurement of cardiac troponin T (cTnT) [12] and our research group in Jack Ladenson's laboratory at Washington University concentrated on cardiac troponin I (cTnI) [13]. Phase one clinical studies demonstrated that both cTnI and cTnT will be elevated after a heart attack in patients [13], [14]. The early studies had also proven that the cardiac troponin assays show similar early sensitivities for myocardial damage to that of CK-MB but the cardiac troponins would remain elevated (positive) for longer than CK-MB thus eliminating the need for LD measurement in late-arriving patients.
Both markers demonstrated excellent cardiac specificity allowing for biochemical detection of myocardial infarction in clinical situations where CK-MB could not perform due to concurrent skeletal muscle damage such as after accidental chest contusion or in postoperative situations [15], [16]. While evidence accumulated that cardiac troponins may become the new gold standard for the diagnosis of myocardial damage, doubt also emerged regarding tissue specificity of cTnT [17]. Patients with regenerative skeletal muscle diseases but without recognizable cardiac damage would have elevated cTnT concentration without similar increase in cTnI values. Using immunohistochemistry staining of newborn, and healthy and diseased adult human skeletal and cardiac muscle tissue, we have demonstrated that the difference between cTnI and cTnT expression is real and it is related to re-expression of the cardiac TnT gene by skeletal muscle during muscle regeneration [18, 19]. Our findings were confirmed by RNA expression studies in 1999 [20]. Because the human cTnT mRNA undergoes differential splicing that produces different forms of cTnT protein molecules in heart or skeletal muscle, the cTnT immunoassay was later redesigned and eliminated the unwanted cross reactivity with cTnT of skeletal muscle origin [21]. The current generations of cTnT assays are no longer hindered by compromised cardiac specificity.
As cTnI and cTnT immunoassays started appearing in clinical practice additional questions were raised regarding the performance of the troponin methods. Both cTnI and cTnT were detected in patients who did not satisfy the definition of MI according to the WHO criteria. These non-MI chest pain patients had elevated cardiac troponins and were diagnosed with unstable angina (UA) or non-ischemic cardiac diseases, but they had increased odds of later cardiac events [22], defined as a later AMI, re-infarction or death, depending on the clinical trial. The increased odds for later events were demonstrated by short term (14-30 days) and long term (up to a year) follow up clinical trials. The patients who had elevated admission cTns without an AMI could have 10-fold increase in the odds of developing later cardiac complications versus those who didn't have measurable troponin elevation. It was also discovered that non-MI patients with elevated cTn would benefit from invasive therapy but those without cTnI elevation would do better with conservative therapy [23], [24], [25, 26]. False positive results due to skeletal troponin interference could be ruled out because troponins were proven to have 100% tissue specificity, therefore new models were needed to explain these findings.
THE NEW ERA OF DIAGNOSIS OF ISCHEMIC CARDIAC INJURY – THE ACUTE CORONARY SYNDROMES
The recognition of increased risk of later myocardial damage after increased troponin blood concentrations in non-MI patients were incorporated into two different practice recommendations that were published within a year of each other. The National Academy of Clinical Biochemistry (NACB), International Federation of Clinical Chemistry and Laboratory Medicine (IFCC), American Heart Association (AHA) and American College of Cardiology (ACC) published their recommendations in 1999 [27], and the European Society of Cardiology (ESC), ACC and AHA consensus document was released in 2000 [28]. Both documents recommended the revision of diagnosis of AMI and offered guidance for testing of cardiac markers. One of the most important changes in the diagnosis of myocardial ischemic injury, as put forward by these documents, recommended the replacement of cardiac enzymes with cardiac troponins in the WHO's “two out of three” criteria. The second major change was incorporating stable angina, unstable angina (UA), non-Q wave MI (NQWMI), ST-elevation MI (STEMI) and Q-wave MI (QWMI) on a continuum. These diagnoses were now representatives of the same ischemic process, collectively called the Acute Coronary Syndromes (ACS). The NACB document recommended the use of two diagnostic cut-off marker concentrations, one (lower concentration) for the diagnosis of ACS, and the second (higher) concentration as the diagnostic cut-off for AMI. The joint ECS/ACC/AHA document clearly stated that CK-MB was no longer required for the diagnosis of AMI because of the availability of cTns, and any elevation of cardiac troponin I or T would establish the diagnosis of an ischemic cardiac injury [28]. ECG was required to sub-classify cTn positive patients depending on the presence or absence of ECG abnormalities. Later interpretations of the ECS/ACC/AHA document recommended the elimination of the need for ECG abnormalities for the diagnosis of some forms of AMI in cTn positive patients by defining NSTEMI and NQWMI diagnoses that were characterized by chest pain and cTn positivity only. To diagnose STEMI and Q-wave myocardial infarction required cTn positivity in the presence of new ECG findings (Figure 1.) Elevated cTn concentration was defined by the ECS/AHA/ACC consensus document as a concentration that exceeded the 99th percentile of the assay-specific reference limit of a healthy population on at least one occasion within the first 24 hours after the onset of symptoms [28].
Figure 1.
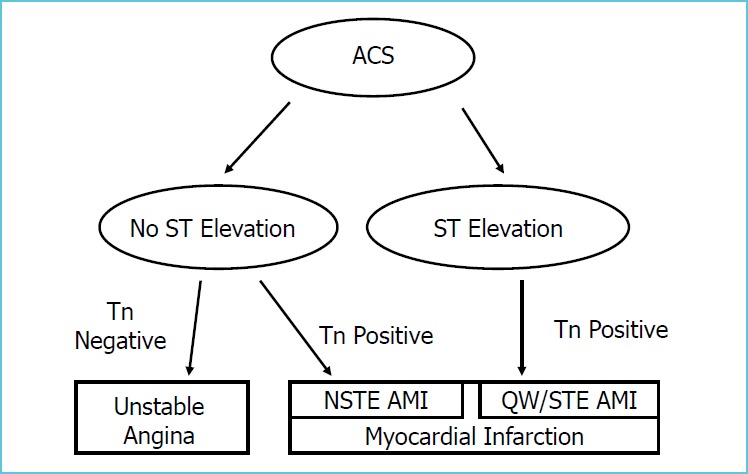
Schematic representation of diagnosis of ischemic myocardial injury, using cardiac troponin measurement and ECG findings
Application of the new classification in clinical practice soon revealed the increased financial and social consequences of the new diagnostic algorithm [29] that was the end result of the increased number of patients diagnosed with MI due to the increased sensitivity of cTns for minor myocardial damage. Because the new guidelines did not completely invalidate the WHO “two out of three” diagnostic criteria, the time of myocardial damage was still considered to be defined by the onset of characteristic chest pain. However, because cardiac marker elevation could be present without the traditional diagnosis of an MI, i.e., without the presence of chest pain and ECG abnormalities, this discrepancy had to be reconciled and resulted in research in three separate directions: The search for ischemia versus infarction markers, the development of “cardiac risk assessment” markers, and the eventual redefinition of MI.
Proponents of the 'ischemia versus infarction theory' postulated that there could be myocardial ischemia without the development of a traditional infarction causing only reversible myocardial damage, but if ischemia existed for an extended period of time it could lead to irreversible damage. The logical conclusion of this theory was to try to find biochemical markers that would signal the presence of ischemia before the permanent damage occurred and use cTns or CK-MB for the diagnosis of MI. The best representative of the ischemia markers was the ischemia modified albumin (IMA), measured by the albumin-cobalt binding assay [30-32]. After many clinical trials IMA did not fulfill the expectations for its diagnostic role as an ischemia marker and it lost its significance after the redefinition of myocardial infarction.
Cardiac risk assessment by biochemical markers is based on the observation that certain biochemical markers can be detected at various concentrations in an asymptomatic (healthy) population or may be found during admission for suspected MI even when the diagnosis of AMI can be ruled out. However, on follow up, patients with detectable cardiac marker concentration may do worse after hospital discharge than patients without elevated markers. It was also recognized that higher concentrations of these biochemical markers confer increasing risk for short and long term cardiac events. The risk markers could be either cardiac troponins I or T [33] and CK-MB [34], but they could be inflammatory markers such as myeloperoxidase [35] or C-reactive protein (CRP) [36] also. In addition to these examples several other markers, associated mostly with inflammation or platelet activation, have been described in the literature and were commercially available. One important characteristic of these risk markers is the fact that they are somewhat independent predictors of cardiac risk, therefore simultaneous measurement of the concentrations of more than one marker can provide a more precise forecast of risk of future cardiac events and patient outcome [37-39]. Because the between- and within-individual variation of the risk markers can be great, their concentration is assessed in the general population then the population values are divided into quartiles or quintiles. The relative risks of an event for each segment of the population is determined from clinical studies. The individual's own result of the risk marker concentration is compared to the age- and gender-related reference ranges and the risk associated with the quartile (or quintile) where the test result falls is assigned to the individual. It is important to recognize that using risk markers is not to establish a diagnosis but to calculate the relative risk of a cardiac event and to offer appropriate risk-mitigating intervention to the patient in the form of life style changes or medical intervention. The major shortcoming of most of the cardiac risk markers is that they are acute phase reactants therefore they can be present at elevated concentrations during minor illnesses. Interpretation of the results of cardiac risk markers requires careful evaluation of the patient's clinical history.
UNIVERSAL DEFINITIONS OF MYOCARDIAL INFARCTION
The most significant impact of cTn measurement is the realization that myocardial cell damage can occur before clinical signs and symptoms of “traditional” myocardial infarction and that this damage may have long term consequences for the patient. Investigating cTn release characteristics revealed that myocardial cell damage may be due to coronary artery occlusion as hypothesized in the WHO model but myocardial cell damage can also happen for other reasons than a coronary artery occlusion. To establish the correct mechanism of myocardial damage, other diagnostic modalities than cTn measurement may have to used. These additional diagnostic modalities includes ECG but imaging techniques are also valuable. This newly acquired insight into the mechanism of myocardial damage had to be incorporated into a new definition of myocardial infarction.
The expert consensus document, entitled Universal Definition of Myocardial Infarction, published by the Joint International Task Force in 2007 developed the first universally accepted practice guidelines that incorporated the new knowledge gained from clinical and research use of cardiac troponins [40]. This document addressed the diagnosis of myocardial infarction due to acute ischemia as well as for other causes, established the decision limits for diagnosis using biochemical markers and recommended different diagnostic cut-offs based on the clinical circumstances around the time the myocardial cell death occurred. The document defined six different types of myocardial infarction, including spontaneous MI, sudden cardiac death and several types of MI that developed during or immediately after an invasive cardiac procedure. All diagnostic cut-offs were based on the 99th percentile of the particular cTn assay in use but different circumstances required different multiples of the 99th percentile cut-off [40]. The document extensively discussed the use of ECG and imaging for diagnosis and differential diagnosis of myocardial damage [40]. The diagnosis of reinfarction, previously hotly debated as to whether it could be assessed by the use of cTns, was also addressed by this document and the expert panel presented recommendations for the use of cTns for this purpose. They proposed that a rise of cTns by more than 20%, or by more than 3 standard deviations for the assay in use, in two sample within 3-6 hours after decreasing cTn concentration had been observed would be diagnostic of a reinfarction [40].
The 2007 consensus document [40] was followed by a revision in 2012 [41]. The 2012 document retained previous recommendations and added a new MI diagnosis to the previous six (Table 1). As in previous documents it also provided a list of diseases that may present with elevated cardiac markers but are not MI or may not even be cardiac diseases. It also listed common abnormalities that could cause false negative or false positive ECG changes that could be misinterpreted as ischemic myocardial damage. Table 2 is an abbreviated list of common non-MI diseases that may present with cardiac marker elevation. This list is intended to illustrate the fact that tissue specificity of a biochemical marker will not necessarily define the mechanism of tissue injury and to caution against too simplistic interpretation of an elevated cTn result.
Table 1.
Types of MI and most significant criteria for diagnosis as recommended by the third universal definition of myocardial infarction [41]
| Type of MI | Cause or circumstance defining Type of MI | Multiples of 99th percentile of cardiac marker required for diagnosis | Notes |
|---|---|---|---|
| Type 1 | Spontaneous | >1x | MI related to spontaneous rapture of plaque and subsequent coronary artery occlusion |
| Type 2 | Secondary | >1x | MI is due to ischemic imbalance (oxygen supply-demand mismatch) |
| Type 3 | Sudden cardiac death | >10x or undefined | Antemortem blood may not be available for cardiac marker testing. Clinical history is strongly suggestive of cardiac event. Autopsy may be required for diagnosis. |
| Type 4a | PCI | >5x | >5 x 99th percentile URL after initial normal marker values or >20% increase above stable or decreasing baseline. ECG or imaging may be required for diagnosis |
| Type 4b | Stent thrombosis | >1x | Stent thrombosis detected by coronary angiography or autopsy |
| Type 4c | Restenosis | >1x | >50% stenosis on coronary angiography |
| Type 5 | CABG | >10x | ECG and imaging evidence is required in addition to cardiac marker elevation |
Notes: For all diagnosis by biochemical markers characteristic rise and fall of marker concentration is required.
Positive marker concentration is defined as at least one result above the 99th percentile (or above the relevant multiple) of the upper reference limit (URL) for the assay in use.
Table 2.
Elevation of cardiac troponin values due to myocardial injury but not due to MI
| tachy/bradycardia | |
| aortic dissection | |
| hypertrophic cardiomyopathy | |
| congestive heart failure | |
| shock | |
| respiratory failure | |
| cardiac contusion | |
| cardiac surgery or ablation | |
| myocarditis, endocarditis, pericarditis | |
| pulmonary embolism | |
| rhabdomyolisis | |
| sepsis, viral illness | |
| stroke | |
| amyloidosis, sarcoidosis, hemochromatosis | |
| strenuous exercise | |
| renal failure | |
| burns of large body surface area |
ANALYSIS OF BIOCHEMICAL MARKERS OF CARDIAC INJURY
CK-MB was the first cardiac marker that could be measured with a practical immunoassay. The experience gained with CK-MB established the fact that immunoassays, also called “mass assays”, would be superior to activity based assays used earlier for cardiac enzyme measurement. The use of monoclonal antibodies provided the necessary analytical specificity and analytical sensitivity could be enhanced to detect marker release from arbitrarily small tissue damage. Even first generation cTn assays were capable of detecting the death of as little as 1 gram of cardiac muscle through the measurement of marker concentration in the circulation and newer, more sensitive assays could detect the destruction of even a smaller volume of heart tissue.
In spite of impressive analytical performance, cardiac troponin assays were not without problems. We mentioned the nonspecificity that plagued the first generation cTnT immunoassay and that later was corrected [17, 21]. Cardiac troponin I assays, and, to a smaller extent, cardiac troponin T assays, too, had other problems in the form of large discrepancies between results from different manufacturers or between different generations of the same manufacturer's assay. The concentration difference between two tests producing the lowest and highest result on the same sample could be as much as 10- to 30-fold. These differences could be caused by multiple reasons. There was no agreement between how manufacturers established their calibrator values and there was no standardized calibrator available for use by assay manufacturers. Post translational modification of cTnI in the form of proteolysis at both the N- and C-terminal of the troponin molecule, oxidation, phosphorylation or complexation with troponin C could alter or eliminate epitopes that were recognized by the different monoclonal antibodies (MoAb) in the different assays [42]. The final outcome of the many factors in assay differences translated into approximately ten- to 20-fold differences in the limit of detection (LoD) and greater than ten-fold differences at the 99th percentile concentration [43]. False negative results could be also seen if an epitope was deleted because of proteolysis or if autoantibodies completely blocked attachment of the MoAb in the test kit. These large differences made application of practice guidelines difficult and required the establishment of assay-dependent cut-offs for each assay individually, leading to confusion in clinical practice. The troponin T assays, being produced by a single manufacturer, had fewer problems but they were not free of the similar differences between successive assay generations.
In addition to false negative and false positive cTnI results due to cTn specific autoantibodies against some epitopes, human anti-mouse antibodies (HAMA) could lead to incorrect results in assay formulation using mouse MoAbs and other heterophile antibodies could produce similar interference in other assays using polyclonal antibodies. Recently developed assays are less prone to these interferences because of manufacturers' effort to incorporate blocking agents in their reagents but the unwanted influence of heterophile and autoantibodies must be kept in mind when investigating false negative or false positive cTn results.
cTnI assay standardization was proposed and the international Subcommittee for Cardiac Troponin I Standardization was established by the American Association for Clinical Chemistry (AACC). This committee identified and validated cTnI candidate reference materials [44], [45] to be used as primary standards by test kit manufacturers and the final calibration standard is now available from the US National Institute of Standards and Technology (NIST) for cTnI assay developers or troubleshooting in the clinical laboratory. Unfortunately the availability of reference standard did not fully eliminate cTnI assay differences although it minimized them, therefore establishing LoD and 99th percentile cut-off is still required for each cTnI immunoassay.
HIGH SENSITIVITY TROPONIN ASSAYS (HS-TN)
As the clinical importance of even small amounts cTn in the circulation was recognized, successively newer generation cTn assays were developed with the intention of providing improved analytical sensitivity, but many assays demonstrated only marginal improvement in LoD or better precision at low analyte concentration. Second, third or fourth generation cTn assays were marketed as “sensitive”, “high-sensitive”, “highly sensitive”, “high performance” or “high-sensitivity” without an exact definition of this terminology. Most of the time the operational characteristics, such as LoD or 99th percentile cut-off, of the various assay generations remained within the ng/mL concentration range, essentially unchanged from previous generations. Starting around 2010 true high-sensitivity troponin assays started appearing for clinical laboratory use. The main defining characteristic of these high-sensitivity assays was an increase in analytical sensitivity of two orders of magnitude as compared to traditional cTn methods [46], creating new challenges for the user. Traditional cTn immunoassays reported marker concentration in ng/mL (nanogram/milliliter) units, numerically equivalent to microgram/L (microgram/liter) measurement units. The new, high-sensitivity cTn assays, therefore, had to express cTn concentrations using multiple zeros in the traditional unit of measure, a potentially very error prone procedure. The other option was to use different measurement units for traditional and high-sensitivity assays, a confusing proposition. Although no solution has been found for this problem, recommendation has been made to use ng/L (nanogram/liter) measurement unit for all cTn assay results [46], regardless if it is traditional or high-sensitivity method. If this recommendation is adopted by both assay manufacturers and clinicians traditional and high-sensitivity results can be easily distinguished. For example, a traditional cTn assay that has a 99th percentile cut-off of 0.05 ng/mL (in traditional units) and a hs-cTn assay that has a 99th percentile cut-off of 5 ng/L would be reported as 50 ng/L and 5 ng/L, respectively. As a practicing laboratory physician I strongly agree with this recommendation and encourage its adoption.
The emergence of hs-cTn assays created new challenges for the laboratory and clinical community. The significantly lower LoD made it possible to measure cTn concentration in up to 95% of healthy control subjects [47] contrary to the approximately 15% with the traditional assays. Later studies indicated that age and gender specific reference ranges may be needed both for hs-cTnT [48] and hs-cTnI [49], but other investigators did not confirm these findings [47]. Whether gender specific reference ranges are truly necessary must be decided by additional clinical trials, but using sex-related hs-cTnI reference range was found to improve detection in women, but not in men, of major cardiac events within one year after an initial presentation for ACS [50]. The benefits of improved early detection of ACS by hs-cTns are offset by reduced specificity for ACS because more patients are detected with myocardial injury not due to ischemia [51].
The low 99th percentile cut-off of hs-cTn assays and their even lower LoD when combined with high precision (low coefficient of variation or CV) at the decision limit provide speedy diagnosis and accurate identification of patients who can be safely discharged from the emergency room [52, 53] and produce better than 99% negative predictive value for subsequent MI or cardiac death at 30 days after initial hospital visit [53]. If patients with symptoms suggestive of ACS can be safely and rapidly discharged from the hospital, it can reduce inpatient admissions, cost of hospitalization and have major benefits for patients also.
Because an inherent characteristic of cardiac marker release in acute ischemic events is the rise and fall of marker concentration, non-ACS, non-MI diagnoses may be ruled out by repeat measurements of cardiac marker concentrations by hs-cTn assays. The improved precision and the very low LoD of hs assays allows for repeat marker testing within a shorter time frame than with traditional troponin assays. Both absolute concentrations and percent change from baseline or admission values (delta values) within the first six hours have been evaluated for this diagnostic algorithm and found to be valuable to 'rule in' ACS or to 'rule out' significant stenosis, recurrent infarction or death within one year after initial admission in patients with non-ST elevation chest pain [54]. Jeager et al. investigated the diagnostic utility of absolute concentration change of hs-cTnI from baseline within one hour of admission in patients with suspected AMI [55]. They have reported their one hour delta hs-cTnI protocol could rule out AMI with 100 % sensitivity (negative predictive value = 100%), and it could rule in AMI with 96 % specificity (positive predictive value = 70 %) [55]. Their one hour delta hs-cTnI protocol exhibited better diagnostic accuracy than a combination of ECG and hs-cTnI measurement while providing the improved diagnostic performance faster.
Only a handful of hs-cTn assays are on the market at this time but their number is growing. Laboratory professionals and clinicians may be still digesting results of recent research and may have just started adopting practice recommendations when newer discoveries are published. Professional organizations have attempted to further this process via various publications that distill the essence of the detailed practice guidelines. A recent publication by the IFCC Task Force on Clinical Applications of Cardiac Bio-Markers (TF-CB) is a valuable educational aid to help with adopting hs-cTn assays in every day practice [56].
SUMMARY AND CONCLUSIONS
Sixty years of research into the physiology and pathology of ischemic and non-ischemic cardiac injury has greatly increased our understanding of the events taking place during and after myocardial cell death. Clinical signs and symptoms, ECG, combined with measurement of biochemical markers and imaging studies improved our capacity to detect and respond to an acute coronary event. Successively newer biochemical markers of these injuries have evolved from cytosolic enzymes to tissue specific structural proteins, culminating in our current, best biochemical markers of cardiac troponins I and T. Improved analytical techniques of cardiac troponin measurement produced high-sensitivity cTn assays with detection limits two orders of magnitude better than the first methods and enhanced our diagnostic sensitivity and specificity of cardiac damage. The same cTn assays forced us to reassess our understanding and practices of diagnosing and treating myocardial injury, but did not close the chapter on biochemical diagnosis of these clinical entities.
The fast pace of change in our understanding of myocardial injury and the use of biochemical markers for the diagnosis of these diseases provide challenges for practicing laboratory professionals, emergency medicine physicians and cardiologist to keep up with the new recommendation and practice guidelines that are the result of research in this field. Hopefully this review article presents information on the most significant aspects of the use of biochemical markers for the diagnosis of myocardial injury. The historical approach was elected in the hope of providing additional explanation why certain markers or practices are preferred over others when dealing with the patient who has cardiac damage.
It is impossible to discuss all the biochemical markers of myocardial damage in a single review article. The number of markers are too numerous and many of them were short lived. This article attempted to present the most significant milestones on this field with emphasis on the recent discoveries and issues related to high-sensitivity cTn assays. It is expected that this area of laboratory medicine and cardiology will experience additional growth in the near future as results of new clinical trials get published, leading to further refinements in our understanding of ACS and MI.
REFERENCES
- 1.CDC N. Underlying Cause of Death 1999-2013 on CDC WONDER Online Database, released 2015. Data are from the Multiple Cause of Death Files, 1999-2013, as compiled from data provided by the 57 vital statistics jurisdictions through the Vital Statistics Cooperative Program. 2015. [cited 2015 December 1, 2015]; Available from: http://www.cdc.gov/heartdisease/facts.htm. [Google Scholar]
- 2.Ladue J.S., Wroblewski F., Karmen A., Serum glutamic oxaloacetic transaminase activity in human acute transmural myocardial infarction. Science, 1954. 120(3117): p. 497-499. [DOI] [PubMed] [Google Scholar]
- 3.Paloheimo J.A., Pitkanen E., Elevated lactic dehydrogenase (LDH) activity and the lactic dehydrogenase isoenzyme pattern of serum. Ann Med Intern Fenn, 1965. 54(3): p. 129-136. [PubMed] [Google Scholar]
- 4.Hammermeister K.E., Merendino K.A., Confusion due to similar LDH isoenzyme patterns in myocardial infarction and prosthetic valve hemolytic anemia. Ann Thorac Surg, 1971. 11(5): p. 431-437. [DOI] [PubMed] [Google Scholar]
- 5.Rej R., Immunochemical quantitation of isoenzymes of aspartate aminotransferase and lactate dehydrogenase. Clin Biochem, 1983. 16(1): p. 17-9. [DOI] [PubMed] [Google Scholar]
- 6.Vaidya H.C., et al. , Direct measurement of creatine kinase-MB activity in serum after extraction with a monoclonal antibody specific to the MB isoenzyme. Clin Chem, 1986. 32(4): p. 657-663. [PubMed] [Google Scholar]
- 7.Siegel A.J., Silverman L.M., Holman B.L., Elevated creatine kinase MB isoenzyme levels in marathon runners. Normal myocardial scintigrams suggest noncardiac source. Jama, 1981. 246(18): p. 2049-2051. [PubMed] [Google Scholar]
- 8.Wu A.H., et al. , Creatine kinase MB isoforms in patients with skeletal muscle injury: ramifications for early detection of acute myocardial infarction. Clin Chem, 1992. 38(12): p. 2396-2400. [PubMed] [Google Scholar]
- 9.Vretou-Jockers E., Vassilopoulos D., Skeletal muscle CK-B activity in neurogenic muscular atrophies. Journal of Neurology, 1989. 236(5): p. 284-287. [DOI] [PubMed] [Google Scholar]
- 10.Kagen L.J., Myoglobin: methods and diagnostic uses. CRC Crit Rev Clin Lab Sci, 1978. 9(4): p. 273-302. [DOI] [PubMed] [Google Scholar]
- 11.Farah C.S., Reinach F.C., The troponin complex and regulation of muscle contraction. FASEB Journal, 1995. 9(9): p. 755-767. [DOI] [PubMed] [Google Scholar]
- 12.Katus H.A., et al. , Enzyme linked immuno assay of cardiac troponin T for the detection of acute myocardial infarction in patients. Journal of Molecular & Cellular Cardiology, 1989. 21(12): p. 1349-1353. [DOI] [PubMed] [Google Scholar]
- 13.Bodor G.S., et al. , Development of monoclonal antibodies for an assay of cardiac troponin-I and preliminary results in suspected cases of myocardial infarction. Clinical Chemistry, 1992. 38(11): p. 2203-2214. [PubMed] [Google Scholar]
- 14.Gerhardt W., et al. , S-troponin T in suspected ischemic myocardial injury compared with mass and catalytic concentrations of S-creatine kinase isoenzyme MB [see comments]. Clinical Chemistry, 1991. 37(8): p. 1405-1411. [PubMed] [Google Scholar]
- 15.Adams J.E.r., et al. , Diagnosis of perioperative myocardial infarction with measurement of cardiac troponin I [see comments]. New England Journal of Medicine, 1994. 330(10): p. 670-674. [DOI] [PubMed] [Google Scholar]
- 16.Eikvar L., et al. , Serum cardio-specific troponin T after open heart surgery in patients with and without perioperative myocardial infarction. Scandinavian Journal of Clinical & Laboratory Investigation, 1994. 54(4): p. 329-335. [DOI] [PubMed] [Google Scholar]
- 17.Braun S.L., et al. , Discrepant results for cardiac troponin T and troponin I in chronic myopathy, depending on instrument and assay generation. Clin Chem, 1996. 42(12): p. 2039-2041. [PubMed] [Google Scholar]
- 18.Bodor G.S., et al. , Cardiac troponin-I is not expressed in fetal and healthy or diseased adult human skeletal muscle tissue. Clin Chem, 1995. 41(12 Pt 1): p. 1710-1715. [PubMed] [Google Scholar]
- 19.Bodor G.S., et al. , Cardiac troponin T composition in normal and regenerating human skeletal muscle [see comments]. Clin Chem, 1997. 43(3): p. 476-484. [PubMed] [Google Scholar]
- 20.Ricchiuti V., Apple F.S., RNA expression of cardiac troponin T isoforms in diseased human skeletal muscle [published erratum appears in Clin Chem 2000 Mar;46(3):437]. Clin Chem, 1999. 45(12): p. 2129-2135. [PubMed] [Google Scholar]
- 21.Ricchiuti V., et al. , Cardiac troponin T isoforms expressed in renal diseased skeletal muscle will not cause false-positive results by the second generation cardiac troponin T assay by Boehringer Mannheim. Clin Chem, 1998. 44(9): p. 1919-1924. [PubMed] [Google Scholar]
- 22.Newby L.K., et al. , Value of serial troponin T measures for early and late risk stratification in patients with acute coronary syndromes. The GUSTO-IIa Investigators. Circulation, 1998. 98(18): p. 1853-1859. [DOI] [PubMed] [Google Scholar]
- 23.Cannon C.P., et al. , Invasive versus conservative strategies in unstable angina and non-Q-wave myocardial infarction following treatment with tirofiban: rationale and study design of the international TACTICS-TIMI 18 Trial. Treat Angina with Aggrastat and determine Cost of Therapy with an Invasive or Conservative Strategy. Thrombolysis In Myocardial Infarction. Am J Cardiol, 1998. 82(6): p. 731-736. [DOI] [PubMed] [Google Scholar]
- 24.Kontny F., Improving outcomes in acute coronary syndromes--the FRISC II trial. Clin Cardiol, 2001. 24(3 Suppl): p. I3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morrow D.A., et al. , Cardiac troponin I for stratification of early outcomes and the efficacy of enoxaparin in unstable angina: a TIMI-11B substudy. J Am Coll Cardiol, 2000. 36(6): p. 1812-1817. [DOI] [PubMed] [Google Scholar]
- 26.Morrow D.A., et al. , Ability of minor elevations of troponins I and T to predict benefit from an early invasive strategy in patients with unstable angina and non-ST elevation myocardial infarction: results from a randomized trial. JAMA, 2001. 286(19): p. 2405-2412. [DOI] [PubMed] [Google Scholar]
- 27.Wu A.H., et al. , National Academy of Clinical Biochemistry Standards of Laboratory Practice: recommendations for the use of cardiac markers in coronary artery diseases. Clin Chem, 1999. 45(7): p. 1104-1121. [PubMed] [Google Scholar]
- 28.Myocardial infarction redefined--a consensus document of The Joint European Society of Cardiology/American College of Cardiology Committee for the redefinition of myocardial infarction. J Am Coll Cardiol, 2000. 36(3): p. 959-969. [DOI] [PubMed] [Google Scholar]
- 29.Collinson P.O., et al. , Impact of European Society of Cardiology/American College of Cardiology guidelines on diagnostic classification of patients with suspected acute coronary syndromes. Ann Clin Biochem, 2003. 40(Pt 2): p. 156-160. [DOI] [PubMed] [Google Scholar]
- 30.Sinha M.K., et al. , Ischemia modified albumin is a sensitive marker of myocardial ischemia after percutaneous coronary intervention. Circulation, 2003. 107(19): p. 2403-2405. [DOI] [PubMed] [Google Scholar]
- 31.Morrow D.A., et al. , The search for a biomarker of cardiac ischemia. Clin Chem, 2003. 49(4): p. 537-539. [DOI] [PubMed] [Google Scholar]
- 32.Roy D., et al. , Ischemia Modified Albumin for the assessment of patients presenting to the emergency department with acute chest pain but normal or non-diagnostic 12-lead electrocardiograms and negative cardiac troponin T. Int J Cardiol, 2004. 97(2): p. 297-301. [DOI] [PubMed] [Google Scholar]
- 33.Hartmann F., et al. , Risk stratification and therapeutic decision making in patients with acute coronary syndrome--the role of cardiac troponin T. Clin Chem Lab Med, 1999. 37(11-12): p. 1107-1111. [DOI] [PubMed] [Google Scholar]
- 34.Kleiman N., et al. , Prospective analysis of creatine kinase muscle-brain fraction and comparison with troponin T to predict cardiac risk and benefit of an invasive strategy in patients with non-ST-elevation acute coronary syndromes. J Am Coll Cardiol, 2002. 40(6): p. 1044. [DOI] [PubMed] [Google Scholar]
- 35.Baldus S., et al. , Myeloperoxidase Serum Levels Predict Risk in Patients With Acute Coronary Syndromes. Circulation, 2003. 2: p. 2. [DOI] [PubMed] [Google Scholar]
- 36.Ridker P.M., et al. , Prospective study of C-reactive protein and the risk of future cardiovascular events among apparently healthy women [see comments]. Circulation, 1998. 98(8): p. 731-733. [DOI] [PubMed] [Google Scholar]
- 37.de Winter R.J., et al. , Independent prognostic value of C-reactive protein and troponin I in patients with unstable angina or non-Q-wave myocardial infarction. Cardiovasc Res, 1999. 42(1): p. 240-245. [DOI] [PubMed] [Google Scholar]
- 38.Morrow D.A., et al. , C-reactive protein is a potent predictor of mortality independently of and in combination with troponin T in acute coronary syndromes: a TIMI 11A substudy. Thrombolysis in Myocardial Infarction. J Am Coll Cardiol, 1998. 31(7): p. 1460-1465. [DOI] [PubMed] [Google Scholar]
- 39.deFilippi C., et al. , Cardiac troponin T and C-reactive protein for predicting prognosis, coronary atherosclerosis, and cardiomyopathy in patients undergoing long-term hemodialysis. Jama, 2003. 290(3): p. 353-359. [DOI] [PubMed] [Google Scholar]
- 40.Thygesen K., et al. , Universal definition of myocardial infarction. Circulation, 2007. 116(22): p. 2634-2653. [DOI] [PubMed] [Google Scholar]
- 41.Thygesen K., et al. , Third universal definition of myocardial infarction. J Am Coll Cardiol, 2012. 60(16): p. 1581-1598. [DOI] [PubMed] [Google Scholar]
- 42.Wu A.H., et al. , Characterization of cardiac troponin subunit release into serum after acute myocardial infarction and comparison of assays for troponin T and I. American Association for Clinical Chemistry Subcommittee on cTnI Standardization. Clin Chem, 1998. 44(6 Pt 1): p. 1198-1208. [PubMed] [Google Scholar]
- 43.Apple F.S., et al. , Plasma 99th percentile reference limits for cardiac troponin and creatine kinase MB mass for use with European Society of Cardiology/American College of Cardiology consensus recommendations. Clin Chem, 2003. 49(8): p. 1331-1336. [DOI] [PubMed] [Google Scholar]
- 44.Christenson R.H., et al. , Standardization of Cardiac Troponin I Assays: Round Robin of Ten Candidate Reference Materials. Clin Chem, 2001. 47(3): p. 431-437. [PubMed] [Google Scholar]
- 45.Christenson R.H., et al. , Toward standardization of cardiac troponin I measurements part II: assessing commutability of candidate reference materials and harmonization of cardiac troponin I assays. Clin Chem, 2006. 52(9): p. 1685-1692. [DOI] [PubMed] [Google Scholar]
- 46.Apple F.S., Collinson P.O., Biomarkers I.T.F.o.C.A.o.C., Analytical characteristics of high-sensitivity cardiac troponin assays. Clin Chem, 2012. 58(1): p. 54-61. [DOI] [PubMed] [Google Scholar]
- 47.Venge P., et al. , Normal plasma levels of cardiac troponin I measured by the high-sensitivity cardiac troponin I access prototype assay and the impact on the diagnosis of myocardial ischemia. J Am Coll Cardiol, 2009. 54(13): p. 1165-1172. [DOI] [PubMed] [Google Scholar]
- 48.Saenger A.K., et al. , Multicenter analytical evaluation of a high-sensitivity troponin T assay. Clin Chim Acta, 2011. 412(9-10): p. 748-754. [DOI] [PubMed] [Google Scholar]
- 49.Apple F.S., Simpson P.A., Murakami M.M., Defining the serum 99th percentile in a normal reference population measured by a high-sensitivity cardiac troponin I assay. Clin Biochem, 2010. 43(12): p. 1034-1036. [DOI] [PubMed] [Google Scholar]
- 50.Cullen L., et al. , Sex-specific versus overall cut points for a high sensitivity troponin I assay in predicting 1-year outcomes in emergency patients presenting with chest pain. Heart, 2016. 102(2): p. 120-126. [DOI] [PubMed] [Google Scholar]
- 51.Mair J., High-sensitivity cardiac troponins in everyday clinical practice. World J Cardiol, 2014. 6(4): p. 175-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vafaie M., et al. , Prognostic Value of Undetectable hs Troponin T in Suspected Acute Coronary Syndrome. Am J Med, 2015. pii: S0002-9343(15)01020-7. doi: 10.1016/j.amjmed.2015.10.016. [DOI] [PubMed] [Google Scholar]
- 53.Shah A.S., et al. , High-sensitivity cardiac troponin I at presentation in patients with suspected acute coronary syndrome: a cohort study. Lancet, 2015. 386(10012): p. 2481-2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sanchis J., et al. , Usefulness of delta troponin for diagnosis and prognosis assessment of non-ST-segment elevation acute chest pain. Eur Heart J Acute Cardiovasc Care, 2015. pii: 2048872615593534. [DOI] [PubMed] [Google Scholar]
- 55.Jaeger C., et al. , One-hour rule-in and rule-out of acute myocardial infarction using high-sensitivity cardiac troponin I. Am Heart J, 2016. 171(1): p. 92-102 e5. [DOI] [PubMed] [Google Scholar]
- 56.Apple F.S., et al. , IFCC educational materials on selected analytical and clinical applications of high sensitivity cardiac troponin assays. Clin Biochem, 2015. 48(4-5): p. 201-203. [DOI] [PubMed] [Google Scholar]
- 57.Kelley W.E., Januzzi J.L., Christenson R.H., Increases of cardiac troponin in conditions other than acute coronary syndrome and heart failure. Clin Chem, 2009. 55(12): p. 2098-2112. [DOI] [PubMed] [Google Scholar]