

Abstract
Objectives
To review the clinical classification, diagnosis and pathophysiology of pulmonary hypertension in children, emphasizing the role of right ventricular function, ventricular interaction and congenital heart disease in the evolution and progression of disease, as well as management strategies and therapeutic options.
Data Source
MEDLINE, PubMed
Conclusion
Critically ill children with pulmonary hypertension associated with congenital heart disease are a high-risk population. Congenital cardiac defects resulting in either increased pulmonary blood flow or impaired pulmonary venous drainage predispose patients to developing structural and functional aberrations of the pulmonary vasculature. Mortality from pulmonary hypertension is most directly related to right ventricular failure.
Keywords: pulmonary hypertension, congenital heart disease, cardiac surgery, post-operative management, right ventricular function, critical care
Introduction
The simple definition of pulmonary hypertension (PH), which is a mean pulmonary artery pressure (PAP) of greater than or equal to 25 mmHg at rest, belies the complexity of the underlying pathophysiology. In the critical care setting, particularly in patients with congenital heart disease, pulmonary vascular disease can complicate the clinical course even if the resting PAP does not meet the definition of PH (1). Indeed, patients with congenital cardiac defects resulting in either increased pulmonary blood flow or impaired pulmonary venous drainage are prone to episodes of acute reactive pulmonary vasoconstriction that can result in catastrophic cardiopulmonary collapse, especially after exposure to cardiopulmonary bypass (CPB) (2).
Clinical Classification
The most recent clinical classification, which followed the 5th World Symposium on PH in 2013, divided PH into five groups, with twenty-nine subgroups (3). Registries in France, the United Kingdom, and the United States indicate that the most common causes of PH in the pediatric population are idiopathic PH and PH associated with congenital heart disease (4–6). In the neonatal population, persistent pulmonary hypertension of the newborn (PPHN) is relatively common with an incidence estimated to be approximately 2 per 1000 live birth. Furthermore, PH associated with chronic lung disorders, such as bronchopulmonary dysplasia, is increasingly recognized. This most recent update aligned the classification of pulmonary arterial hypertension (PAH) in association with congenital heart disease with the Nice Pediatric classification and included 4 categories: Eisenmenger syndrome, left-to-right shunts, PAH with coincidental heart disease, and post operative PH. Furthermore, the classification proposed criteria for closing intracardiac shunts based upon pulmonary vascular resistance (PVR), as this is an important clinical dilemma (3).
Diagnosis
Indwelling pulmonary artery catheters provide the most direct information, allowing for measurement of vascular pressures, cardiac output and calculation of PVR. The routine use of these catheters has been called into question, but may be important in the management of the highest risk critically ill pediatric patients. The most common noninvasive study used in the intensive care unit is transthoracic echocardiography. The important data that may be obtained by echocardiography are: an estimate of systolic pulmonary arterial pressure, right and left ventricular function, and cardiac anatomy, including determinations of chamber sizes, valvular function and intracardiac shunts. In the absence of a measurable tricuspid regurgitant jet, parameters related to right ventricular outflow patterns and time intervals may be assessed by Doppler echocardiography with demonstrated accuracy compared to right-heart catheterization. Arkles and colleagues found that the shape of the right ventricular Doppler envelope predicted hemodynamics and right heart function in adult PH patients (7). The same group also demonstrated that another echocardiographic estimate of right heart function, the tricuspid annular plane systolic excursion (TAPSE), was reflective of RV function when compared to right heart catheterization, and predicted survival in a cohort of adult PH patients (8).
Cardiac catheterization remains the gold standard for the diagnosis of pulmonary hypertension. In addition to measuring pulmonary hemodynamics, cardiac catheterization can assess for intra- and extracardiac shunts, pulmonary vascular anatomy and reactivity, which can provide information that guides therapeutic decisions. Children who are responsive to acute vasodilator therapy, which is defined as a ≥ 20% decrease in PAP without a decrease in cardiac output, have been shown to have improved survival (9). However, the proper timing of cardiac catheterization is often less clear, in part because the risks increase in patients with acute disease. In fact, a recent study found that the risks of cardiac catheterization are higher in pediatric patients with PH compared to adults, but that it can be performed safely in these patients when undertaken with a programmatic approach (10).
Pathophysiology
Patients with PH have increased right ventricular afterload due to increased PVR and decreased pulmonary vascular compliance. Recent data have demonstrated value in measuring pulmonary vascular impedance, which combines resistance and compliance. Increased impedance relates to several basic mechanisms. There is heterogeneity in the clinical manifestations of pulmonary vascular disease, with advanced disease characterized by increased pulmonary vascular reactivity, sustained pulmonary vasoconstriction, vascular remodeling, and luminal obstruction due to in situ thrombosis and/or obstructive neointimal and plexiform lesions. In 1958, Heath and Edwards first described the histopathology of pulmonary vascular changes associated with congenital cardiac defects, and devised a six grade classification (11). Rabinovitch and colleagues followed with a morphometric classification system, based on lung biopsies taken from patients (aged 2 days to 30 years, with a median age of 1 year) with congenital cardiac defects (12). This morphometric analysis showed progression of disturbed arterial growth and remodeling of the pulmonary vascular bed that correlated with the aberrant hemodynamic state of the pulmonary circulation. These changes were characterized by: (i) abnormal extension of vascular smooth muscle into small peripheral pulmonary arteries and mild medial hypertrophy of normally muscular arteries (Grade A), (ii) severe medial hypertrophy of normally muscular arteries (Grade B) and (iii) decreased pulmonary arterial number (Grade C). The degree to which these changes are reversible remains unclear, but likely depends in part upon the etiology, and may be influenced by age (13). In a seminal study, Rabinovitch and colleagues found that age at surgery, lung morphometric analysis, and the Heath-Edwards system grade predicted the reversibility of structural and functional pulmonary vascular changes secondary to congenital cardiac defects with increased pulmonary blood flow after surgical repair (14). In addition, it must be remembered that even early reversible pulmonary vascular disease can contribute to morbidity and mortality. An important study by Celermajer and colleagues, for example, demonstrated that children with increased pulmonary blood flow due to intracardiac shunting had a selective impairment of endothelium-dependent pulmonary vascular relaxation, before their baseline PAP or PVR increased significantly (1).
Aberrant endothelial function, characterized by a decrease in endogenous vasodilators, such as nitric oxide (NO) and prostacyclin (PGI2) and an increase in endogenous vasoconstrictors, such as endothelin (ET-1), contributes significantly to the increased pulmonary vasoconstriction and impaired relaxation in PH. NO is produced in the vascular endothelium by the enzyme endothelial NO synthase (eNOS), from the precursor L-arginine. Once formed, NO diffuses into the adjacent smooth muscle cell and activates soluble guanylate cyclase (sGC), producing cyclic guanosine monophosphate (cGMP). cGMP results in smooth muscle cell relaxation through protein kinase G (PKG). cGMP is broken down by a family of phosphodiesterases (PDE), with PDE5 being prominent in the pulmonary vasculature. Human studies have demonstrated that NO signaling is perturbed following CPB (15, 16). For example, in a seminal study, Wessel and colleagues examined children with congenital heart disease and PH undergoing repair with CPB (15). They administered acetylcholine, an endothelium-dependent vasodilator, before and after CPB, and inhaled NO, a selective endothelium-independent pulmonary vasodilator postoperatively. They found that PAP and PVR fell in response to acetylcholine before CPB – but following CPB the response to acetylcholine was lost, while the response to inhaled NO was intact – indicating that CPB caused selective pulmonary vascular endothelial dysfunction.
Arachidonic acid metabolism within vascular endothelial cells, results in the production of PGI2 and thromboxane (TXA2). PGI2 activates adenylate cyclase, resulting in increased cyclic adenosine monophosphate (cAMP) production, activation of protein kinase A, and subsequent vasodilation, whereas TXA2 results in vasoconstriction via phospholipase C signaling. PGI2 also binds to platelet receptors, which inhibits their activation. Adatia and colleagues demonstrated an increased TXA2 to PGI2 ratio in children with cyanotic congenital heart disease and in children with increased pulmonary blood flow from left-to-right shunts (17). Furthermore, these investigators showed that this ratio decreased following correction of the left-to-right shunts (18).
ET-1 is a 21 amino acid polypeptide also produced by vascular endothelial cells. The vasoactive properties of ET-1 are complex. However, its most striking property is its sustained hypertensive action. The hemodynamic effects of ET-1 are mediated by at least two distinct receptor populations, ETA and ETB. The ETA receptors are located on vascular smooth muscle cells, and mediate vasoconstriction, whereas the ETB receptors are located on endothelial and smooth muscle cells, and thus may mediate both vasodilation and vasoconstriction respectively. In addition, ETB receptors are involved in the clearance of ET-1. Animal and human studies indicate that CPB-induced increases in ET-1 participate in increased PVR and reactivity post-operatively (19, 20). For example, Schulze-Neick and colleagues demonstrated that PVR could be decreased in 7 patients with post-operative PH by the infusion of BQ123 an ETA-receptor selective antagonist – a reduction that was associated with arterial and venous plasma ET-1 levels – and that was not amenable to a further reduction by inhaled NO (20).
Management Strategies and Therapeutic Options
In the critical care setting, avoidance, recognition and treatment of pulmonary hypertensive crises are paramount (Figure 1). In 1979, John Wheller and colleagues described 2 infants and 1 child with the previously unreported complication of pulmonary hypertensive crisis and right-heart failure after closure of large unrestricted ventricular septal defects (21). It is now recognized that pulmonary hypertensive crises are most likely to occur in susceptible patients after cardiac surgery. Bando and colleagues reviewed a 14-year experience of just over 2400 patients and found that those with total anomalous pulmonary venous return, truncus arteriosus, transposition of the great arteries, ventricular septal defect, hypoplastic left heart syndrome, and atrioventricular septal defect had the highest incidence of postoperative PH crises (22). Mortality in this group was 8.5%; overall 23% of patients with PH events died. By multivariable analysis, the absence of PAP monitoring, venous oximetry, and preoperative PH, were independent risk factors for death associated with PH crises.
Figure 1.
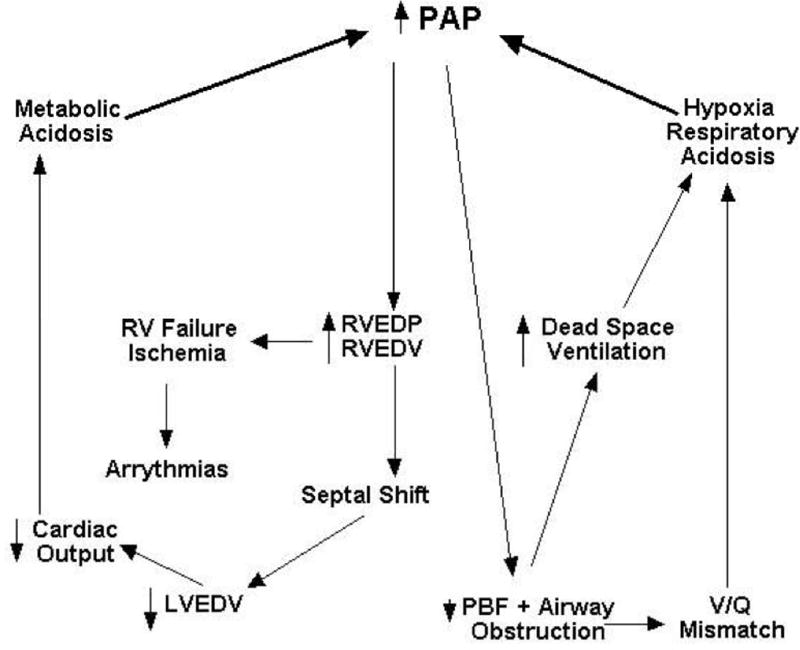
Pulmonary hypertensive crisis. An acute increase in pulmonary arterial pressure (PAP) results in a transient decrease in pulmonary blood flow (PBF) and airway obstruction due, in part, to the compression of small airways by hypertensive pulmonary arterioles. Respiratory acidosis results from an increase in dead space ventilation and ventilation-perfusion (V/Q) mismatch. Simultaneously, right ventricular end-diastolic pressure (RVEDP) and volume (RVEDV) increase, compromising right coronary perfusion, which can result in ischemia and failure of the right ventricle (RV). Also, the interventricular septum is shifted leftward decreasing left ventricular filling (decreased left ventricular end-diastolic volume [LVEDV]) and cardiac output, resulting in metabolic acidosis. Without intervention, this physiology spirals into circulatory collapse.
PH crises involve acute elevations in pulmonary vascular impedance that cause an increase in right ventricular afterload, right ventricular ischemia, and right heart output (Figure 1). Contributing to a decrease in cardiac output is an associated increase in right ventricular end diastolic volume and pressure that shifts the interventricular septum to the left, which decreases left ventricular end diastolic volume and stroke volume (i.e., ventricular interdependence). Decreased cardiac output results in decreased systemic oxygen delivery and metabolic acidosis and decreased pulmonary blood flow increases dead space ventilation, leading to impaired respiratory acidosis, both of which cause pulmonary vasoconstriction. Distention of the pulmonary arteries and perivascular edema produce large and small airway obstruction, respectively, which further impairs ventilation-perfusion matching and decreases lung compliance. In fact, the decrease in lung compliance can be so dramatic that chest wall movement is impaired, even with manual ventilation. A cycle of worsening hypoxemia, hypercapnia, and acidosis (metabolic and/or respiratory) that results in further increases pulmonary vascular impedance develops that ultimately ends with right heart failure and death if left untreated.
In 1966 Dr. Rudolph elucidated the response of the pulmonary vasculature to hypoxia and hydrogen ion concentration (23). Today, oxygenation through delivery of high-inspired oxygen concentration and alkalinization are first line maneuvers in the treatment of PH, and the active avoidance of hypoxia and acidosis is fundamental to the management of patients at risk for PH. In addition, minimizing catecholamine-mediated α1-adrenergic stimulation associated with pain and/or agitation and avoiding excessive lung volumes in patients receiving positive pressure ventilation, which increases PVR, are important components of management. The most widely used therapies for PH work by altering one of three endothelial signaling cascades: NO-cGMP, PGI2, and ET-1. Figure 2 is a simplified depiction of the various sites of action of these therapies. In the critical care setting, augmentation of NO-cGMP signaling is most common, but the use of PGI2 analogs is increasing. With severe disease combination therapy is often required
Figure 2.
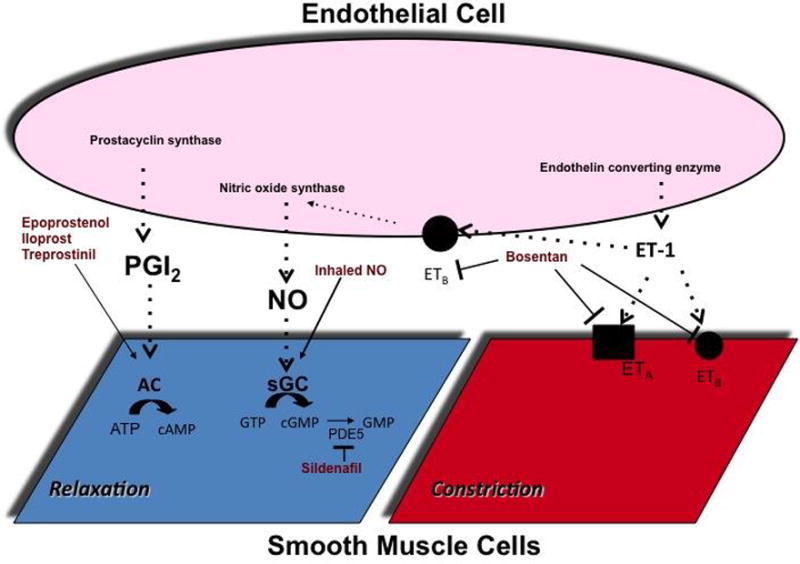
A simplified schematic of the sites of action of selected therapies. Arrows indicate activation and (T) indicate inhibition. PGI2 = prostaglandin I2, ET-1 = endothelin-1, ETA = endothelin A receptor, ETB = endothelin B receptor, NO = nitric oxide, sGC = soluble guanylate cyclase, GTP = guanosine-5′-triphosphate, cGMP = guanosine-3′-5′cyclic monophosphate, GMP = guanosine monophosphate, AC = adenylate cyclase, cAMP = adenosine-3′-5′-monophosphate, AMP = adenosine monophosphate, PDE5 = phosphodiesterase type 5.
Inhaled NO is the best-studied and most widely used agent for acute selective pulmonary vasodilation. When delivered by inhalation, NO diffuses across the alveolus into the smooth muscle of the accompanying capillary, resulting in relaxation. NO then diffuses into the blood vessel where it is rapidly inactivated by its interaction with hemoglobin. In this way, the effects of inhaled NO are relatively confined to the pulmonary circulation and to ventilated areas of the lung, optimizing ventilation-perfusion matching. Russell and colleagues found that inhaled NO decreased mean PAP in patients that emerged from CPB with PH (35%), but not in patients without PH after CPB. Miller and colleagues evaluated the efficacy of inhaled NO for the prevention of post-operative PH crises in a randomized double blind study of 124 infants with large ventricular septal defects or atrioventricular septal defects undergoing repair (2). They found that compared to placebo, patients receiving inhaled NO had fewer PH crises and shorter times to meet extubation criteria. There was no difference in mortality. Likewise, investigators have examined the utility of inhaled NO in patients after bidirectional cavopulmonary connections and Fontan completions (24–26). In these patients, inhaled NO decreased central venous and transpulmonary pressure gradients, and increased oxygen saturations. In addition, the pulmonary vascular response to inhaled NO has been studied as a part of the assessment for operability in patients with PH associated with congenital heart disease (27–30). These studies found that the combination of 100% oxygen and inhaled NO (80 ppm) produced maximal pulmonary vasodilation and was more predictive than either treatment alone for postoperative outcome (27–30).
PDE5 inhibitors (such as sildenafil) augment NO-cGMP signaling by inhibiting the degradation of cGMP. Three small studies found that enteral sildenafil facilitated weaning from inhaled NO in pediatric patients with congenital heart disease undergoing therapy for postoperative PH (31–33). Two studies examined the effects of intravenous sildenafil in pediatric patients after cardiac surgery (34, 35). Both studies found that intravenous sildenafil decreased PAP and PVR either to a greater extent than inhaled NO or synergistically, but that its use was associated with increased intrapulmonary shunting and decreased systemic arterial pressures.
Intravenous PGI2, epoprostenol, has proven to be effective therapy for chronic PH. The inhalational route allows for more selective pulmonary vascular relaxation, which is particularly useful in the intensive care setting. Ivy and colleagues studied iloprost, a PGI2 analog that is FDA approved for administration by nebulization, in 22 children with PH (36). They found that inhaled iloprost decreased PAP to a degree equivalent to inhaled NO with oxygen. Likewise, Rimensberger and colleagues administered inhaled iloprost and NO, alone and in combination, to 15 children with PH secondary to congenital cardiac defects (37). Both agents decreased the PVR to systemic vascular resistance (SVR) ratio to a similar degree, and there was no added benefit from a combination of the treatments. Limsuwan and colleagues found that inhaled iloprost decreased mean PAP and increased systemic saturations without decreasing systemic blood pressure in 8 children suffering from acute increases in PAP after repair of congenital heart disease (38).
Inhibition of ET-1 signaling (at least with currently available enteral formulations) does not reliably cause significant pulmonary vascular relaxation acutely, and thus ET receptor antagonists are generally used for more chronic therapy. However, in high-risk patients, these agents are started in the intensive care unit. Bosentan, an enteral dual ET receptor antagonist, is the most common ET receptor antagonist. A number of studies have demonstrated the efficacy of bosentan in patients with chronic PAH, including children. Newer agents include selective ETA-receptor antagonists.
Right Ventricular Function in Pulmonary Hypertension
Mortality from PH is most directly related to right ventricular function. Pulmonary vasodilation supports right ventricular function to the extent that afterload decreases. When challenged with increased afterload, the contractility of the right ventricular increases, due to changes in the sarcomere length-tension relationship, increased Ca+2 sensitivity, and alterations in the force-frequency relationship of the cardiomyocyte. In addition, the time course over which right ventricular afterload increases and the state of the right ventricle (in particular, right ventricular mass) influences the degree to which the right ventricle can compensate. For example, patients with Eisenmenger’s syndrome tolerate elevated right ventricular afterload far better than patients with normal right ventricles who suffer an acute pulmonary embolism.
If PH persists, compensatory mechanisms fail leading to elevations in right ventricular end-diastolic volume and decreased output. As described for PH crises, due to ventricular interdependence, increases in right ventricular end-diastolic volume and pressure result directly in decreased left ventricular filling and output (39). In fact, indicators of diastolic ventricular interaction, such as decreases in left ventricular end-diastolic volume, are more closely related to stroke volume than PAP in patients with PAH. Right and left ventricular contractility are intimately related. The ventricles share muscle fibers, the interventricular septum, and the pericardial space. In fact, animal studies estimate that upwards of 40% of right ventricular systolic pressure generation results from left ventricular contraction and pressure (systolic ventricular interaction) (40). In addition, right coronary artery perfusion is largely dependent on the pressure gradient between the aortic root and right ventricle.
Taken together, the principles of right ventricular support are: a reduction in right ventricular afterload, optimization of right ventricular volume, augmentation of right ventricular contractility, and maintenance of left ventricular contractility and pressure. Importantly, this strategy requires adequate left ventricular function. Fluid management that optimizes right ventricular volume presents a significant clinical challenge. Although volume loading may be necessary in some situations, excessive volume may provoke adverse diastolic ventricular interactions. In some situations, diuresis to decrease right ventricular volume may be necessary. The ideal agent or combination of agents for inotropic support of right ventricular contractility is not known. Dopamine has been shown to increase cardiac output in patients with PH, but Liet and colleagues found that dopamine increased the PVR:SVR ratio in preterm infants with a widely patent ductus arteriosus (41). Based on animal studies, epinephrine may have a superior hemodynamic profile in the setting of PH compared to dopamine (42). Dobutamine at low doses may result in a reduction in PVR, while increasing right ventricular contractility. Several clinical studies have demonstrated the efficacy of dobutamine in adult patients with PH (43). Likewise milrinone a PDE3 inhibitor that augments ventricular contractility while decreasing PVR and SVR has been shown to improve right ventricular output in adult patients with PH (44). The decrease in SVR may not be desirable and thus may need to be addressed by the addition of a vasopressor. Levosimendan, a Ca2+ sensitizing agent and PDE3 inhibitor approved for use in Europe but not the United States, has been shown to decrease PVR and improve right ventricular output in adult patients with RV failure secondary to a number of conditions including PH (45).
Vasopressors increase SVR and systemic blood pressure and therefore may augment right ventricular output due to the associated increase in left ventricular pressure. Increased systemic pressure can also help maintain right coronary perfusion. Norepinephrine has performed well in animal studies (46, 47). Tourneux and colleagues demonstrated that norepinephrine increased left ventricular output, systemic arterial pressure, and pulmonary blood flow, while decreasing the pulmonary to systemic pressure ratio in 18 newborns with PPHN (48). Phenylephrine has been shown to increase right coronary blood flow in the setting of increased right ventricular pressures, but may also increase PVR (49). Vasopressin, a systemic vasoconstrictor and pulmonary vasodilator, is a promising therapy for PH. Mohamed and colleagues administered vasopressin to 10 infants with PPHN for refractory hypotension and hypoxemia (50). All infants received inhaled NO prior to vasopressin. The administration of vasopressin was associated with a reduction in the oxygenation index, the ability to reduce the inhaled NO dose, and an increase in the systemic arterial pressure. Urine output actually increased after 24 hours.
Atrial septostomy is sometimes considered as a part of the management strategy for chronic PH in order to allow for right-to-left atrial shunting, decompression of the right ventricle and left ventricular filling (51). In the acute setting, severe hypoxemia may limit the success of this approach. Labombarda and colleagues described favorable results with the placement of a Potts anastomosis (descending aorta to left pulmonary artery) in two children with severe idiopathic PH, thereby directing desaturated blood to the lower body (52).
Conclusions
Critically ill pediatric patients with PH associated with congenital heart disease are a high-risk population. Understanding the pathophysiology of PH and the therapeutic approach is necessary for intensive care physicians caring for these vulnerable patients. Supporting ventricular function is essential.
Acknowledgments
Copyright form disclosures: Dr. Oishi received support for article research from the National Institutes of Health (NIH). Dr. Fineman received support for article research from the NIH.
Footnotes
Conflict of interest: There are no conflicts of interest to note.
References
- 1.Celermajer DS, Cullen S, Deanfield JE. Impairment of endothelium-dependent pulmonary artery relaxation in children with congenital heart disease and abnormal pulmonary hemodynamics. Circulation. 1993;87:440–6. doi: 10.1161/01.cir.87.2.440. [DOI] [PubMed] [Google Scholar]
- 2.Miller OI, Tang SF, Keech A, et al. Inhaled nitric oxide and prevention of pulmonary hypertension after congenital heart surgery: a randomised double-blind study. Lancet. 2000;356:1464–9. doi: 10.1016/S0140-6736(00)02869-5. [DOI] [PubMed] [Google Scholar]
- 3.Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D34–41. doi: 10.1016/j.jacc.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 4.Fraisse A, Jais X, Schleich JM, et al. Characteristics and prospective 2-year follow-up of children with pulmonary arterial hypertension in France. Arch Cardiovasc Dis. 2010;103:66–74. doi: 10.1016/j.acvd.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Haworth SG, Hislop AA. Treatment and survival in children with pulmonary arterial hypertension: the UK Pulmonary Hypertension Service for Children 2001–2006. Heart. 2009;95:312–7. doi: 10.1136/hrt.2008.150086. [DOI] [PubMed] [Google Scholar]
- 6.Barst RJ, McGoon MD, Elliott CG, et al. Survival in childhood pulmonary arterial hypertension: insights from the registry to evaluate early and long-term pulmonary arterial hypertension disease management. Circulation. 2012;125:113–22. doi: 10.1161/CIRCULATIONAHA.111.026591. [DOI] [PubMed] [Google Scholar]
- 7.Arkles JS, Opotowsky AR, Ojeda J, et al. Shape of the right ventricular Doppler envelope predicts hemodynamics and right heart function in pulmonary hypertension. American journal of respiratory and critical care medicine. 2011;183:268–76. doi: 10.1164/rccm.201004-0601OC. [DOI] [PubMed] [Google Scholar]
- 8.Forfia PR, Fisher MR, Mathai SC, et al. Tricuspid annular displacement predicts survival in pulmonary hypertension. American journal of respiratory and critical care medicine. 2006;174:1034–41. doi: 10.1164/rccm.200604-547OC. [DOI] [PubMed] [Google Scholar]
- 9.Kawut SM, Horn EM, Berekashvili KK, et al. New predictors of outcome in idiopathic pulmonary arterial hypertension. The American journal of cardiology. 2005;95:199–203. doi: 10.1016/j.amjcard.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 10.Bobhate P, Guo L, Jain S, et al. Cardiac catheterization in children with pulmonary hypertensive vascular disease. Pediatr Cardiol. 2015;36:873–9. doi: 10.1007/s00246-015-1100-1. [DOI] [PubMed] [Google Scholar]
- 11.Heath D, Edwards JE. The pathology of hypertensive pulmonary vascular disease; a description of six grades of structural changes in the pulmonary arteries with special reference to congenital cardiac septal defects. Circulation. 1958;18:533–47. doi: 10.1161/01.cir.18.4.533. [DOI] [PubMed] [Google Scholar]
- 12.Rabinovitch M, Haworth SG, Castaneda AR, et al. Lung biopsy in congenital heart disease: a morphometric approach to pulmonary vascular disease. Circulation. 1978;58:1107–22. doi: 10.1161/01.cir.58.6.1107. [DOI] [PubMed] [Google Scholar]
- 13.Yamaki S, Wagenvoort CA. Comparison of primary plexogenic arteriopathy in adults and children. A morphometric study in 40 patients. Br Heart J. 1985;54:428–34. doi: 10.1136/hrt.54.4.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rabinovitch M, Keane JF, Norwood WI, et al. Vascular structure in lung tissue obtained at biopsy correlated with pulmonary hemodynamic findings after repair of congenital heart defects. Circulation. 1984;69:655–67. doi: 10.1161/01.cir.69.4.655. [DOI] [PubMed] [Google Scholar]
- 15.Wessel DL, Adatia I, Giglia TM, et al. Use of inhaled nitric oxide and acetylcholine in the evaluation of pulmonary hypertension and endothelial function after cardiopulmonary bypass. Circulation. 1993;88:2128–38. doi: 10.1161/01.cir.88.5.2128. [DOI] [PubMed] [Google Scholar]
- 16.Schulze-Neick I, Penny DJ, Rigby ML, et al. L-arginine and substance P reverse the pulmonary endothelial dysfunction caused by congenital heart surgery. Circulation. 1999;100:749–55. doi: 10.1161/01.cir.100.7.749. [DOI] [PubMed] [Google Scholar]
- 17.Adatia I, Barrow SE, Stratton PD, et al. Thromboxane A2 and prostacyclin biosynthesis in children and adolescents with pulmonary vascular disease. Circulation. 1993;88:2117–22. doi: 10.1161/01.cir.88.5.2117. [DOI] [PubMed] [Google Scholar]
- 18.Adatia I, Barrow SE, Stratton PD, et al. Effect of intracardiac repair on biosynthesis of thromboxane A2 and prostacyclin in children with a left to right shunt. Br Heart J. 1994;72:452–6. doi: 10.1136/hrt.72.5.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reddy VM, Hendricks-Munoz KD, Rajasinghe HA, P, et al. Post-cardiopulmonary bypass pulmonary hypertension in lambs with increased pulmonary blood flow. A role for endothelin 1. Circulation. 1997;95:1054–61. doi: 10.1161/01.cir.95.4.1054. [DOI] [PubMed] [Google Scholar]
- 20.Schulze-Neick I, Li J, Reader JA, et al. The endothelin antagonist BQ123 reduces pulmonary vascular resistance after surgical intervention for congenital heart disease. J Thorac Cardiovasc Surg. 2002;124:435–41. doi: 10.1067/mtc.2002.121492. [DOI] [PubMed] [Google Scholar]
- 21.Wheller J, George BL, Mulder DG, et al. Diagnosis and management of postoperative pulmonary hypertensive crisis. Circulation. 1979;60:1640–4. doi: 10.1161/01.cir.60.7.1640. [DOI] [PubMed] [Google Scholar]
- 22.Bando K, Turrentine MW, Sharp TG, et al. Pulmonary hypertension after operations for congenital heart disease: analysis of risk factors and management. J Thorac Cardiovasc Surg. 1996;112:1600–7. doi: 10.1016/S0022-5223(96)70019-3. discussion 1607–9. [DOI] [PubMed] [Google Scholar]
- 23.Rudolph AM, Yuan S. Response of the pulmonary vasculature to hypoxia and H+ ion concentration changes. J Clin Invest. 1966;45:399–411. doi: 10.1172/JCI105355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goldman AP, Delius RE, Deanfield JE, et al. Pharmacological control of pulmonary blood flow with inhaled nitric oxide after the fenestrated Fontan operation. Circulation. 1996;94:II44–8. [PubMed] [Google Scholar]
- 25.Gamillscheg A, Zobel G, Urlesberger B, et al. Inhaled nitric oxide in patients with critical pulmonary perfusion after Fontan-type procedures and bidirectional Glenn anastomosis. J Thorac Cardiovasc Surg. 1997;113:435–42. doi: 10.1016/S0022-5223(97)70355-6. [DOI] [PubMed] [Google Scholar]
- 26.Cai J, Su Z, Shi Z, et al. Nitric oxide and milrinone: combined effect on pulmonary circulation after Fontan-type procedure: a prospective, randomized study. Ann Thorac Surg. 2008;86:882–8. doi: 10.1016/j.athoracsur.2008.05.014. discussion 882–8. [DOI] [PubMed] [Google Scholar]
- 27.Balzer DT, Kort HW, Day RW, et al. Inhaled Nitric Oxide as a Preoperative Test (INOP Test I): the INOP Test Study Group. Circulation. 2002;106:I76–81. [PubMed] [Google Scholar]
- 28.Adatia I, Perry S, Landzberg M, et al. Inhaled nitric oxide and hemodynamic evaluation of patients with pulmonary hypertension before transplantation. J Am Coll Cardiol. 1995;25:1656–64. doi: 10.1016/0735-1097(95)00048-9. [DOI] [PubMed] [Google Scholar]
- 29.Atz AM, Adatia I, Lock JE, et al. Combined effects of nitric oxide and oxygen during acute pulmonary vasodilator testing. J Am Coll Cardiol. 1999;33:813–9. doi: 10.1016/s0735-1097(98)00668-8. [DOI] [PubMed] [Google Scholar]
- 30.Barst RJ, Agnoletti G, Fraisse A, et al. Vasodilator testing with nitric oxide and/or oxygen in pediatric pulmonary hypertension. Pediatr Cardiol. 2010;31:598–606. doi: 10.1007/s00246-010-9645-5. [DOI] [PubMed] [Google Scholar]
- 31.Atz AM, Wessel DL. Sildenafil ameliorates effects of inhaled nitric oxide withdrawal. Anesthesiology. 1999;91:307–10. doi: 10.1097/00000542-199907000-00041. [DOI] [PubMed] [Google Scholar]
- 32.Namachivayam P, Theilen U, Butt WW, et al. Sildenafil prevents rebound pulmonary hypertension after withdrawal of nitric oxide in children. Am J Respir Crit Care Med. 2006;174:1042–7. doi: 10.1164/rccm.200605-694OC. [DOI] [PubMed] [Google Scholar]
- 33.Lee JE, Hillier SC, Knoderer CA. Use of sildenafil to facilitate weaning from inhaled nitric oxide in children with pulmonary hypertension following surgery for congenital heart disease. J Intensive Care Med. 2008;23:329–34. doi: 10.1177/0885066608321389. [DOI] [PubMed] [Google Scholar]
- 34.Schulze-Neick I, Hartenstein P, Li J, et al. Intravenous sildenafil is a potent pulmonary vasodilator in children with congenital heart disease. Circulation. 2003;108(Suppl 1):II167–73. doi: 10.1161/01.cir.0000087384.76615.60. [DOI] [PubMed] [Google Scholar]
- 35.Stocker C, Penny DJ, Brizard CP, et al. Intravenous sildenafil and inhaled nitric oxide: a randomised trial in infants after cardiac surgery. Intensive Care Med. 2003;29:1996–2003. doi: 10.1007/s00134-003-2016-4. [DOI] [PubMed] [Google Scholar]
- 36.Ivy DD, Doran AK, Smith KJ, et al. Short- and long-term effects of inhaled iloprost therapy in children with pulmonary arterial hypertension. J Am Coll Cardiol. 2008;51:161–9. doi: 10.1016/j.jacc.2007.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rimensberger PC, Spahr-Schopfer I, Berner M, et al. Inhaled nitric oxide versus aerosolized iloprost in secondary pulmonary hypertension in children with congenital heart disease: vasodilator capacity and cellular mechanisms. Circulation. 2001;103:544–8. doi: 10.1161/01.cir.103.4.544. [DOI] [PubMed] [Google Scholar]
- 38.Limsuwan A, Wanitkul S, Khosithset A, A, et al. Aerosolized iloprost for postoperative pulmonary hypertensive crisis in children with congenital heart disease. Int J Cardiol. 2008;129:333–8. doi: 10.1016/j.ijcard.2007.08.084. [DOI] [PubMed] [Google Scholar]
- 39.Belenkie I, Dani R, Smith ER, et al. Ventricular interaction during experimental acute pulmonary embolism. Circulation. 1988;78:761–8. doi: 10.1161/01.cir.78.3.761. [DOI] [PubMed] [Google Scholar]
- 40.Damiano RJ, Jr, La Follette P, Jr, Cox JL, et al. Significant left ventricular contribution to right ventricular systolic function. Am J Physiol. 1991;261:H1514–24. doi: 10.1152/ajpheart.1991.261.5.H1514. [DOI] [PubMed] [Google Scholar]
- 41.Liet JM, Boscher C, Gras-Leguen C, et al. Dopamine effects on pulmonary artery pressure in hypotensive preterm infants with patent ductus arteriosus. J Pediatr. 2002;140:373–5. doi: 10.1067/mpd.2002.123100. [DOI] [PubMed] [Google Scholar]
- 42.Barrington KJ, Finer NN, Chan WK. A blind, randomized comparison of the circulatory effects of dopamine and epinephrine infusions in the newborn piglet during normoxia and hypoxia. Crit Care Med. 1995;23:740–8. doi: 10.1097/00003246-199504000-00024. [DOI] [PubMed] [Google Scholar]
- 43.Acosta F, Sansano T, Palenciano CG, et al. Effects of dobutamine on right ventricular function and pulmonary circulation in pulmonary hypertension during liver transplantation. Transplant Proc. 2005;37:3869–70. doi: 10.1016/j.transproceed.2005.10.045. [DOI] [PubMed] [Google Scholar]
- 44.Fukazawa K, Poliac LC, Pretto EA. Rapid assessment and safe management of severe pulmonary hypertension with milrinone during orthotopic liver transplantation. Clin Transplant. 2010;24:515–9. doi: 10.1111/j.1399-0012.2009.01119.x. [DOI] [PubMed] [Google Scholar]
- 45.Russ MA, Prondzinsky R, Carter JM, et al. Right ventricular function in myocardial infarction complicated by cardiogenic shock: Improvement with levosimendan. Crit Care Med. 2009;37:3017–23. doi: 10.1097/CCM.0b013e3181b0314a. [DOI] [PubMed] [Google Scholar]
- 46.Kerbaul F, Rondelet B, Motte S, et al. Effects of norepinephrine and dobutamine on pressure load-induced right ventricular failure. Crit Care Med. 2004;32:1035–40. doi: 10.1097/01.ccm.0000120052.77953.07. [DOI] [PubMed] [Google Scholar]
- 47.Hirsch LJ, Rooney MW, Wat SS, et al. Norepinephrine and phenylephrine effects on right ventricular function in experimental canine pulmonary embolism. Chest. 1991;100:796–801. doi: 10.1378/chest.100.3.796. [DOI] [PubMed] [Google Scholar]
- 48.Tourneux P, Rakza T, Bouissou A, et al. Pulmonary circulatory effects of norepinephrine in newborn infants with persistent pulmonary hypertension. J Pediatr. 2008;153:345–9. doi: 10.1016/j.jpeds.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 49.Vlahakes GJ, Turley K, Hoffman JI. The pathophysiology of failure in acute right ventricular hypertension: hemodynamic and biochemical correlations. Circulation. 1981;63:87–95. doi: 10.1161/01.cir.63.1.87. [DOI] [PubMed] [Google Scholar]
- 50.Mohamed A, Nasef N, Shah V, McNamara PJ. Vasopressin as a rescue therapy for refractory pulmonary hypertension in neonates: case series. Pediatr Crit Care Med. 2014;15:148–54. doi: 10.1097/PCC.0b013e31829f5fce. [DOI] [PubMed] [Google Scholar]
- 51.Kerstein D, Levy PS, Hsu DT, Hordof AJ, et al. Blade balloon atrial septostomy in patients with severe primary pulmonary hypertension. Circulation. 1995;91:2028–35. doi: 10.1161/01.cir.91.7.2028. [DOI] [PubMed] [Google Scholar]
- 52.Labombarda F, Maragnes P, Dupont-Chauvet P, et al. Potts anastomosis for children with idiopathic pulmonary hypertension. Pediatr Cardiol. 2009;30:1143–5. doi: 10.1007/s00246-009-9485-3. [DOI] [PubMed] [Google Scholar]