

Abstract
We combined two tuberculosis (TB) genome-wide association studies (GWAS) from Ghana and The Gambia with subsequent replication totalling 11,425 participants. A significant association with disease was observed at SNP rs4331426 located in a gene-poor region on chromosome 18q11.2 (P=6.8×10−9, OR=1.19, 95%CI=1.13-1.27). Our finding shows that GWAS can identify novel loci for infectious causes of mortality even in Africa where levels of linkage disequilibrium are particularly low.
Tuberculosis (TB) causes significant morbidity and mortality world-wide1,2 with a high disease burden in sub-Saharan Africa. Although previous studies have indicated that susceptibility to pulmonary TB has a substantial genetic component3,4, progress in the determination of contributing genetic variants has been slow. While genome-wide association studies (GWAS) have successfully identified many common variants associated with a variety of diseases5, studies of infectious diseases have so far largely failed to identify novel causative variants, partly due to the small sample sizes studied6,7. We here present a combined analysis of two West African genome-wide association studies, a new data set from Ghana and The Wellcome Trust Case Consortium (WTCCC) tuberculosis study of Gambians (WTCCC, unpublished) with further replication series analyzing in all 11,425 African individuals.
The two GWA studies were performed with TB cases and controls recruited from Ghana and The Gambia (study flow-chart in Supplementary Methods) The Ghanaian GWA study included 921 cases and 1,740 controls genotyped using the Affymetrix SNP Array 6.0 with 743,635 autosomal SNPs included in the analysis (Supplementary Methods). The Gambian WTCCC GWA study included 1,316 cases and 1,382 controls who were genotyped using the Affymetrix GeneChip 500K array with 354,607 autosomal SNPs (Supplementary Methods) (WTCCC, unpublished). In total, 333,754 SNPs were included in the combined analysis of 2,237 cases and 3,122 controls from Ghana and The Gambia, achieving 90% power to detect significant association at genotype relative risks of 1.4 or greater. Multidimensional scaling (MDS) analysis was carried out to investigate the population structure (Supplementary Methods)8. The first six MDS components were incorporated as covariates into the logistic regression association analysis in the combined study. The subsequent quantile-quantile plot showed inflation at a level comparable to other GWA studies after correction with the MDS components (λ=1.05, Supplementary Methods).
In order to look at these findings more globally we subjected the Ghanaian and Gambian results to an MDS analysis, including available data from the Nigerian Yoruba population, and also non-African populations (Han Chinese, Japanese, CEPH) from the HapMap Project. As expected, differences to the Chinese/Japanese and CEPH populations were large, but the three populations originating from West Africa could also be distinguished clearly (Supplementary Methods). These population-specific differences complicate accurate imputation of the Ghanaian or Gambian genotypes using the Nigerian Yoruba HapMap data set. Fst values for genetic distances for each pair of Ghanaian populations, Gambian populations and between Ghanaian and Gambian populations as a whole are given in Supplementary Methods.
In the combined analysis, we identified 17 SNPs with P≤10−5 with the same direction of effect in both the Ghanaian and WTCCC Gambian study groups (Supplementary Table 1). We attempted to replicate these 17 SNPs in an additional 1,076 cases and 1,611 controls from Ghana (Replication I). The addition of the results obtained in Replication I to the two GWA studies revealed two SNPs (rs2335704 and rs4331426) with P<5×10−7 (Supplementary Table 1). To corroborate the findings, we further genotyped these two SNPs in additional cohorts from Ghana (150 cases, 2,214 controls) and Malawi (236 cases, 779 controls), (Replication II), as well as in 332 family trios/duos from Ghana, whereby cases of nuclear families were part of the complete association analysis. For the two hit regions, neighbouring SNPs were studied in an attempt to fine-map the association signals (Supplementary Table 2).
When combining the GWA study data with those from Replications I and II, SNP rs4331426, which maps to chromosome 18q11.2, obtained the highest association signal with an overall P=6.8×10−9 (OR=1.19, 95%CI=1.13-1.27), or PGC =1.6×10−8 after genomic control correction (Fig. 1, Table 1). Consistent estimates of ORs as assessed by the Breslow Day test were observed (P=0.95) across the studies and ethnic groups, and no difference in ORs was seen between the logistic regression and Mantel-Haenszel tests. Heterogeneity between the studies was negligible with I2=0.0%. The analysis of Ghanaian nuclear families supported the association, P=0.016 (OR=1.33, 95%CI=1.05-1.68), although this statistic was not included in the final P value because the cases were part of the association analyses (Table 1).
Figure 1.
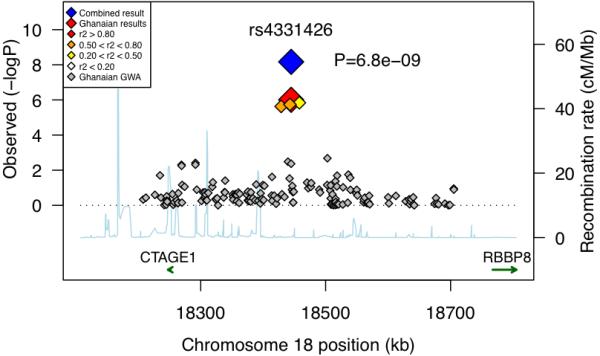
Association plot with fine mapping markers on chromosome 18 in the combined analysis (r2 values between rs4331426 and adjacent SNPs derived from the Ghanaian population; P uncorrected for λGC).
Table 1.
Association statistics of rs4331426 in the combined analysis
| rs4331426 – G allele | Controls | Controls | Cases | Cases | OR 95% CI | P value |
|---|---|---|---|---|---|---|
| GWA study scan | N | Freq | N | Freq | ||
| Ghana | 1740 | 0.448 | 921 | 0.491 | 1.18 (1.05-1.32) | 4.3E-03 |
| The Gambia (WTCCC) | 1377 | 0.476 | 1309 | 0.521 | 1.18 (1.06-1.31) | 2.9E-03 |
| Replication I | ||||||
| Ghana | 1609 | 0.429 | 1076 | 0.477 | 1.19 (1.06-1.33) | 2.8E-03 |
| Replication II | ||||||
| Ghana | 2199 | 0.442 | 148 | 0.476 | 1.18 (0.92-1.51) | 1.9E-01 |
| Malawi | 576 | 0.525 | 178 | 0.563 | 1.15 (0.91-1.45) | 2.3E-01 |
| Combined analysis | 7501 | 3632 | 1.19 (1.12-1.26) | 6.8E-09 | ||
| λGC corrected combined P value | 1.6E-08 | |||||
| Ghanaian nuclear families* | 1.33 (1.05-1.68) | 1.6E-02 |
Cases from nuclear families were part of the Ghanaian GWA case-control study; the TDT statistic is not included in the combined analysis.
The associated chromosome 18 variant (SNP rs4331426) is common in African populations but much rarer in all other populations and has a remarkably consistent OR across African cohorts, including the East African samples from Malawi. Additional work is required to ascertain the causative variant, its functional significance and any possible counterbalancing selective pressure. The nearest genes to this SNP are GATA6, CTAGE1, RBBP8, CABLES1, as well as a number of, as yet, un-annotated open reading frames. However, the generally low linkage disequilibrium on 18q11.1-q11.2 suggests that rs4331426 is within a gene-desert region that is punctuated by evolutionarily conserved domains with regulatory potential.
In addition to rs4331426, a second variant, rs2335704 on chromosome 2 was found associated after Replication I (P=3×10−7, OR=1.23, 95%CI=1.14 -1.34). However, the significance decreased to P=2.1×10−6 (OR=1.29, 95%CI=1.11-1.28) after adding results of Replication II (Supplementary Fig. 1, Supplementary Table 3). The Mantel-Haenszel test, stratified for ethnic groups, revealed heterogeneity in ORs between the Ghanaian ethnic group of the Ga-Adangbe compared to all other ethnicities. This heterogeneity suggests that the result should be interpreted with caution.
GWA studies in African populations are, in general, limited by extensive genetic diversity and shorter LD ranges, and to date no novel loci of genome wide statistical significance (P<5×10−8) have been reported using the GWA approach in African studies (WTCCC, unpublished). A recent malaria GWA study in The Gambia found substantial population complexity6, indicating marked diversity among African ethnicities and emphasizing the need for careful population structure correction and ethnicity stratification.
In general, finding convincing non-MHC susceptibility loci across populations for infectious diseases has been difficult, even in individuals of European ancestry. A recent GWA study on HIV viral set-point revealed strong signals within the HLA-B and HLA-C loci, but not any novel non-MHC loci7. Pathogen variation may underlie some of the difficulties in finding loci for a given infectious disease and M. tuberculosis displays substantial geographic variation in genotype frequencies9. However, as we have shown previously and also here9-13, combined analyses with increased aggregate sample sizes may help in the identification of novel genetic variants, perhaps particularly those less sensitive to pathogen variation. Analyzing African individuals poses specific challenges, and genetic differences between populations, even within West Africa, are large enough to complicate standard imputation procedures. Assessing imputation accuracy using the YRI as reference we found a mean genotype error rate across all chromosomes of 8.2% for the combined Ghanaian and Gambian sample, which raises some concern about the validity of imputation in this setting. We performed a genome wide imputation analysis with stringent quality criteria (Rsq>0.7) followed by association analysis of the imputed SNPs (Supplementary Table 4). In these calculations, no variant reached a significance of P<10−6. However, many variants did reach the significance level of the rs4331326 SNP, and these will be carried forward in future studies.
We analyzed variants of the Affymetrix SNP arrays located in candidate genes previously found to be associated with resistance or susceptibility to TB. We observed weak to moderate signals at several genes including the HLA-DQ region (rs9469220, P=0.0017) (Supplementary Table 5). Considering the a priori associations for these regions there appears to be an over-representation of nominally significant results, suggesting that a number of the analyzed SNPs might be true TB susceptibility loci. However, more work will be necessary to replicate positive findings of previous association studies and to identify the true causative variants of these candidate gene analyses.
This work demonstrates that a novel non-MHC locus can be identified for a major fatal infectious disease caused by a highly polymorphic pathogen and suggests that many further loci may be identifiable with GWA studies of sufficient sample size, even in African populations, who suffer the greatest burden of communicable diseases.
Supplementary Material
Acknowledgements
We would like to thank the patients and families, field workers, nurses and physicians who contributed to these studies. We thank Dr Mark McCarthy and Dr Chiea Khor for critical assessment of the manuscript.
Footnotes
URLs: The International HapMap Project (http://hapmap.ncbi.nlm.nih.gov); The Wellcome Trust Case Control Consortium (www.wtccc.uk.org)
Competing interests statement: The authors declare that they have no competing financial interests.
References
- 1.The World Health Organization Crunch time for tuberculosis control. Lancet. 2009;373:1145. doi: 10.1016/S0140-6736(09)60662-0. [DOI] [PubMed] [Google Scholar]
- 2.The World Health Organization WHO annual report on global TB control--summary. Wkly Epidemiol. Reports. 2003;78:122–8. [PubMed] [Google Scholar]
- 3.Comstock GW. Tuberculosis in twins: a re-analysis of the Prophit survey. Am. Rev. Respir. Dis. 1978;117:621–4. doi: 10.1164/arrd.1978.117.4.621. [DOI] [PubMed] [Google Scholar]
- 4.Stead WW, Senner JW, Reddick WT, Lofgren JP. Racial differences in susceptibility to infection by Mycobacterium tuberculosis. N. Engl. J. Med. 1990;322:422–7. doi: 10.1056/NEJM199002153220702. [DOI] [PubMed] [Google Scholar]
- 5.WTCCC Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–78. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jallow M, et al. Genome-wide and fine-resolution association analysis of malaria in West Africa. Nat. Genet. 2009 doi: 10.1038/ng.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fellay J, et al. A whole-genome association study of major determinants for host control of HIV-1. Science. 2007;317:944–7. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ziegler A. Genome-wide association studies: quality control and population-based measures. Genet. Epidemiol. 2009;33:S45–50. doi: 10.1002/gepi.20472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gagneux S, et al. Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. 2006;103:2869–2873. doi: 10.1073/pnas.0511240103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fitness J, et al. Large-scale candidate gene study of tuberculosis susceptibility in the Karonga district of northern Malawi. Am. J. Tro. Med. Hyg. 2004;71:341–9. [PubMed] [Google Scholar]
- 11.Lienhardt C, et al. Investigation of environmental and host-related risk factors for tuberculosis in Africa. I. Methodological aspects of a combined design. Am. J. Epidemiol. 2002;155:1066–73. doi: 10.1093/aje/155.11.1066. [DOI] [PubMed] [Google Scholar]
- 12.Thye T, et al. MCP-1 promoter variant −362C associated with protection from pulmonary tuberculosis in Ghana, West Africa. Hum. Mol. Genet. 2009;18:381–8. doi: 10.1093/hmg/ddn352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Intemann CD, et al. Autophagy gene variant IRGM −261T contributes to protection from tuberculosis caused by Mycobacterium tuberculosis but not by M. africanum strains. PLoS Pathog. 2009;5:e1000577. doi: 10.1371/journal.ppat.1000577. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.