

Controlled adsorption and phase behavior of colloids at liquid interface.
Keywords: Smart emulsions, colloidal aggregation, self-assembly, DNA functionalization
Abstract
We report a study of reversible adsorption of DNA-coated colloids on complementary functionalized oil droplets. We show that it is possible to control the surface coverage of oil droplets using colloidal particles by exploiting the fact that, during slow adsorption, compositional arrest takes place well before structural arrest occurs. As a consequence, we can prepare colloid-coated oil droplets with a “frozen” degree of loading but with fully ergodic colloidal dynamics on the droplets. We illustrate the equilibrium nature of the adsorbed colloidal phase by exploring the quasi–two-dimensional phase behavior of the adsorbed colloids under the influence of depletion interactions and present simulations of a simple model that illustrates the nature of the compositional arrest and the structural ergodicity.
INTRODUCTION
One of the key challenges in complex self-assembly is the creation of ordered, quasi–two-dimensional (2D) patterns of many distinct colloids or nanoparticles. To achieve this objective, it is important that the substrate be smooth and clean, and that different species of colloidal particles be able to bind independently and reversibly to the surface. Moreover, the surface-bound particles should be sufficiently mobile to ensure that the structure that is most stable is also kinetically accessible.
Most solid substrates are less than ideal for this purpose: the surfaces often contain defects or impurities that trap colloids. Moreover, bound colloids diffuse slowly on such surfaces. Liquid interfaces would be less susceptible to the above problems; however, the strength with which colloids bind to liquid-liquid interfaces (through the Pickering mechanism) may be a hundred or thousand times stronger than the thermal energy and, as a consequence, controlled and reversible adsorption of different species is difficult to achieve (1–5). In addition, control of the interactions between surface-incorporated colloids is difficult because long-range capillary forces tend to dominate (6–8).
Here, we introduce a new strategy to assemble colloids reversibly at a liquid-liquid interface. To achieve this, we functionalize both the liquid-liquid interface and the colloidal surface such that colloids can bind reversibly to the interface through complementary DNA interactions. Of course, DNA has been used extensively as selective glue between nanometer- and micrometer-sized solid colloids (9–12). Other groups have explored functionalizing vesicles or fluid membranes with single-stranded DNA (ssDNA) (13, 14). A hybrid approach (15) involved functionalizing fluid membranes enveloping hard spherical colloids, and Feng et al. (16) have explored the use of DNA-functionalized oil droplets (ODs). The motivation for using such fluid substrates was to ensure that the grafted ssDNA could diffuse on the surface. The DNA can then accumulate into “rafts” at the point of contact between two particles with complementary functionalization. A drawback of the above strategies to achieve mobile grafting is that the amount of ssDNA that can be incorporated into the phospholipid bilayers or monolayers is quite limited (13, 16). Here, we present an approach that overcomes this drawback, thus allowing us to create ODs that are densely grafted with mobile ssDNA.
In our approach, we use ODs with diameters of 20 to 30 μm, stabilized by SDS, onto which we adsorb polylysine-g[3.5]–polyethylene glycol–biotin (PLL-PEG-bio), a comb-like polyelectrolyte. As illustrated in Fig. 1, the positively charged PLL backbone adsorbs in thin patches onto the negatively charged SDS head groups. To every third to fourth lysine repeat unit, a 2- or 3.4-kD PEG chain is attached. Half of the nearly 40 PEG chains per PLL backbone carry a biotin group, which we functionalized with streptavidin. Finally, biotinylated ssDNA, denoted A, was brought onto the OD’s surfaces via the streptavidin. Thus, we created small, flat patches of PLL molecules that diffuse freely with their DNA on the oil-water interface. Adding to the ODs 0.57-μm large polystyrene (PS) particles with the complementary ssDNA, A′, these anchor reversibly to the interfaces via DNA hybridization. Depending on the solvent conditions, this system exhibits an unexpectedly rich quasi-2D phase diagram of colloid aggregation.
Fig. 1. DNA functionalization of oil droplets and colloids.
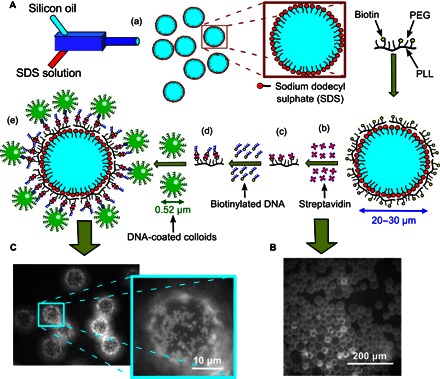
(A) Cartoon representing various stages of the sample preparation: (a) SDS-stabilized droplets are prepared by mixing 10 mM SDS and silicone oil in a microfluidic device; (b) PLL-PEG-biotin adsorbs at the SDS-stabilized ODs that are negatively charged because of the sulfate head group of the SDS surfactant; (c) Texas Red–labeled streptavidin linkers are then attached to the biotin heads on the ODs from the solution; (d) biotinylated ssDNA (A DNA) is then added, attaching to these streptavidin linkers; and (e) green fluorescent PS colloids coated with complementary A′ DNA are then allowed to bind via DNA hybridization. (B) Fluorescence image of the ODs after attaching the fluorescent streptavidin from solution. (C) Typical image showing the fluorescent colloids hybridized to the OD surfaces with a zoom onto the south pole of the droplet.
RESULTS
DNA functionalization of ODs
The SDS-stabilized silicon-OD preparation method is detailed in the Supplementary Materials. The coverage of PLL-PEG-bio on the OD was tested by binding fluorescently labeled streptavidin to the biotinylated PEG ends (Fig. 1A, iii). Although a lysine monomer carries one positive charge on the amino group, the repeat units linked to a PEG chain via that amino group are neutral. Hence, each PLL backbone made of 137 repeat units will carry ~40 positive charges (17), causing the backbone to adsorb flat on the negatively charged SDS head groups. The hydrophilic, uncharged, and flexible PEG side chains align perpendicular to the interface, forming a comb-like geometry (17, 18). The biotin-terminated PEG chains are longer than the others, thus rendering the biotin accessible. After removing excess PLL-PEG-bio from the OD solution, Texas Red–labeled streptavidin was adsorbed onto the biotinylated PEG end groups. The strong fluorescence on the OD surfaces (Fig. 1B) indicates that the PLL-PEG-bio chains are located at the oil-water interface. We used fluorescence intensity measurements to estimate the maximum PLL-PEG-bio grafting density for a mixed volume of ODs (fig. S1). All experiments presented here were performed at the onset of the saturated grafting regime to avoid free, unbound chains in solution. The observed fluorescence intensity was several orders of magnitude stronger than that of droplets coated with a biotinylated lipid monolayer (16). The observed density of streptavidin linkers (~104 μm2) on the OD surface corresponds to an average distance of 10 nm between biotin-streptavidin ends. After removing excess streptavidin from the continuous aqueous phase, we grafted biotinylated ssDNA, A, to the OD solution—again, any unbound DNA was removed before green fluorescent, 0.52-μm-large, A′ DNA–coated PS colloids were added. Care was taken to keep the final aqueous condition constant with an added NaCl concentration of ~50 mM. The samples were studied using epifluorescence video microscopy after they were incubated overnight, allowing for DNA equilibrium hybridization. In most samples, we used colloidal bulk volume fractions of Φc ≤ 0.05%. Focusing on the “south pole” of the ODs that are located at the top surface of the sealed capillaries (Fig. 1C), it becomes apparent that the PS particles have hybridized to the ODs, whereas a considerable fraction of the colloids remained in the aqueous bulk phase.
Colloidal 2D aggregation at the oil-water interface
While keeping the PLL-PEG-bio and A DNA coverage approximately constant for all experiments, we studied the effect of varying the SDS concentration on the aggregation behavior of the PS colloids hybridized to the ODs (Fig. 2). As we increased the excess SDS concentration in bulk, we observed a transition from a gas-like distribution of hybridized PS colloids (SDS concentrations, <2 mM; video S1) to the formation of 2D “continents” with crystalline order at higher concentrations of SDS (>5 mM; video S2). Between 2 and 4 mM SDS concentrations, a two-phase region was observed, where individual hybridized colloids coexisted with more disordered fluid-like colloidal islands containing more than just a few colloids (Fig. 2B). We attribute this behavior to depletion attraction induced by SDS micelles. All samples showed a considerable fraction of free, nonhybridized colloids in solution. These did not aggregate because of the steric stabilization provided by the A′ DNA brush on the colloids. However, at SDS concentrations above 2 mM, we also observed weak colloidal aggregation in bulk, again presumably because of depletion forces.
Fig. 2. Comparison between experimental and numerical results for packing of colloids at the interface.

(A to C) Fluorescence micrographs (under green fluorescent protein illumination) showing change in the packing of the 0.52-μm PS colloids on the surface of ODs as a function of SDS concentration in the bulk of the sample. The colloids go from a colloidal gas-like phase (A) to a liquid-like cluster (B) to a well-aligned hexagonal packing at the interface (C). (D to F) Snapshots for different strengths of the Asakura-Oosawa (AO) potential: VAO = 0 (D), 6 (E), and 8 (F) (simulation details are in the Supplementary Materials). These images were modulated with a Gaussian blur emulating the experimental situation.
Melting colloids off of the OD surfaces
One of the most striking features of the colloid-OD system is that the loading of ODs by colloids appears to depend on the preparation protocol, even though the colloids on the ODs are highly mobile. As we argue below, this protocol dependence is due to the fact that, during slow deposition, colloids have more time to collect the PLL-PEG–bound DNA (PLL-PEG-DNA) “receptors” than during fast deposition. During slow deposition, the bound colloids deplete the remaining PLL-PEG-bio domains from the interface so that no further colloids can bind.
We first verified whether it was possible to remove the colloids from the OD-water interface by heating the sample well above the hybridization (“melt”) temperature, Tm = 32°C, for the sequence used here. The width of the melt region for complementary ssDNA free in solution is typically ΔT = 10° to 20°C, depending on the solvent conditions and the DNA concentration (19), but it narrows down to ~1°C when the same DNA strands are attached to colloids (12); moreover, Tm shifts to higher values. In a control experiment in which we mixed 0.52-μm particles functionalized with A or with A′ in a 1:1 ratio, using the same solvent condition as those used in the OD experiments, we measured Tm ≃ 35°C (fig. S2).
We first mixed A′-functionalized 0.52-μm particles with the A-coated ODs [~2-mM SDS, 4 mM tris (pH 7 to 8), 50 mM NaCl] and allowed the colloids to hybridize to the ODs overnight at room temperature. The fluorescence images in Fig. 3A show that, at room temperature, liquid colloidal islands coexist with nonaggregated colloids hybridized to the interface at this SDS concentration. As we increased the temperature (using a computer-controlled piezoelectric heating stage on the microscope), these islands became smaller and more mobile, but even when heating beyond 35°C, the colloids did not detach from the droplet surface; rather, the particles diffused even faster. Even at 80°C, very few particles came off. We hypothesize that the reason for the strong binding of the colloids is that, because the A DNA on the PLL-PEG is mobile, A DNA will accumulate in the contact region, and hence, the binding strength will increase with time. Consequently, the final number of A-A′ bonds in the contact region will be larger (and Tm will be higher) than for the contact region between two hard spheres where the A DNA is fixed. This effect will be enhanced by the fact that the effective contact region between colloids and ODs is larger than that between two colloids with the same curvature. However, the binding strength should still decrease as we decrease the overall A′ DNA concentration. To test this hypothesis, we reduced the total number of A′ strands on the PS particles by replacing 70% of the A′ strands with noncomplementary B strands (see Materials and Methods). The images for increasing temperatures in Fig. 3B show that now particles start to melt off the surface at ~56°C; at ~66°C, almost all bound particles have left the OD surfaces. This supports our assumption that the colloid-OD binding is due to DNA hybridization and that the very strong binding experienced by colloids with a dense A′ coating is due to the accumulation of complementary PLL-PEG-DNA in the contact region. We also note that the melting region is not as sharp as for hard particles, although a larger number of binding DNA in the contact area would suggest a narrowing of the melt region. This widening can be understood by considering that the distribution of PLL-PEG-DNA over the different colloidal contacts is determined by kinetics: some colloids will accumulate more than others. However, once bound, redistribution of PLL-PEG-DNA domains over different colloidal contacts is very slow. Hence, some colloids are more strongly bound than others. This broadening will only occur if the deposition of colloids on the ODs is relatively fast. Otherwise, every colloid that lands on the surface has time to accumulate its full share of PLL-PEG-DNA. But of course, this also means that less PLL-PEG-DNA remains for any subsequent colloids.
Fig. 3. Thermal recyclability of colloidal binding at the interface.

(A and B) Sequence of fluorescence images showing heating of a sample of 0.57-μm colloids grafted to ODs using colloids fully covered with A′ DNA (~1.5 mM SDS and 50 mM NaCl concentrations) (A) and where three-fourths of the A′ DNA is replaced by nonbinding B (~2.5 mM SDS and 50 mM SDS) (B). In all images, we focus on the south pole of the ODs using the same illumination and exposure time. (A) For full coverage, most colloids remained on the OD surface even when heating to 80°C, although they became increasingly mobile, which is expressed in the increasingly blurry appearance of the images. (B) For samples with reduced number of binding sites on the colloids, melting was set at ~56°C, and at ~70°C, almost all colloids have come off the ODs. The image taken shortly after cooling the sample (over a period of 1 hour) to 14°C showed only a few colloids bound to the ODs. The same sample recovered similar coverage and aggregation state as the initial samples after 1 day (right-most bottom image).
When starting with a high Φc, there is no time for the colloids to collect their share of PLL-PEG-DNA, and hence, more remains for subsequent colloids. If we start with a high Φc, a higher density of colloids on the ODs is observed, which automatically leads to fewer DNA bridges between any given colloid and the OD, and therefore to a reduced Tm. Note that after melting off the particles, the initial particle grafting density on the ODs is recovered upon cooling to 20°C, although that the process is very slow. Few colloids have hybridized back to the ODs 1 hour after they were cooled initially to 14°C, and we recovered the initial colloid density only after 1 day at room temperature (Fig. 3B).
Simulations
Because it is difficult to probe the distribution of colloid-OD bonds directly in experiments, we used kinetic Monte Carlo simulations (see the Supplementary Materials) to investigate the factors that affect the protocol-dependent colloid-OD binding. In these simulations, the interface is described as a 2D discrete square lattice with a given density of square patches, ρp. A patch represents a single DNA-functionalized PLL-PEG-biotin chain of length lp, in units of lattice sites, and an interfacial diffusion coefficient Dp. We represent the contact region of the colloids with the interface by a square of length lC. The number of colloids that arrive per unit time at the oil-water interface is denoted by F (the “flux”). F increases monotonically with the bulk concentration Φc. In our simulations, we assume that the colloids bind irreversibly if they overlap with at least one site of the patch. Both colloids and patches are assumed to be impenetrable. Because we are interested in the initial stage of binding, we ignore the diffusion of colloid-patch complexes. However, free patches can diffuse and thus accumulate in accessible sites underneath a colloid-patch complex, forming a raft. The simulations confirm that as F decreases, the limiting density of surface-bound colloids decreases because of the depletion of accessible patches (see the Supplementary Materials).
We also used numerical simulations to study the 2D aggregation behavior of the bound colloids on the SDS concentration. To this end, we used Brownian dynamics simulations on a curved interface, describing the pairwise colloid interactions by the superposition of a repulsive Yukawa and the short-ranged, attractive AO potential (see the Supplementary Materials for further details). Keeping the fraction of bound colloids per OD constant, we increased the strength of the attraction—it scales linearly with the micelle concentration. Figure 2 (D to F) shows snapshots for different strengths of the AO potential. As observed experimentally, by increasing the density of the depletant, we observe a transition from a fluid-like structure to crystalline-like order (Fig. 2).
Particle tracking, differential dynamic microscopy, and microrheology
To understand how binding to the OD interface affects the dynamics of the colloids compared to free particle motion in bulk, we performed particle tracking and differential dynamic microscopy (DDM) measurements. For particle tracking, we used 1.2-μm PS colloids coated with a grafting density of A′ DNA comparable to the one used on the smaller particles. We also lowered the overall added colloid concentration, thus assuring low particle concentrations on the OD surface. We only track trajectories close to the south pole such that the particles move mostly in a plane perpendicular to the viewing direction. We study root mean square displacements that are much smaller than the radius of the OD, and hence, we assume that the curvature of the ODs can be neglected in the analysis of the diffusive motion (see Fig. 4A and video S3; video S4 shows tracking in the bulk). The averaged diffusion coefficient DOD ~ 4.0 × 10−14 m2s−1 obtained for 1.2-μm colloids bound to the ODs was found to be reduced by one order of magnitude compared with that of free particles in solution, DOD ~ 3.37 × 10−13 m2s−1; the latter value is close to the theoretical estimate Dtheo ~ 4.11 × 10−13 m2s−1 (Fig. 4B). Particle tracking of free PS particles was done in the absence of ODs but under equivalent solvent conditions and temperature. Feeding the displacements extracted from the same particle tracks into an in situ analysis program detailed in the Supplementary Materials (20), we also extracted the effective viscoelastic properties the bound colloids experienced. These suggest that the colloids experience a viscosity of (9.5 ± 0.6) mPa s, which is roughly 10 times larger than that of water.
Fig. 4. Comparing dynamics of colloids bound to the interface and free in solution.
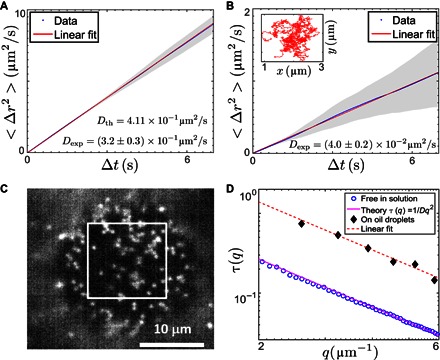
(A and B) Mean square displacement versus delay time for 1.2-μm colloids diffusing freely in solution (A) and for DNA-bound colloids diffusing on the OD surfaces averaged over five tracks (B). The inset shows a typical single-particle track. (C) Microscope image of 0.57-μm colloids bound to an OD surface, focusing on its south pole. The square selection was used for DDM analysis. (D) Decay time τ (q) as a function of the scattering vector q extracted from DDM analysis for both colloids diffusing on OD surfaces [as shown in (C)] and from microscope movies taken from free colloids in solution; the theoretical line passing through the data for the free colloids was obtained, assuming a colloid diameter of 0.57 μm, a buffer solution viscosity of 0.9 mPa s, and room temperature.
For samples with only 0.52-μm particles present, we used DDM to extract the particle diffusivities on the ODs and in bulk (21, 22). Similar to the larger particles, a considerable reduction in the diffusivity (~ 2.4 × 10−13 m2s−1) of bound colloids is found, whereas the value Dsol ~ 9.52 × 10−13 m2s−1 measured for the free particles is again similar to the theoretical one (Fig. 4, C and D, and video S1). Note that the DDM results for the particles on the OD surface have a larger error because of the reduced statistics, as we limited the sampling to the small region on the south pole to avoid systematic errors because of the curvature of the droplets.
DISCUSSION
The experimental results demonstrate three facts: first, the attachment of our colloids was driven by DNA binding; second, the hybridization between colloid and complementary DNA on the surfaces of the ODs was thermally reversible; and third, the colloids attached to the surface underwent aggregation, which was purely driven by depletion attraction caused by the surfactant micelles present (23–25). All our experimental findings are supported by the simulation results.
Further proof that our colloids were indeed anchored to the OD surfaces purely via DNA hybridization and not due to surface tension effects (like in Pickering-Ramsden emulsions or other nonspecific interactions) came from several different control experiments. In the first control sample, we mixed our OD solutions with colloids coated with noncomplementary B DNA, using the standard solvent conditions with an SDS concentration of ~2 mM. No colloidal adsorption to the ODs was observed, even after a week. In the second control sample, we replaced the A DNA on the ODs by PEG-biotin that formed a similar steric stabilization layer as the DNA linkers, whereas the colloids were grafted with either A′ or B DNA. These samples also did not show any signs of binding between the ODs and the colloids. Even the mixtures of DNA-coated colloids and bare SDS-stabilized ODs did not display any colloidal adsorption on the oil-water interfaces at all bulk SDS concentrations used.
In all OD-colloid mixtures, irrespective of the bulk SDS concentration, a large number of colloids remained in solution. As described in the section on melting colloids off of the OD surfaces, we propose that the amount of colloids hybridized to the ODs depends on the total colloid volume fraction. Starting with a Φc ≃ 0.05 wt%, which corresponded roughly to a full DNA coverage of the ODs in solution, we obtained relatively high colloid coverage (Figs. 1 and 2), but still a number of colloids remained free in bulk solution. Using a 10 times lower Φc, such as that used for tracking purposes, we observed far fewer colloids on the OD surfaces. We argue that most of the A DNA bound via the PLL-PEG-bio chains was recruited by the hard colloids, forming small rafts of several chains in the contact region. However, when using higher initial colloid concentrations, these rafts would be smaller. Once all A DNA was bound, no further colloids were able to bind to the oil-water interface. The interfacial regions free of colloids can thus be thought of as regions purely stabilized by SDS. That is, the colloid hybridization leads to segregation into regions of a pure SDS monolayer and others holding the PLL-PEG-bio with the DNA. Because the fluorescence of the streptavidin attached to the biotins on the PLL-PEG was much weaker than that on the colloids, we could not observe this segregation in experiments. However, our simulation studies strongly support our assumptions.
The strong slowing down of the colloidal diffusion on the ODs also supports our hypothesis that several mobile PLL-PEG-bio chains form rafts. It appears that the measured colloidal diffusion coefficients are dominated by the diffusion coefficient of the rafts. We have shown that the diffusion coefficient of colloids sedimented onto either a “soft” cross-linked polymer surface or a “hard” sterically stabilized support surface does not deviate more than some 10% from their bulk diffusion value (26), whereas here, we observed a reduction of one order of magnitude.
Saffman and Delbrück (27) analyzed the mobility b of proteins in a lipid bilayer. They derived an analytical expression for a mobility of a cylindrical disc embedded in a double layer. This mobility depends on both the viscosity of the bulk and that of the layer. The approach of Saffman and Delbrück (27) was generalized to monolayers at interfaces by Fischer et al. (28) and Fuller and Vermant (29). These theories could be compared with the experiments of Sickert and Rondelez (30) on the diffusion of colloidal particles at monolayer-coated water-air interfaces.
Here, we give a rough estimate of the diffusivity of the PLL-PEG-bio rafts. We argue that because of steric hindrance between the PEG chains, the PLL chains are in a stretched configuration on the surface (fig. S3) (17, 31). The positively charged PLL will be bound to the SDS surfactants at the water-oil interface, where the hydrophobic tails of the surfactants extend into the roughly 50 times more viscous oil phase. We can estimate the mobility of such a chain by b = (6π(ηoil + ηwater)R)−1, where the length of the stretched PLL chains is ~48 nm, assuming that the length of a lysine monomer is 0.355 nm (17). This gives us a translational diffusion coefficient of DPPL ~ 1.8 × 10−13 m2s−1, which is about 1/10th of that of the free 0.52-μm particles (Dsol = 9.52 × 10−13 m2s−1) and close to those bound to the surface (DOD ~ 2.4 × 10−13 m2s−1). A slightly larger reduction is observed for the 1.2-μm particles, which agrees with our assumption that a larger raft of several PLL-PEG-bio chains may be bound in the contact region. Hence, the colloidal motion is dominated by the viscous drag of the monolayer raft with the oil. This finding agrees with the microrheology data, which suggest that the larger colloids experience an apparent viscosity of about 9.0 mPa s, whereas we extract a viscosity of ~3.5 mPa s from the DDM measurements of the bound smaller colloids.
Our results presented in Fig. 2 demonstrate that the surfactant concentration is an important ingredient in our system’s aggregation behavior. In the final OD-colloid mixture, we adjusted the SDS concentration in the aqueous bulk. For the buffer solution with 50 mM added NaCl concentration, a minimum of 1 to 2 mM SDS concentration was required to prevent coalescence of the ODs. This SDS concentration is just below the critical micelle concentration (CMC) for SDS, which is around 2.5 mM for the ionic strength used in our experiments. Hence, at higher SDS concentrations, we create more and more micelles, which induce increasingly stronger depletion attraction between the DNA-stabilized colloids that do not aggregate in the same salty buffer solution in the absence of any surfactant. Depletion attractions between large colloids in bulk solution, induced by small colloids or polymers, have been studied extensively in theory, simulations, and experiments (32–35). Fewer studies used charged surfactant micelles as the depleting agent. Iracki et al. (36) observed the aggregation behavior of hard, negatively charged silica beads sedimented onto a nonsticking glass surface in the presence of SDS. Similar to our experiments, they showed that below the CMC hard sphere, repulsion dominated, whereas an increasingly deeper attractive minimum emerged as the SDS concentration increased beyond the CMC. Again, our simulation results confirm that scenario.
Also, when we go to higher SDS concentrations, we see ordered 2D crystals with faceted boundaries. Similar to the observations by Meng et al. (37) made on depletion-driven colloidal aggregation on the inside of emulsion droplets, these crystals remain of a finite size and “fracture” because of the competition between elastic deformation of the 2D crystals that prefer a flat arrangement and the binding strength of the colloids to the interface via DNA. As the melting experiments demonstrate, any clusters formed at room temperature will become smaller and more mobile upon heating. To summarize, we have introduced a new colloid aggregation mechanism on emulsion droplets that allows reversible detachment from the OD surface in a controlled manner using DNA hybridization. The fact that the hybridization recruits the mobile linkers between the colloids and the OD may be used as a model system to understand and study the dynamics of particle or protein adsorption to biological molecules in a crowded environment. The very slow dynamics of the aggregation process may help in understanding how this aggregation can be controlled (18, 38).
MATERIALS AND METHODS
Coating droplets and colloids with DNA
The DNA strands used in experiments were purchased from Integrated DNA Technologies. The three sticky ends are as follows: A, 5′-CCGGCC-3′; A′, 5′-GGCCGG-3′; and B, 5′-CGCAGCACC-3′. The XX′ pair acted as rigid double-stranded spacer, as described in earlier work (39). They were hybridized before being attached to either the ODs (A) or the colloids (A′). The complete DNA strands are reported in table S1.
To achieve a similar DNA binding capacity/μm2 on the OD as that on the colloids, 50 μl of the freshly prepared ODs stabilized in 10 mM SDS solutions was taken from the creamed layer and mixed with 22.5 μl (1 mg/ml) of PLL (20)-g[3.5]–PEG(2)/PEG(3.4)–biotin(50%) (purchased from SuSoS AG) and 250 μl of 1 mM SDS, 4 mM tris buffer (pH 7.5), maintaining an overall 50 mM concentration of NaCl and >2 mM concentration of SDS. This mixture was incubated on rollers overnight, washed twice with a wash solution [5 mM SDS, 50 mM NaCl, 4 mM tris (pH 7 and 8)], and then dispersed in a suspending solution [2 mM SDS, 4 mM tris (pH 7 to 8), 50 mM NaCl]. Because the droplets float, we used a syringe to remove the buffer beneath them. Then, 3 μl (1 mg/ml) of Texas Red–labeled streptavidin (Sigma-Aldrich) per 50 μl of the droplet solution was added and incubated on rollers for another hour. Again, the droplets were washed twice with the wash solution and suspended in suspending solution. This procedure provided ODs with approximately the same streptavidin coverage (~104/μm2) as that on the colloids.
The hybridized DNA constructs with double-stranded spacers were grafted onto the ODs and the colloids using the biotin-streptavidin binding (40). A DNA was added to the suspension of streptavidin-coated ODs in 10× excess. They were allowed to bind on rollers in a 50 mM NaCl and 2 mM SDS solution. After 4 to 6 hours, the salt concentration was raised to 100 mM while maintaining the SDS concentration. After another 3 hours on the rollers, the Eppendorf tubes were allowed to stand vertically so that the ODs could float to the top and excess DNA and salt could be removed. The DNA-coated ODs were then washed three times using the wash solution and resuspended in the suspending solution.
Green fluorescent colloids (5 μl; D ≈ 0.5 μm; Microparticles GmbH) were diluted to 400 μl by adding TE buffer and then sonicated for 30 min before A′ DNA was added. The NaCl solution concentrate (1 M) was then added to this mixture, establishing an overall salt concentration of 100 mM. After this solution was incubated on the rollers for 4 to 6 hours, more NaCl solution was added, raising the NaCl concentration to 300 mM. This was allowed to stay on the rollers for another 4 hours. The colloids were then pelleted using a microcentrifuge, the supernatant was removed, and the DNA-coated colloids were then resuspended in fresh TE buffer (heated to 40°C). This washing protocol was repeated four to six times, thus removing excess DNA and salt present in the system (19, 40). The last two washes were done in TE containing 100 mM NaCl, thereby ensuring steric stability of the DNA brush attached to the colloids.
Finally, OD and colloid solutions were mixed and allowed to bind overnight on rollers—the final NaCl concentration used in all experiments was 50 mM. The desired SDS concentration was adjusted at this stage.
Sample chambers
Capillary chambers purchased from CM Scientific (0.2 × 4–mm inner diameter) were irradiated under an ultraviolet lamp for 30 min and then plasma-cleaned in an oxygen plasma oven for 2 min (Diener Electronics Femto). About 40 μl of the sample was filled into the capillary chambers using pipettes and then sealed and glued onto a glass slide using a two-component epoxy glue.
Imaging and temperature cycling
For imaging, we used a Nikon Ti-E inverted epifluorescence microscope with either a Nikon Plan Fluor E40×/0.75 dry objective or a Nikon Plan APO 60×/1.20 water immersion objective, a Grasshopper3 (Point Grey Research Inc.), Sony IMX174 CMOS sensor CCD camera, and a “perfect focus system,” allowing for long tracking measurements. A blue light-emitting diode source (485 nm; Cree XPEBLU) and white light in combination with a filter cube were used to excite the fluorescence on the colloids and ODs. The sample temperature was controlled with a computer using a home-made Peltier stage with a thermocouple. Typical heating and cooling rates were 1°C/min.
Image analysis and diffusivity measurements
Image conversion and analysis were done using customized ImageJ and Matlab routines. Single-particle diffusivity movies were analyzed, and tracks were generated using customized Matlab scripts and the Analyze subroutines of ImageJ. These tracks were subsequently analyzed in Matlab, generating mean square displacements to calculate the diffusivities. Several bright-field time series of 1- of 2-min durations were recorded and evaluated using a Matlab routine for DDM analysis developed by S. H. Nathan (22, 41). In DDM, bright-field (or fluorescence) microscope images separated by a given time lag are subtracted such that only the dynamic information on colloid motion remains. Fourier-transforming these difference images for varying time lags and correlating them provides the system’s relaxation time, τ = (Dq2)−1, as a function of the scattering wavelength q.
Supplementary Material
Acknowledgments
E.E. thanks M. Muthukumar for discussions. A.S.N., D.E.P.P., and N.A.M.A. acknowledge discussions with M. Telo da Gama. Funding: A.C. acknowledges support from the ETN-COLLDENSE (H2020-MCSA-ITN-2014, grant no. 642774). E.E. and J. Burelbach thank the Winton Programme for the Physics of Sustainability for the Pump Prime Grant and the scholarship award, respectively. D.J. thanks the Udayan Care-VCare grant, the Nehru Trust for Cambridge University, the Schlumberger Foundation’s Faculty for the Future Program, and Hughes Hall Santander Bursary Scholarship. Z.X. thanks the National University of Defense Technology Scholarship at Cambridge. A.S.N., D.E.P.P., and N.A.M.A. acknowledge financial support from the Portuguese Foundation for Science and Technology (FCT) (grants EXCL/FIS-NAN/0083/2012, UID/FIS/00618/2013, and IF/00255/2013). J. Brujic thanks the Materials Research Science and Engineering Center program of the National Science Foundation under Award DMR-1420073 and L. L. Pontani. Author contributions: D.J. did most of the experiments and contributed in writing the manuscript, whereas D.B. prepared the ODs and tested the DNA grafting to the ODs. D.J. and A.C. analyzed all experimental results. J. Burelbach supported the particle tracking analysis, and Z.X. contributed in determining the stability and the melt properties of the DNA linkers. A.S.N., D.E.P.P., and N.A.M.A. performed all simulation studies. J. Brujic advised on the OD preparation and contributed in writing the manuscript. E.E. designed and directed the research and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/8/e1600881/DC1
Supplementary Materials and Methods
fig. S1. Calibration curve for the surface coverage of PLL-PEG-bio.
fig. S2. Phase transition of the DNA-functionalized PS spheres.
fig. S3. Schematics of the oil-water interface.
fig. S4. Microrheology on the oil-water interface.
fig. S5. Schematic representation of the stochastic model.
fig. S6. Dependence of the binding density on the flux and diffusion coefficient.
fig. S7. Pairwise potential for different strengths AO potential.
table S1. Various DNA constructs used in the experiments.
video S1. Dynamics of DNA-anchored colloids in gas phase imaged with 30 frames/s, focusing on the south pole.
video S2. Dynamics of colloids aggregated into 2D crystals on OD surfaces imaged with 5 frames/s, focusing on the south pole.
video S3. Single 1-μm large colloids DNA-anchored to the OD imaged with 50 frames/s.
video S4. Single 1-μm large colloid diffusing freely in solution (kept in focus with a feedback mechanism), imaged with 50 frames/s.
Reference (42)
REFERENCES AND NOTES
- 1.Ramsden W., Separation of solids in the surface-layers of solutions and ‘suspensions’ (observations on surface-membranes, bubbles, emulsions, and mechanical coagulation)—Preliminary account. Proc. R. Soc. Lond. 72, 156–164 (1903). [Google Scholar]
- 2.Binks B. P., Murakami R., Phase inversion of particle-stabilized materials from foams to dry water. Nat. Mater. 5, 865–869 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Pickering, S. U. CXCVI.—Emulsions. J. Chem. Soc. Trans. 91, 2001–2021 (1907). [Google Scholar]
- 4.Dinsmore A. D., Hsu M. F., Nikolaides M. G., Marquez M., Bausch A. R., Weitz D. A., Colloidosomes: Selectively permeable capsules composed of colloidal particles. Science 298, 1006–1009 (2002). [DOI] [PubMed] [Google Scholar]
- 5.Herzig E. M., White K. A., Schofield A. B., Poon W. C. K., Clegg P. S., Bicontinuous emulsions stabilized solely by colloidal particles. Nat. Mater. 6, 966–971 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Lewandowski E. P., Bernate J. A., Tseng A., Searson P. C., Stebe K. J., Oriented assembly of anisotropic particles by capillary interactions. Soft Matter 5, 886–890 (2009). [Google Scholar]
- 7.Cavallaro M. Jr, Botto L., Lewandowski E. P., Wang M., Stebe K. J., Curvature-driven capillary migration and assembly of rod-like particles. Proc. Natl. Acad. Sci. U.S.A. 108, 20923–20928 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Madivala B., Vandebril S., Fransaer J., Vermant J., Exploiting particle shape in solid stabilized emulsions. Soft Matter 5, 1717–1727 (2009). [Google Scholar]
- 9.Mirkin C. A., Letsinger R. L., Mucic R. C., Storhoff J. J., A DNA-based method for rationally assembling nanoparticles into macroscopic materials. Nature 382, 607–609 (1996). [DOI] [PubMed] [Google Scholar]
- 10.Alivisatos A. P., Johnsson K. P., Peng X., Wilson T. E., Loweth C. J., Bruchez M. P. Jr, Schultz P. G., Organization of ‘nanocrystal molecules’ using DNA. Nature 382, 609–611 (1996). [DOI] [PubMed] [Google Scholar]
- 11.Nykypanchuk D., Maye M. M., van der Lelie D., Gang O., DNA-guided crystallization of colloidal nanoparticles. Nature 451, 549–552 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Geerts N., Eiser E., DNA-functionalized colloids: Physical properties and applications. Soft Matter 6, 4647–4660 (2010). [Google Scholar]
- 13.Hadorn M., Boenzli E., Sørensen K. T., Fellermann H., Hotz P. E., Hanczyc M. M., Specific and reversible DNA-directed self-assembly of oil-in-water emulsion droplets. Proc. Natl. Acad. Sci. U.S.A. 109, 20320–20325 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parolini L., Mognetti B. M., Kotar J., Eiser E., Cicuta P., Di Michele L., Volume and porosity thermal regulation in lipid mesophases by coupling mobile ligands to soft membranes. Nat. Commun. 6, 5948 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van der Meulen S. A. J., Leunissen M. E., Solid colloids with surface-mobile DNA linkers. J. Am. Chem. Soc. 135, 15129–15134 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Feng L., Pontani L.-L., Dreyfus R., Chaikin P., Brujic J., Specificity, flexibility and valence of DNA bonds guide emulsion architecture. Soft Matter 9, 9816–9823 (2013). [Google Scholar]
- 17.Rossetti F. F., Reviakine I., Csúcs G., Assi F., Vörös J., Textor M., Interaction of poly(L-lysine)-g-poly(ethylene glycol) with supported phospholipid bilayers. Biophys. J. 87, 1711–1721 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruiz-Taylor L. A., Martin T. L., Zaugg F. G., Witte K., Indermhule P., Nock S., Wagner P., Monolayers of derivatized poly(l-lysine)-grafted poly(ethylene glycol) on metal oxides as a class of biomolecular interfaces. Proc. Natl. Acad. Sci. U.S.A. 98, 852–857 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Di Michele L., Mognetti B. M., Yanagishima T., Varilly P., Ruff Z., Frenkel D., Eiser E., Effect of inert tails on the thermodynamics of DNA hybridization. J. Am. Chem. Soc. 136, 6538–6541 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Yanagishima T., Frenkel D., Kotar J., Eiser E., Real-time monitoring of complex moduli from micro-rheology. J. Phys. Condens. Matter 23, 194118 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Cerbino R., Trappe V., Differential dynamic microscopy: Probing wave vector dependent dynamics with a microscope. Phys. Rev. Lett. 100, 188102 (2008). [DOI] [PubMed] [Google Scholar]
- 22.E. Eiser, in Multi Length-Scale Characterisation, D. W. Bruce, D. O’Hare, R. I. Walton, Eds. (John Wiley and Sons Ltd., New York, 2014), pp 233–282. [Google Scholar]
- 23.Buzzaccaro S., Rusconi R., Piazza R., “Sticky” hard spheres: Equation of state, phase diagram, and metastable gels. Phys. Rev. Lett. 99, 098301 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Buzzaccaro S., Colombo J., Parola A., Piazza R., Critical depletion. Phys. Rev. Lett. 105, 198301 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Li W., Ma H. R., Depletion potential near curved surfaces. Phys. Rev. E 66, 061407 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Di Michele L., Yanagishima T., Brewer A. R., Kotar J., Eiser E., Fraden S., Interactions between colloids induced by a soft cross-linked polymer substrate. Phys. Rev. Lett. 107, 136101 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Saffman P. G., Delbrück M., Brownian motion in biological membranes. Proc. Natl. Acad. Sci. U.S.A. 72, 3111–3113 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fischer T. M., Dhar P., Heinig P., The viscous drag of spheres and filaments moving in membranes or monolayers. J. Fluid Mech. 558, 451–475 (2006). [Google Scholar]
- 29.Fuller G. G., Vermant J., Complex fluid-fluid interfaces: Rheology and structure. Annu. Rev. Chem. Biomol. Eng. 3, 519–543 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Sickert M., Rondelez F., Shear viscosity of Langmuir monolayers in the low-density limit. Phys. Rev. Lett. 90, 126104 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Kawaguchi S., Imai G., Suzuki J., Miyahara A., Kitano T., Ito K., Aqueous solution properties of oligo- and poly(ethylene oxide) by static light scattering and intrinsic viscosity. Polymer 38, 2885–2891 (1997). [Google Scholar]
- 32.Oosawa F., Asakura S., Surface tension of high-polymer solutions. J. Chem. Phys. 22, 1255 (1954). [Google Scholar]
- 33.Gast A. P., Hall C. K., Russel W. B., Polymer-induced phase separations in nonaqueous colloidal suspensions. J. Colloid Interface Sci. 96, 251–267 (1983). [Google Scholar]
- 34.Hagen M. H. J., Frenkel D., Determination of phase diagrams for the hard-core attractive Yukawa system. J. Chem. Phys. 101, 4093–4097 (1994). [Google Scholar]
- 35.Pusey P. N., Pirie A. D., Poon W. C. K., Dynamics of colloid-polymer mixtures. Physica A 201, 322–331 (1993). [Google Scholar]
- 36.Iracki T. D., Beltran-Villegas D. J., Eichmann S. L., Bevan M. A., Charged micelle depletion attraction and interfacial colloidal phase behavior. Langmuir 26, 18710–18717 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Meng G., Paulose J., Nelson D. R., Manoharan V. N., Elastic instability of a crystal growing on a curved surface. Science 343, 634–637 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Marenduzzo D., Finan K., Cook P. R., The depletion attraction: An underappreciated force driving cellular organization. J. Cell Biol. 175, 681–686 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Varrato F., Di Michele L., Belushkin M., Dorsaz N., Nathan S. H., Eiser E., Foffi G., Arrested demixing opens route to bigels. Proc. Natl. Acad. Sci. U.S.A. 109, 19155–19160 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Di Michele L., Varrato F., Kotar J., Nathan S. H., Foffi G., Eiser E., Multistep kinetic self-assembly of DNA-coated colloids. Nat. Commun. 4, 2007 (2013). [DOI] [PubMed] [Google Scholar]
- 41.S. Nathan, thesis, University of Cambridge (2015). [Google Scholar]
- 42.Ramos L., Fabre P., Swelling of a lyotropic hexagonal phase by monitoring the radius of the cylinders. Langmuir 13, 682–686 (1997). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/8/e1600881/DC1
Supplementary Materials and Methods
fig. S1. Calibration curve for the surface coverage of PLL-PEG-bio.
fig. S2. Phase transition of the DNA-functionalized PS spheres.
fig. S3. Schematics of the oil-water interface.
fig. S4. Microrheology on the oil-water interface.
fig. S5. Schematic representation of the stochastic model.
fig. S6. Dependence of the binding density on the flux and diffusion coefficient.
fig. S7. Pairwise potential for different strengths AO potential.
table S1. Various DNA constructs used in the experiments.
video S1. Dynamics of DNA-anchored colloids in gas phase imaged with 30 frames/s, focusing on the south pole.
video S2. Dynamics of colloids aggregated into 2D crystals on OD surfaces imaged with 5 frames/s, focusing on the south pole.
video S3. Single 1-μm large colloids DNA-anchored to the OD imaged with 50 frames/s.
video S4. Single 1-μm large colloid diffusing freely in solution (kept in focus with a feedback mechanism), imaged with 50 frames/s.
Reference (42)