

Abstract
Context
There is no current standard among myopathologists for reporting muscle biopsy findings. The National Institute of Neurological Disorders and Stroke has recently launched a common data element (CDE) project to standardize neuromuscular data collected in clinical reports and to facilitate their use in research.
Objective
To develop a more-uniform, prospective reporting tool for muscle biopsies, incorporating the elements identified by the CDE project, in an effort to improve reporting and educational resources.
Design
The variation in current biopsy reporting practice was evaluated through a study of 51 muscle biopsy reports from self-reported diagnoses of genetically confirmed or undiagnosed muscle disease from the Congenital Muscle Disease International Registry. Two reviewers independently extracted data from deidentified reports and entered them into the revised CDE format to identify what was missing and whether or not information provided on the revised CDE report (complete/incomplete) could be successfully interpreted by a neuropathologist.
Results
Analysis of the data highlighted showed (1) inconsistent reporting of key clinical features from referring physicians, and (2) considerable variability in the reporting of pertinent positive and negative histologic findings by pathologists.
Conclusions
We propose a format for muscle-biopsy reporting that includes the elements in the CDE checklist and a brief narrative comment that interprets the data in support of a final interpretation. Such a format standardizes cataloging of pathologic findings across the spectrum of muscle diseases and serves emerging clinical care and research needs with the expansion of genetic-testing therapeutic trials.
The muscle biopsy is an essential, and frequently used, diagnostic tool in the evaluation of pediatric and adult neuromuscular disorders, but its usefulness may be limited by poor communication from ordering clinicians or by incomplete reporting of pathologic findings on the biopsy report.1,2 The diagnostic workup of patients with neuromuscular disease requires a comprehensive evaluation of the patient’s clinical presentation, laboratory abnormalities, and pathologic findings. A thorough clinical history and examination by a neuromuscular expert, complemented by laboratory, and potentially by imaging studies, is critical in formulating the appropriate diagnostic question for the pathologist and in selecting the appropriate muscle to biopsy. The diagnostic question should be formulated before ordering the muscle biopsy to increase the pretest probability of identifying any given neuromuscular disorder and to improve the overall diagnostic yield of the procedure. It is, therefore, critical that this diagnostic question (and the information that led to its formation) be communicated to the pathologist to ensure that all pertinent positive and negative findings are reported and communicated back to the clinician. Even in the absence of a clear pathologic diagnosis, the reporting of both positive and negative findings is essential to guiding the clinician’s additional testing toward achieving a diagnosis.
Uniform and detailed reporting on muscle biopsies beyond a diagnostic impression also supports the common data element (CDE) project currently implemented through the National Institute of Neurological Disorders and Stroke (NINDS).3–8 The purpose of the CDE project is to standardize the collection of clinical data and to facilitate the comparison of results across studies. Standard data collection also supports effective data aggregation from multiple centers and studies, which is often required to adequately power analyses in rare neuromuscular disorders. The CDE standards have recently been developed for muscle biopsy reporting, with the purpose of making the reports maximally useful for supporting diagnostic classification, patient clinical-trial stratification, and research.9 This standard and comprehensive approach to muscle-biopsy reporting will help filter large, genetic-testing data sets according to phenotypic and biopsy markers when whole exome/genome sequencing technology is increasingly applied to neuromuscular disorders.
The lack of standard practices in muscle-biopsy reporting provides challenges when training new myopathologists and when attempting to evaluate patients across multiple institutions. The goal of this project was to develop a prospective format for muscle biopsy reporting that incorporates the features identified by the CDE project. A retrospective study of 51 muscle-biopsy reports from the Congenital Muscle Disease International Registry (CMDIR) was performed to assess the use of diagnostic terminology in muscle biopsies across numerous centers. Our analysis of data-element reporting highlighted (1) the importance of communication among the clinical team involved in the patient’s care (the ordering physician, surgeon/neurologist performing the biopsy, and myopathologist); (2) the need for, and usefulness of, a standardized approach (grounded in a CDE form) to muscle-biopsy reporting in patients with neuromuscular diseases; and (3) the situations in which a standard checklist approach alone may not communicate the information essential to making a diagnosis. This information was used to develop a prospective muscle-biopsy reporting form composed of checklist and narrative elements that will ideally improve physician communication, provide educational resources for myopathologists in training, and facilitate efficient and complete reporting of muscle-biopsy findings.
MATERIALS AND METHODS
Fifty-one deidentified muscle-biopsy reports were selected from the CMDIR.10 The CMDIR is the largest global database of patients with congenital muscle disease, which includes congenital myopathy, congenital muscular dystrophy, and congenital myasthenic syndromes. Of CMDIR-registered patients for whom muscle biopsy reports were available, reports were selected randomly from patients with a self-reported diagnosis of congenital muscular dystrophy (n = 34), congenital myopathy (n = 11), and/or an undiagnosed form of congenital-onset disease (n = 6). Of these 51 reports, 43 reports (84%) were primary muscle biopsy reports and 8 (16%) were consult or second-opinion reports from the same patient pool (Table 1). To prevent overrepresentation by a particular myopathologist or site, only the 43 primary muscle-biopsy reports were used in our statistical analysis.
Table 1.
Primary Muscle Biopsy Review Sites
| Country | Institution | Reports Evaluated, No. |
|---|---|---|
| Australia | Children’s Hospital at Westmead, Westmead, New South Wales | 2 |
| Canada | Calgary Laboratory Services, Calgary, Alberta | 2 |
| Manitoba Health Sciences Centre, Winnipeg, Manitoba | 2 | |
| Germany | Muscle-Centre Gottingen, Germany, Gottingen | 1 |
| Russia | Moscow Research Institute of Pediatrics and Pediatric Surgery, Moscow | 1 |
| South Africa | National Health Laboratory Service–Constantiaberg Medical Centre, Cape Town | 1 |
| United Kingdom | Dubowitz Neuromuscular Centre, London | 1 |
| United States | Arkansas Children’s Hospital, Little Rock, Arkansas | 1 |
| Barrow Neurological Institute, Phoenix, Arizona | 1 | |
| Rady Children’s Hospital, San Diego, California | 4 | |
| University of California, San Francisco Medical Center, San Francisco | 1 | |
| Children’s Hospital Colorado, Denver | 1 | |
| Miami Children’s, Miami, Florida | 1 | |
| Orlando Regional Medical Center, Orlando, Florida | 1 | |
| University of Iowa, Iowa City | 1 | |
| Children’s Boston, Boston, Massachusetts | 1 | |
| Spectrum Health Michigan, Grand Rapids | 1 | |
| University Hospitals and Clinics, Jackson, Michigan | 1 | |
| University of Minnesota Neuromuscular Laboratory, Minneapolis | 1 | |
| Washington University, St. Louis, Missouri | 1 | |
| Nebraska Medical Center, Omaha | 1 | |
| University of Medicine and Dentistry, New Jersey, Newark | 1 | |
| Tricore, Clovis, New Mexico | 1 | |
| Columbia University, New York, New York | 2 | |
| Mount Sinai Laboratory, New York, New York | 1 | |
| Cincinnati Children’s, Cincinnati, Ohio | 1 | |
| Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania | 1 | |
| Wesley Neuromuscular Laboratory, Memphis, Tennessee | 1 | |
| University of Texas Southwestern Medical Center, Dallas | 1 | |
| Scottish Rite Hospital, Dallas, Texas | 4 | |
| Froedtert Memorial Lutheran Hospital, Milwaukee, Wisconsin | 1 | |
| Not reported | Not reported | 2 |
| Total: 43 |
An adapted CDE form was then created; the revised CDE report (CDE-R), and key elements of that form are shown in Figures 1 and 2. The CDE-R form used in this study was derived from the current NINDS CDE form for muscle biopsies and was expanded on, with consensus agreement, among the authors, who were involved in the early stages of this study. Two reviewers subsequently and independently extracted data from the deidentified reports and entered them into the CDE-R format. Elements of the report that lacked the information required by the CDE-R form were also noted. The reviewers then met with a neuropathologist to evaluate discrepancies in how biopsy-report elements were scored. Inconsistencies were then recognized as errors on the part of the reviewer versus difficulty with interpreting what was described in the report. If there was a difficulty with interpretation, a discussion followed in an effort to come to an agreement on what was being stated by the myopathologist/author of the report.
Figure 1.
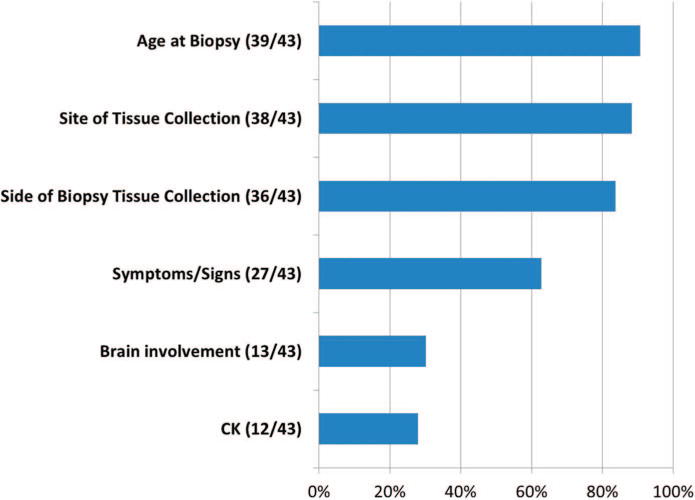
Percentage of clinical findings included in the muscle-biopsy report.
Figure 2.
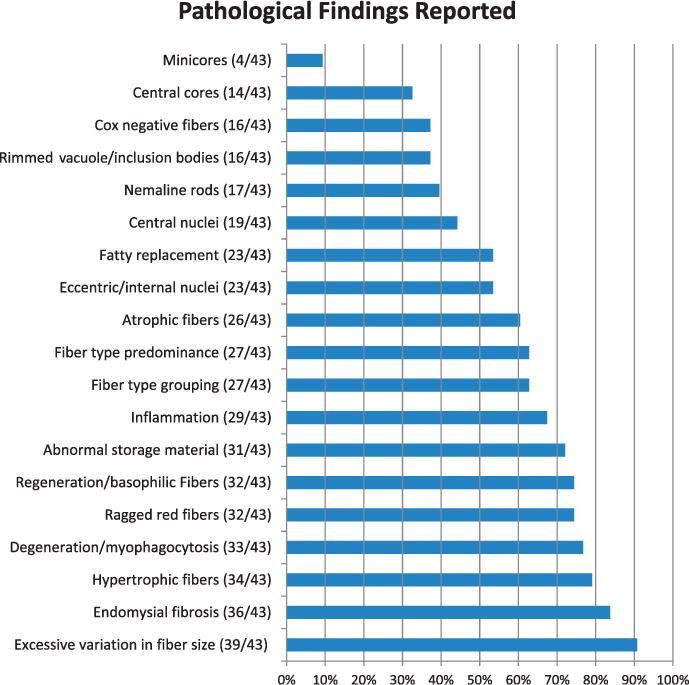
Percentage of key pathologic findings reported. This reflects whether the finding was mentioned in the report, regardless of whether the finding was present or absent within the actual biopsy.
Once an agreement was made on the CDE-R data acquired by the 2 reviewers, the deidentified, extracted data elements (in the absence of the authoring pathologist’s interpretation) were then reviewed independently by a neuropathologist, and a checklist-based interpretation was made. This exercise was performed to assess whether the information required by the CDE-R was sufficient for a neuropathologist to suggest a diagnosis that was consistent with the original report.
The data from this study were presented at several national and international conferences, including the 2013 Muscular Dystrophy Association National Scientific Conference, the 2013 Annual Meeting of the Child Neurology Society, the 2013 Annual Meeting of the American Association of Neuropathologists, the 2013 Annual Meeting of the Canadian Association of Neuropathologists, and the 2013 World Muscle Society Congress. The reporting tool was also made available online as a posting on a neuropathology blog site (http://neuropathologyblog.blogspot.com/; accessed March 23, 2015) and via e-mail to anyone who requested it. Feedback on the details of the form was returned to the corresponding author, and a comprehensive version of the CDE-R form, which includes suggestions from the neuropathology community, is shown in Appendix 1. This additional input focused on suggesting data elements that were not present in either the original CDE form or in the reviewed biopsy reports, and thus, a rereview of the biopsy reports with these additional fields would not have affected the interpretations reported here.
Biopsy-report data and the reporting of individual data elements were evaluated using Microsoft Excel software (version 2010, Microsoft, Redmond, Washington). Agreement analysis using the κ coefficient11,12 was performed by staff at the Quantitative Health Sciences Division of Pediatrics, Medical College of Wisconsin (Milwaukee), using SAS 9.2 software (SAS Institute Inc, Cary, North Carolina). The κ coefficient measures the interrater agreement for categorical variables. The Cohen κ adjusts the proportion of agreement between the 2 raters for the potential agreement that could occur by chance. Its value ranges from −1 to +1, with values of zero or less indicating no agreement and values greater than 0.75 indicating strong agreement.
RESULTS
Biopsy Report Features
Because the 51 biopsy reports reviewed were supplied by the CMDIR, there were notable sampling biases associated with the patient location, age, clinical history, and final diagnosis in these cases.10 A subset of 43 muscle biopsy reports from the primary institutions (rather than the 8 consult reports that were found) were evaluated to prevent overrepresentation of reported elements by a specific site or level of expertise when evaluating the range of reporting. Although most biopsy reports were from institutions in the United States, international locations included Westmead, Australia; London, United Kingdom; Alberta, Canada; Gottingen, Germany; Moscow, Russia; and Cape Town, South Africa (Table 1). There is no “universally accepted,” minimal set of stains that are used for muscle biopsy evaluation in the United States, and so failure to report certain elements may have been affected by the unavailability of stains at some institutions.
Patient ages ranged from 0 to 38 years (mean [SD], 5.4 [8.5] years). The most-common elements of the clinical history provided on the muscle-biopsy report included age, site of tissue collected, side on which the tissue was collected, and symptoms and signs (Figure 1). Evaluation of the reported clinical data, which is often a reflection of the degree of communication between the ordering physician and the pathologist, revealed that demographic information and the features of the specimen (such as the site and side of tissue collected) were far more consistently reported than were other important elements of the patient history.
Diagnoses on the muscle-biopsy reports included a variety of dystrophies, myopathies, and neuropathic conditions (Figure 3). Thirteen of these cases (30%, 13 of 43) had a definitive diagnosis established by the muscle biopsy or subsequent genetic testing, whereas others showed dystrophic or myopathic features without a specific etiology identified.
Figure 3.
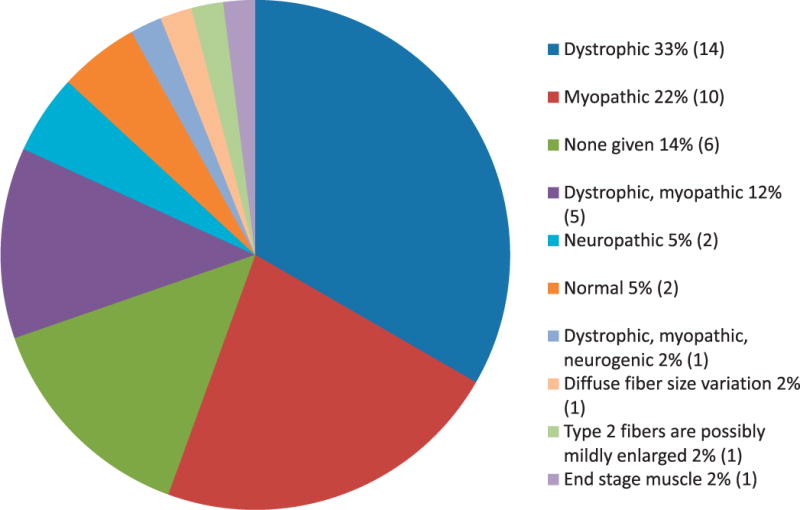
Range of diagnoses in reviewed reports. Note that the numbers reflect the original interpretations of the biopsy reports and do not reflect the degree to which specific elements were reported. The numbers of reports corresponding to each diagnosis are shown in parentheses (n=43 cases reviewed).
Reporting of Data Elements
Biopsy reports were scored for their reference to data elements on the CDE form, regardless of whether the findings were reported as positive or negative. Reporting of both positive and negative findings was highly variable (Figures 1 and 2), with a higher percentage of positive findings reported than negative findings. None of the essential CDEs in the CDE-R were reported in 100% (43 of 43) of the reviewed muscle-biopsy reports.
Evaluation of the CDE Checklist as a Diagnostic Tool
Two medical professionals independently reviewed and extracted data elements from each report to the CDE-R, and disagreements between the 2 observers were evaluated and discussed with a neuropathologist blinded to the ultimate muscle-biopsy diagnosis. Sources of disagreement in the biopsy report evaluation included (1) variable terminology used by myopathologists to signify similar findings (basophilic fibers with central nuclei versus regenerating fibers, myofiber necrosis versus myofiber degeneration), (2) the lack of precise and consistent application of defined nomenclature on the part of the reporting myopathologist (atrophy does not equal hypotrophy, central nuclei do not equal eccentric or internal nuclei), and (3) single reviewer error in identifying a key CDE in the body of report. A diagnostic stratification scheme was developed to account for the range of phenotypes that may be reported using the CDE-R checklist (Table 2), and diagnoses were assigned for each report using that system. After a consensus was reached on the content found in each muscle-biopsy report using the CDE-R, a neuropathologist reviewed the data in a tabular format, blinded to the ultimate reported muscle biopsy diagnosis and provided an interpretation for each case using the categories listed in Table 2. An agreement analysis was then performed to compare the original muscle biopsy diagnosis and a diagnosis made only using CDE-R checklist format.11,12 This analysis found a high degree of concordance when using the CDE-R checklist format alone in 37 of the 43 cases (86%; with an overall κ of 0.796, and a κ for the main category of 0.792). The reasons for disagreements between the report and the checklist are shown in Table 3. Of the 43 primary muscle-biopsy reports reviewed, 13 (30%) belonged to patients with confirmed genetic diagnoses or with a pathologically defined subset of congenital muscle disease (such as nemaline myopathy). Of those 13 patients, the definitive diagnosis was suggested in the narrative reports of 7 patients (54%) and in the checklist interpretation of 6 patients (46%). The single case (8%) in which the narrative report provided better prediction of the involved gene than the checklist did was a centronuclear myopathy case with fibrosis that appeared dystrophic by a checklist interpretation, and in which a mutation in MTM1 was found. The nature of this report review (involving the review of reported results without access to slides), however, is likely to have additionally impaired our “checklist-only” interpretation because the review of actual slides may have resulted in differences in checklist entries by the study neuropathologist.
Table 2.
Diagnosis Stratification Scheme for Agreement Analysis
| Primary Category | Secondary Category | Tertiary Category |
|---|---|---|
| 1. Myopathic changes | A. Muscular dystrophy; cause identified | i(a) Dystrophin deficient |
| B. Muscular dystrophy; unclassified | i(b) a-dystroglycan deficient | |
| C. Minimal changes with abnormal IHC | i(c) Sarcoglycan deficient | |
| i(d) Merosin deficient | ||
| i(e) Collagen VI deficient | ||
| i(f) Dysferlin deficient | ||
| i(g) Caveolin-3 deficient | ||
| i(h) Emerin deficient | ||
| i(i) Normal IHC | ||
| i(j) Nondiagnostic IHC | ||
| D. With specific changes | ii(a) Internalized nuclei | |
| ii(b) Central nuclei | ||
| ii(c) Nemaline rods | ||
| ii(d) Central cores | ||
| ii(e) Multi/minicores | ||
| ii(f) Disorganized myofibrils | ||
| ii(g) Inclusions | ||
| ii(h) Rimmed vacuoles | ||
| ii(i) Ragged, red fibers | ||
| ii(j) Inflammation | ||
| ii(k) Abnormal storage material | ||
| E. With nonspecific changes | ||
| 2. Neuropathic changes | F. Neuropathic changes, sufficient to explain clinical phenotype | |
| G. Neuropathic changes, insufficient to explain clinical phenotype | ||
| 3. Mixed, with myopathic and neuropathic changes | Use categories listed above | Use categories listed above |
| 4. End-stage muscle | ||
| 5. NDAR, with adequate reporting of pathologic findings | ||
| 6. Noninformative report, unclear whether disease was present |
Abbreviations: IHC, immunohistochemistry; NDAR, No diagnostic abnormality recognized.
Table 3.
Discrepancies Between Original Narrative Report Diagnosis and Checklist-Based Diagnosis
| Original Interpretation | Checklist Interpretation | Comment |
|---|---|---|
| Centronuclear myopathy | Dystrophy | A myotendinous insertion site produced focal increases in endomysial fibrosis, making the checklist profile appear more dystrophic. |
| Centronuclear myopathy | Dystrophy | Three cases called centronuclear myopathy on narrative reports also showed diffuse, mild endomysial fibrosis; degree of central nucleation versus internal nucleation was unclear on reports; one of these cases had a confirmed mutation in MTM1. |
| Active myopathy | Dystrophy | A case of dystrophy, in which the narrative report used somewhat nonstandard diagnostic terminology. |
| Dystrophy, α-dystroglycan deficient | Dystrophy, cause unclear | A case with reported decreases in merosin and α-dystroglycan; narrative report seemed to diagnose α-dystroglycanopathy as the relevant abnormality on clinical grounds. |
| Dystrophy, cause unclear | Dystrophy, merosin deficient | Merosin was abnormal per report, but outside consultation discounted the significance of the abnormal finding. |
Development of the Prospective Reporting Form
The muscle biopsy reporting elements listed by the NINDS CDE project formed the basis of our first draft of the prospective reporting form, and the list of important elements was further expanded after reviewing the language observed in the retrospective report review. These elements were then fashioned into a prospective reporting format that combined a checklist and narrative elements, and this form underwent further review by several international experts who each made their own additions. The form was then presented at several international conferences (Muscular Dystrophy Association Meeting 2013, Washington, DC; American Academy of Neuropathology 2013, Charleston, South Carolina; Child Neurology Society 2013, Austin, Texas) and was made available online (http://neuropathologyblog.blogspot.com/; posted June 28, 2013; last comment made July 24, 2013; still accessible March 23, 2015), eliciting further comments and additions. Comments were received via approximately 50 conversations during presentations and approximately 10 e-mail communications. Diagnosticians who were already expert myopathologists generally stated an unwillingness to switch reporting formats but also usually recognized the usefulness of this checklist as a training or reference tool for diagnosticians with limited muscle-biopsy exposure. In contrast, diagnosticians with less muscle-biopsy exposure or test volume responded very favorably to the existence of this resource. The reporting tool was also generally thought to be suitable for research documentation and was potentially highly useful for future informatics studies. The additions to the checklist suggested by the myopathology community served to take a form that was primarily focused on pediatric disease and make it more inclusive. Approximately one-third of myopathologists who provided feedback on the form remarked that it was too long for practical clinical use.
COMMENT
Although muscle biopsies have been available and refined as a diagnostic tool for decades, recent advances in genetic testing and prospects for therapy are changing the way in which muscle-biopsy reports are used. The testing required to achieve a definitive diagnosis may not be known at the time of the initial biopsy reporting because genetic confirmation of the myopathologist’s initial suggestions often requires weeks of additional testing. Clinicians and scientists often depend on the presence or absence of specific muscle-biopsy pathologic findings when deciding on subsequent genetic testing and/or inclusion of patient samples in novel gene-discovery platforms. Because this information is also critical in research, clinical trials, and in the meta-analyses across studies, the NINDS has launched the CDE program to standardize data collection and reporting. An evaluation of the current reporting practices in myopathology is an important initial step to increase the usefulness of muscle-biopsy reporting for both diagnostic and research purposes, particularly as more genetic diagnoses are made in patients with neuromuscular disorders. A standard and unbiased reporting format for muscle biopsies offers the advantage of providing a complete list of core pathologic findings and pertinent negatives that will assist in (1) the establishment of a properly prioritized approach to genetic testing, based on muscle-biopsy core elements; (2) the correlation of broad-spectrum genetic-testing data to muscle histopathologic phenotypes; (3) the establishment of clear-cut pathologic criteria for patient inclusion into research platforms; and (4) the training of new myopathologists in the identification of pertinent pathologic findings.
Advantages and Limitations of a Checklist Muscle Biopsy Report Format
The CDE checklist format for muscle-biopsy reporting offers the advantage of being standard and quick to complete, which increases the likelihood that the full battery of core positive and negative findings will be reported by all myopathologists who use it. Although the list of core findings is not all inclusive, it is composed of features that are identifiable using the standard battery of histochemical tests used by most institutions and which most myopathologists routinely check on each biopsy. Of note, there are situations in which the checklist format falls short in suggesting a diagnosis. In the context of our data set (which was heavily biased toward cases of congenital muscular dystrophy), the main discrepancies occurred when evaluating the significance of fibrosis, internal/central nucleation, and inflammation. Discrepancies occurred in 3 cases of centronuclear myopathy when using the checklist alone because it was unable to distinguish between internal and central nucleation resulting from reparative changes versus the presence of those issues from a primary abnormality of nuclear positioning. Without access to the original slides, we could not determine whether additional data elements or a different narrative description would have assisted with this distinction. Additionally, one report described increased fibrosis in the context of a myotendinous insertion site, which led to the misinterpretation on the checklist review of endomysial fibrosis related to a dystrophic process. Because myotendinous insertion sites can cause local increases in fiber-size variation, increases in internally nucleated fibers, and increases in fibrosis, the presence of such a site in a biopsy specimen and the relationship of observed pathologic findings to this site should be mentioned in a narrative description in the case.1 Lastly, because muscular dystrophy and inflammatory myopathy can both show inflammation, fibrosis, and internal nucleation, there is certainly some potential for difficulty when using a checklist approach to distinguish these entities. In cases in which this differential diagnosis is a concern, it would be necessary to provide some sort of narrative description of these findings to more-clearly describe how these findings contribute to the myopathologist’s overall interpretation.
Use of the CDE-R Form as a Prospective Reporting Tool
Using a combination of the NINDS CDE form, our discussions during this study, and the feedback from the neuropathology community, we have generated a form for prospective muscle-biopsy reporting, which is shown in Appendix 1. This form offers a comprehensive list of the features that may be present on muscle biopsies (defined in Appendix 2) and provides the opportunity to use a checklist to inventory microscopic findings and a narrative segment to provide an interpretation. This combination of reporting formats offers the opportunity to be complete in dataelement reporting, while offering flexibility when providing a diagnostic interpretation. Our approach here was to provide a form that was as comprehensive and editable (to the preference of the myopathologist using it). This reporting format offers the advantages of organization, completeness, and standard terminology but remains untested in its usefulness as a prospective reporting tool. At the very least, the form provides a resource that will be useful in identifying clinical and pathologic features that affect the ability to reach a specific diagnosis. Even if this form is only used as a training or reference tool, it could thus still facilitate better clinical communication and practice. Concerns that have been raised about standardizing muscle-biopsy reporting include (1) the length of time required to report in this fashion, (2) that the checklist format will be too constraining, and (3) that the development of this form may accidentally promote a requirement for its use by third parties or compliance entities. Each of these issues represents a valid concern, but the fact remains that the current variation in current muscle-biopsy reporting practices impairs our capacity to provide expert-level, pathologic evaluations on an international scale. This project is a first, small effort to provide materials for the efficient and complete reporting of muscle-biopsy findings, and our future work will focus on the refinement of this reporting tool based on the input of our own experience and the feedback of the international myopathology community. A long-term, international, collaborative effort on this scale will also facilitate the identification of additional issues in myopathology for which the establishment of additional reporting or training materials would be most useful.
Future Directions
This project represents the first step toward developing a useful, widely distributed, synoptic reporting tool for muscle biopsies. We expect that the tool described here can be further refined and optimized after some field testing in clinical practice, and several groups have offered to use this reporting format on future cases as a means of further development. After several years of using this reporting format, we will request feedback on its usefulness to further refine it and to optimize its usefulness. Ideally, this will offer the opportunity to compare the degree to which this effort has standardized the reporting of pathologic findings while assessing whether the use of this reporting format has improved quantifiable aspects of the diagnostic workup for muscle disease. Additionally, the reporting format proposed here will facilitate the inclusion of muscle-biopsy data into multi-institutional databases, which may allow for the correlation of clinical or genetic data with specific pathologic patterns. This may facilitate the recognition of rare disease phenotypes and allow the contextualization of abnormalities detected in large-scale, genetic data sets to improve the diagnostic process in rare muscle disorders.
Acknowledgments
We thank Steve Moore MD, PhD, for his helpful comments during the course of this project. We also thank Brian Moore, MD, Med, and the American Association of Neuropathologists for distributing and/or publicizing the prospective muscle-biopsy reporting form to the neuropathology community and the numerous members of the neuropathology community who offered advice to improve the usefulness of our reporting form. This work was supported by the US National Institutes of Health [grants K08 AR059750 and L40 AR057721], Cure CMD, and the Children’s Hospital of Wisconsin Research Institute (Milwaukee).
Dr Sewry receives book royalties from Elsevier (Kidlington, Oxford, United Kingdom) and Wiley Blackwell (Oxford, Oxfordshire, United Kingdom) and an honorarium from Audentes Therapeutics, Inc (San Francisco, California).
APPENDIX 1: The Muscle-Biopsy Common Data Element–Revised (MB CDE-R) Form





APPENDIX 2: Definition of Pathologic Findings in the Microscopic Description
Whole-Fiber or Cytoplasmic Changes
Atrophy
Shrinkage of myofibers, usually resulting in a small fiber with a round or angulated shape. This can be difficult to distinguish from hypotrophy, which displays myofiber smallness with fiber shapes that are round or shaped appropriately. Advanced atrophy can cause nuclear bags, clumps, or clusters.
Blood Vessel Deposits Suggestive of Amyloid
Blood vessel wall thickening, including the presence of amorphous glassy material.
Cylindric Spirals
Spiral structures that are morphologically similar to honeycomb structures but more electron dense and are best seen on preparations for electron microscopy. Cylindric spirals stain red on Gomori trichrome stain and display positive staining for NADH and myoadenylate deaminase. They are not associated with a specific disease.
Degenerating Fibers
Degenerative changes in myofibers include loss of basophilic stippling or evidence of myofibrillar disarray.
Endomysial Fibrosis
Fibrosis in the endomysial compartment (between individual muscle fibers).
Excessive Glycogen
Subjective determination of excessive glycogen, usually on periodic acid–Schiff (PAS) staining. Severe glycogen deposition can also be apparent on hematoxylin-eosin (H&E) and other histochemical stains when contractile elements and organelles are displaced by glycogen aggregates.
Excessive Lipid
Subjective determination of excessive lipid, usually on oil red O staining. The appearance of excessive lipid is dependent on the preparation, given that stain exposure times vary between institutions. Comparison to a control specimen and/or correlation with ultrastructural findings can be very helpful. Severe lipid deposition can also be apparent on H&E and other histochemical stains when contractile elements and organelles are displaced by lipid.
Fatty Infiltration
Presence of adipocytes in the endomysial compartment (between individual muscle fibers).
Fiber Type Disproportion
This term refers to a pathologic pattern displaying type-1 fibers that are significantly smaller in diameter than type-2 fibers. The degree of myofiber smallness required varies in the literature between 12% and 25%. There is also usually type-1 fiber predominance in the specimen, but this is not strictly required. The pathologic pattern of fiber type disproportion is present in a number of muscle disorders (particularly congenital myopathies). When this pattern is seen in the absence of findings that would confer a specific pathologic diagnosis (such as nemaline rods or centrally nucleated fibers), then the pathologic diagnosis of congenital fiber type disproportion may be warranted. The recognition of fiber type disproportion is useful because it suggests specific subsets of causative genes in certain clinical and pathologic contexts.
Fiber Type Grouping
The presence of groups of type-1 fibers and groups of type-2 fibers within a single sampled muscle, which is indicative of repetitive rounds of denervation and reinnervation in muscle with neuropathic findings. The precise definition of grouping can vary, but groups of a given fiber type correspond to areas in which a single fiber is completely surrounded by fibers of the same type.
Grouped Atrophy
Atrophy of a cluster of adjacent fibers, usually recognized on H&E stain. This is suggestive of neuropathic disease, particularly when grouped atrophy is coexistent with fiber-type grouping.
Hypertrophic Fibers
Excessively large fibers, usually with a convex, “inflated” shape. The actual caliber of a hypertrophic fiber is dependent on the normal size of myofibers within that muscle (which will vary in different muscles and at different ages).
Hypotrophy
Insufficient fiber growth as a cause of small myofiber size. This usually manifests as small myofibers with round or somewhat normal shapes.
Lobulated/Trabecular Fibers
Fibers with uneven clumping of oxidative enzyme staining.
Moth-Eaten Fibers
Irregular disruption of the myofibrillar network. They can be seen on all stains but may be mistaken for minicores or cores on NADH or succinate dehydrogenase (SDH). They are often seen in dystrophies and various myopathies.
Muscle Spindles
Presence of characteristic muscle spindle apparati within the specimen.
Myophagocytosis
Invasion of the myofiber by a macrophage.
Myotendinous Insertion Sites
Presence of myotendinous insertion sites, which are often marked by the well-organized collagen of the tendon interfacing with myofibers. The myofibers in these areas often show excessive fiber size variation and internal nucleation, which is not a pathological finding in this context.
Necrosis
Necrotic myofibers have lost their basophilic stippling to the extent that mitochondria are no longer visible. Macrophages may also be invading the myofiber.
Nemaline Rods/Bodies
Cytoplasmic aggregates of thin filament material that are best visualized on Gomori trichrome stain (with red-purple staining) or preparations for electron microscopy (as osmiophilic material).
Nonrimmed Vacuoles
Vacuoles within myofibers that do not show red staining at their periphery on Gomori trichrome staining.
Perifascicular Distribution
Distribution of pathology around the edge of a muscle fascicle, with comparative sparing of the interior of the fascicle. Perifascicular myofiber atrophy in conjunction with degenerating and regenerating fibers and inflammatory cells is suggestive of dermatomyositis.
Red Inclusions on Trichrome
A variety, if intracytoplasmic inclusions and myofibrillar aggregates can form red inclusions on Gomori trichrome stain. Electron microscopy, oxidative enzyme stains, or immunohistochemistry can often be useful in identifying the material.
Regenerating Fibers (Basophilic Fibers, Large Nuclei)
These fibers are most frequently regenerative fibers, although there is some overlap with the myofiber disarray that is seen in degenerating fibers.
Rimmed Vacuoles
Areas of clearance on Gomori trichrome stain that are rimmed with red material. These structures are suggestive of the inclusion bodies seen in inclusion body myositis and hereditary inclusion body myopathy.
Ring Fibers/Ringbinden
Myofibers with a center of appropriately arranged, contractile apparatus that is surrounded by contractile elements arranged in a circumferential fashion.
Split Fibers
Fibers that appear to split, particularly when they become hypertrophic. Nuclei are noted to migrate along the split. Split fibers are normally seen at tendinous insertions and may be a feature of dystrophies, myopathies, and some chronic neuropathies.
Targetoid Fibers
An abnormality of oxidative staining (NADH, SDH, cytochrome oxidase [COX]) that shows increased mitochondrial density at the periphery of myofibers and a decreased density of mitochondria in the myofiber interior. This finding is most commonly associated with neuropathic disease.
Tubular Aggregates
Aggregates of tubular membranous material within the cytoplasm of mitochondria that give a honeycomb appearance on electron microscopy. These structures are nonspecific and often increase with age. They can best be visualized at the light microscopic level as red material on Gomori trichrome stain.
Type 1 Fiber Predominance
Occurs when greater than 55% of the fibers in a specimen are type 1 fibers.
Type 2 Fiber Predominance
Occurs when greater than 55% of the fibers in a specimen are type 2 fibers.
Core-Type Changes
Central Cores
Central accumulations of myofibrillar material that produce areas devoid of mitochondria (and thus devoid of staining for oxidative enzymes like NADH, SDH, and COX) at, or near, the center of myofibers. Central cores are typically longer than they are wide.
Corelike Lesions
Areas that are devoid of mitochondria and oxidative staining within myofibers, but that are less-clearly delineated than central cores or minicores.
Minicores
Structures causing similar displacement of mitochondria to that seen with central cores and, thus, providing small areas in myofibers that are devoid of staining for oxidative enzymes, such as NADH, SDH, and COX. Minicores are typically wider than they are long.
Nuclear Changes
Central nuclei
Myofibers with centrally placed nuclei. Where possible, these should be distinguished from internal nuclei in terms of the predominant location of abnormally placed nuclei in the specimen.
Internal nuclei
Internal placement of nuclei within myofibers (generally >1 nuclear diameter from the sarcolemma) but in a placement other than the center of the myofiber. As internal nucleation is often a part of the regenerative process, the presence or absence of additional evidence of tissue damage and regeneration may help determine the reason for the abnormal nuclear placement in the specimen.
Nuclear Bags, Clumps, or Clusters
A dense collection of nuclei, with minimal or no apparent myofiber cytoplasm. This indicates an advanced stage of myofiber atrophy.
Mitochondrial Changes
COX− Fibers
Fibers that are devoid of staining with the cytochrome oxidase (COX) histochemical stain. Fibers that are negative for COX and that overexpress succinate dehydrogenase (SDH) are suggestive of a mitochondrial disorder involving the mitochondrial genome.
Ragged Red Fibers
Myofibers containing aggregates of mitochondria that stain as dense, cytoplasmic material on Gomori trichrome stain. These fibers are suggestive of a mitochondrial disorder.
Strongly SDH-Reactive Blood Vessels (SSVs)
Blood vessels that react strongly to SDH on light microscopy. These are considered abnormal because the increased staining is accompanied by many abnormal mitochondria (confirmed on electron microscopy). They have been reported in cases of mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS) and are thought to be secondary to defective respiratory-chain oxidation.
Patterns of Inflammation
Diffuse Inflammation
Distribution of inflammation throughout the specimen, as opposed to its distribution in a single focus.
Endomysial Inflammation
The presence of inflammation in the endomysial compartment (between individual myofibers.
Fascia Inflammation
Distribution of inflammation within the fascia connected to the muscle.
Focal Inflammation
Distribution of inflammation in one focus, as opposed to its distribution throughout the specimen.
Perimysial/Perifascicular Inflammation
The presence of inflammation in a distribution around the edge or just outside a muscle fascicle.
Perivascular Inflammation
Distribution of inflammation around blood vessels, as opposed to its distribution around myofibers.
Footnotes
The other authors have no relevant financial interest in the products or companies described in this article.
Presented in part at the Muscular Dystrophy Association Scientific Conference; April 22, 2013; Washington, DC; at the Child Neurology Society Meeting; October 30, 2013; Austin, Texas; the World Muscle Society; October 2, 2013; Asilomar, California; the American Academy of Neuropathology; June 21, 2013; Charleston, South Carolina; and the Canadian Association of Neuropathologists; October 18, 2013; Ottawa, Ontario, Canada.
References
- 1.Dubowitz V, Sewry C, Oldfors A. Muscle Biopsy: A Practical Approach Philadelphia. Saunders Elsevier; 2013. pp. 2–15. [Google Scholar]
- 2.Goebel HH, Sewry CA, Weller RO. Muscle Disease: Pathology and Genetics. 2nd. West Sussex, UK: Wiley; 2013. [Google Scholar]
- 3.Biering-Srensen F, Charlifue S, Devivo MJ, et al. Using the spinal cord injury common data elements. Top Spinal Cord Inj Rehabil. 2012;18(1):23–27. doi: 10.1310/sci1801-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grinnon ST, Miller K, Marler JR, et al. National Institute of Neurological Disorders and Stroke common data element project—approach and methods. Clin Trials. 2012;9(3):322–329. doi: 10.1177/1740774512438980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loring DW, Lowenstein DH, Barbaro NM, et al. Common data elements in epilepsy research: development and implementation of the NINDS epilepsy CDE project. Epilepsia. 2011;52(6):1186–1191. doi: 10.1111/j.1528-1167.2011.03018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lynch DR, Pandolfo M, Schulz JB, et al. Common data elements for clinical research in Friedreich’s ataxia. Mov Disord. 2013;28(2):190–195. doi: 10.1002/mds.25201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saver JL, Warach S, Janis S, et al. National Institute of Neurological Disorders and Stroke (NINDS) Stroke Common Data Element Working Group Standardizing the structure of stroke clinical and epidemiologic research data: the National Institute of Neurological Disorders and Stroke (NINDS) stroke common data element (CDE) project. Stroke. 2012;43(4):967–973. doi: 10.1161/STROKEAHA.111.634352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stone K. NINDS common data element project: a long-awaited breakthrough in streamlining trials. Ann Neurol. 2010;68(1):A11–A13. doi: 10.1002/ana.22114. [DOI] [PubMed] [Google Scholar]
- 9.NINDS Common Data Elements for Neuromuscular Diseases. http://www.commondataelements.ninds.nih.gov/NMD.aspx#tab=Data_Standards. Published 2012. Accessed November 17, 2014. Updated April 16, 2015.
- 10.Congenital Muscle Disease International Registry. CMDIR Web page. http://cmdir.org/. Accessed November 17, 2014.
- 11.Cohen J. A coefficient of agreement for nominal scales. Educ Psychol Meas. 1960;20:37–46. [Google Scholar]
- 12.Banerjee M, Capozzoli M, McSweeny L, Sinha D. Beyond kappa: a review of interrater agreement measures. Can J Stat. 1999;27(1):3–23. [Google Scholar]