

Abstract
Cells assemble mitotic spindles during each round of division to insure accurate segregation of their duplicated genome. In animal cells, stereotypical spindles have two poles, each containing one centrosome, from which microtubules are nucleated. In contrast, many cancer cells often contain more than two centrosomes and form transient multipolar spindle structures with more than two poles. In order to divide and produce viable progeny, the multipolar spindle intermediate must be reshaped into a pseudo-bipolar structure via a process called centrosomal clustering. Pseudo-bipolar spindles appear to function normally during mitosis, but they occasionally give rise to aneuploid and transformed daughter cells. Agents that inhibit centrosomal clustering might therefore work as a potential cancer therapy, specifically targeting mitosis in supernumerary centrosome-containing cells.
Graphical Abstract
Introduction
Transformed cells no longer respond to cues that normally suppress cell growth and division. This change, from a normal to a cancerous cell, can arise from mutations in specific genes or be caused by more dramatic alterations in genomic composition in the form of whole or partial chromosome loss or addition [1]. Indeed, chromosomal instability (CIN), defined in a population of cells as a systematic deviation from the normal genomic content, is a hallmark of most solid tumor cancers and likely contributes to the maintenance of transformed cell populations, drug resistance, and disease progression [2, 3]. Though there are several different paths to CIN, in this review we focus on how centrosomal amplification (CA), another hallmark of many cancers, affects the chromosome segregation machinery in cells and leads to CIN. Specifically, we introduce the centrosome and provide a brief overview of how centrosome number is controlled in normal cells. We then describe a mechanistic link between supernumerary centrosomes and CIN. Finally, we discuss how cancers maintain the CIN cycle, and potential therapeutic strategies designed to force cells to exit the cycle.
Centrosome Structure and Function
Overview of Centrosome Structure
Centrosomes are non-membranous organelles that contain a structurally-defined core consisting of a pair of longitudinally orientated mother and daughter centrioles and a surrounding region of electron-dense pericentriolar material (PCM). The centrioles themselves are cylindrical structures with a radially symmetrical arrangement of nine microtubule triplets circumscribing a central cartwheel-like structure ([4]; for a more detailed review of centriole structure see[5]). It is important to note that centriolar microtubules differ from their spindle counterparts. Evidence suggests that they are heavily modified by post-translational modifications [6, 7] and thus are behaviorally distinct from spindle microtubules in terms of both inherent dynamics as well as resistance to commonly used cytotoxic drugs [8]. In contrast to centrioles, the surrounding PCM is structurally ill-defined and until recently has been described as an amorphous, proteinaceous matrix ([9]; for a more detailed review of PCM organization see [10]). The PCM serves as a recruiting center for several proteins [11] involved in the nucleation of microtubules, including the yTURC ring complex [12], and provides a structural scaffold to which newly nucleated microtubules can be anchored [13].
Overview of Centrosome Function in Mitotic Spindle Assembly: A Numbers Game?
At the onset of mitosis, duplicated centrosomes occupy a singular site on the surface of the nucleus. Signaling events in the cell cause the pair to split, and each centrosome migrates along the surface of the nucleus until directly opposite the other. This geometry establishes the eventual pole-to-pole axis of the mitotic spindle, with each centrosome forming an important component of each individual pole [14, 15]. The spindle takes shape when the nuclear envelope breaks down and the centrosome-nucleated microtubules invade the now accessible nucleoplasmic space, eventually making connections with kinetochores as well as with chromosome-derived microtubules [14]. The formation of the bipolar spindle is very important because the spindle is responsible for accurately segregating chromosomes during cellular division, and because defects can lead to chromosomal instability [16].
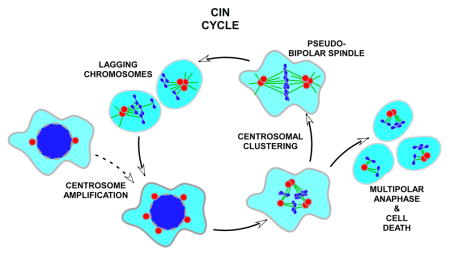
The spindle is typically maintained until all chromosomes are aligned on the metaphase plate, satisfying the spindle assembly checkpoint (SAC; [17]) and allowing anaphase to ensue. Due to the dynamic nature of spindle microtubules (t1/2 ~ 10–100secs [18]), this maintenance requires continuous microtubule nucleation and spatial organization via microtubule-based motors such as dynein, kinesin-5, and kinesin-14 (HSET in human cells; [19, 20]), whose integrated activities shape the spindle by cross-linking and sliding microtubules [21]). Despite the seemingly prominent role of centrosomes during this process, spindle assembly (and even chromosome segregation) occurs unabated in mammalian tissue culture cells following laser ablation of one or both centrosomes [22]. Centrosome-independent spindle assembly is akin to the behavior of several well-characterized meiotic systems, which are capable of assembling bipolar spindles in the absence of centrosomes entirely [23], and similar mechanisms are likely responsible for the assembly of pseudo-bipolar spindles in cells that contain more than two centrosomes. In the presence of supernumerary centrosomes (i.e. >2), spindle assembly typically proceeds through a multipolar intermediate with centrosomes located at >2 spatially distinct foci of microtubule ends. This transient morphology is often resolved prior to anaphase onset by clustering extra centrosomes together into two independent spindle poles (see graphical abstract). Emerging spindles appear deceivingly normal (i.e. are the right size and shape), but are prone to contain erroneous kinetochore attachments that can increase the likelihood of chromosome missegregation [24], the prevalence of aneuploid daughter cells [16, 25], and malignant transformation (see discussion below [26–28]). It makes sense then that the control of centrosome numbers in cells is tightly regulated.
Control of Centrosome Number in Cells
A detailed discussion about the centrosome duplication cycle, beyond the scope of this review, is provided in excellent and recent reviews on the topic [29, 30]. In broad strokes, the centrosome replication cycle can be divided into six major events that occur in concert with the cell division cycle: duplication, elongation, linker dissolution, separation, maturation, and disengagement [29, 30]. Though each of these processes is distinct, involving a specific set of structural and regulatory proteins, there are critically important proteins and overarching control mechanisms that merit special consideration.
Coordination between centrosome duplication and other events of the cell cycle is achieved in part through the activity of cyclins and cyclin-dependent kinases (CDKs; [31, 32]). Cyclins are a family of proteins whose levels rise and fall during the cell cycle [32, 33], and when their intracellular levels are sufficiently high, they bind to and activate cyclin-dependent kinases (CDKs). At the G1/S transition for example, both mother and daughter centrioles serve as a template for procentriole formation [34], and this event is tightly controlled by levels of cyclin E/CDK2 [35], the same cyclin-CDK pair responsible for establishing the pre-initiation complexes required for DNA replication [36]. Similarly, centrosome linker dissolution, whereby the proteinaceous linkages between duplicated centrosome pairs are degraded, is regulated by cyclin B/CDK1 at the G2/M transition [37]. Many additional kinases are involved in the centrosome duplication cycle [38], including Polo-like Kinase 4 (Plk4). Often described as the master regulator of centrosome duplication [39], Plk4 activity is essential for the initiation of procentriole formation and its levels/activity have been shown to correlate with centrosome amplification in several model systems [40, 41]. The levels of Plk4 are strictly regulated as well, in part, through an auto-phosphorylation event that marks itself both for ubiquitination and destruction [42].
Dysregulation of the centrosome duplication cycle can lead to aberrant centrosome numbers. Indeed, experimental perturbation of the levels and/or activities of several of the proteins involved results in CA, and many cancers exhibit aberrant expression levels of these same proteins [43–45]. Though targeting of the centrosome duplication cycle might ultimately prove to be of therapeutic benefit, recent evidence has shown that transformed cells are actually refractory to centrosome loss – for example, transformed cells treated with the Plk4 inhibitor centrinone were able to divide even after drug-induced centrosome loss [46]. Thus, once cells are transformed, complete loss of centrosomes may do little to stop their proliferation.
Mechanistic Links between Centrosome Number and Cancer
In animal cells, the mitotic spindle is comprised of dynamic microtubules that are nucleated around several spatially distinct sources including chromosomes [23] and centrosomes [47]. A normal cell contains exactly two centrosomes at the onset of mitosis: one for each spindle pole. How might dysregulation of centrosome number result in malignant transformation? During mitosis, cells with supernumerary centrosomes often assemble mitotic spindles with supernumerary (i.e. >2) poles [44, 48]. In the late 19th century, Theodore Boveri hypothesized that these multipolar spindles could undergo a multipolar anaphase, resulting in transformed, aneuploid daughter cells with a malignant potential [49, 50]. Recent experimental evidence, however, has shown that multipolar anaphases are relatively uncommon and almost always produce inviable daughter cells [2, 26, 51, 52]. Instead, cells with CA tend to reshape transient multipolar spindles into pseudo-bipolar structures prior to anaphase onset via a dynamic process called centrosomal clustering [27, 51, 53]. This remodeling of spindle morphology requires many of the same proteins that are involved in maintaining and positioning the spindle pole, including dynein, kinesin-14, microtubules, and the actin cytoskeleton [27, 51]. Furthermore, activation of the spindle assembly checkpoint (SAC) appears to facilitate centrosomal clustering by delaying the onset of anaphase [28, 54], and proteins that are closely monitored by the SAC, such as NDC80 and its binding partners, are important for centrosomal clustering processes as well [55]. In summary, centrosomal clustering is a conserved mechanism that allows cells with CA to continue to proliferate.
Resolution of multipolar intermediates through centrosomal clustering results in a higher incidence of mis-attached kinetochores, especially merotelic attachments [24, 26]. These erroneous attachments, defined as an attachment of a single kinetochore to multiple spindle poles, can lead to lagging chromosomes during anaphase, which then are frequently damaged or mis-segregated [24, 52]. As such, merotelic attachments are a major driver of CIN [2, 26]. Because only a small percentage of spindles with supernumerary centrosomes contain merotelic attachments, random proliferative advantages are not quickly lost due to continued mis-segregation or cell death. This type of infrequent, low-level CIN can contribute to, and may possibility initiate, the development of cancer [16, 56], likely by altering the copy number of oncogenes [1]. The relationship between centrosomal clustering, chromosomal instability, and cancer is further described in Figure 1.
Figure 1.

Cartoon schematic of the CIN cycle. Cancer cells often possess supernumerary centrosomes due to dysregulation of the centrosome duplication cycle or failure of cytokinesis (A). Spindles that assemble in these cells typically form transient intermediates with more than two poles (B) leading to multipolar anaphase and apoptosis (C) or centrosomal clustering and the formation of pseudo-bipolar spindles (D). The latter path causes merotelic chromosome attachments and lagging chromosomes (E) sometimes resulting in aneuploidy and CIN. Several well-characterized targets for selective inhibition of centrosomal clustering and forced exit from the CIN cycle are listed in D.
While it is clear that CA can contribute to the development of cancer, there are contrasting data in the literature. For example, two recent studies have demonstrated that supernumerary centrosomes are insufficient to initiate tumorigenesis in mice [40, 57]. We interpret these results to suggest that the promotion of cancer requires levels of centrosomal amplification within an acceptable range that confers sustainable genetic instability [16, 56]. It has been shown, for example, that increasing levels of genetic instability in cells already displaying CIN results in a higher incidence of apoptosis and therefore lower proliferation rates [56]. Instead, levels of CIN must be at a tolerably low level in order to promote the stable genetic changes that are necessary for malignant transformations and tumorigenesis.
Preventing Centrosomal Clustering and Promoting Multipolar Anaphase as an Anticancer Strategy
Definitive evidence that establishes a causal link between CA and tumorigenesis remains elusive, undermining confidence in the potential efficacy of novel anti-cancer therapies designed to reduce the incidence of CA. Regardless, most cancers exhibit CA and therefore require centrosomal clustering as a means to divide and proliferate [45]. As such, CA can serve as a “pan” marker of cancerous cells, and centrosomal clustering can serve as a target for anti-proliferative agents. Several screens have recently been used to identify proteins required for centrosomal clustering [58–60]. Candidates with mitosis-specific functions are of particular interest as they may be used to specifically target dividing cells with CA. However, many candidate targets identified so far also function to insure proper spindle assembly and chromosome segregation fidelity in normal cells and likely will suffer from the same issues that plague other commonly used anti-mitotic cytotoxic agents. Furthermore, centrosomes and their associated microtubule asters are involved in diverse non-mitotic functions such as nuclear expansion [61], chromosomal organization [62, 63], pronuclear migration [64], nuclear/spindle positioning [14, 65, 66] and ciliogenesis [67]. What, if any, contribution that these functions might make to the transformed cell state is now only poorly understood. As such, the challenge to the field is to identify and characterize biomolecules that are critical for centrosomal clustering, but have negligible roles in other critical processes in normal cells.
Currently, several centrosomal clustering inhibitors have been shown to be antiproliferative and partially selective for cells with CA. These include inhibitors of kinesin-14 family members [68], as well as compounds with unknown targets, such as the microtubule binding, anti-fungal griseofulvins [69–71] and nitrofuramides [60]. Inhibition of kinesin-14s is a particularly promising approach because these motors have been shown to be essential for clustering centrosomes in cancer cells, but are not required for division normal cells [28, 60, 72]. Results from experiments using derivatives of griseofulvin and/or nitrofuramide are also encouraging, as these drugs inhibit mitosis selectively in cancer cells [60, 70, 71]. The griseofulvin derivative, GF-15, was also found to inhibit tumorigenesis in mice [71]. Small molecule therapeutics that target centrosomal clustering therefore represent a promising strategy to selectively target populations of cells that require centrosomal clustering for division, but currently, only a disparagingly small number have been identified.
Recent advances in high-throughput data acquisition and data analysis have the potential to significantly improve upon existing screening technologies, enabling more efficient, and perhaps less costly, discovery of new centrosomal clustering inhibitors. For example, small molecule screens designed to examine the effects of compounds on tissue culture cell division have proven powerful, but are inherently limited in that they require the use of cell-permeable effectors. To expand the utility of these approaches, several labs have recently begun to explore the potential of combining cell-free systems with microfluidic-based encapsulation [73–76]. This experimental platform allows for the recapitulation of cellular processes such as spindle assembly and chromosome segregation in discrete volumes of defined size and composition. However, since these “open” systems lack membranes, they can be used to study the effects of a broader range of therapeutic compounds, including those not cell-permeable. Other microfluidic-based approaches, such as flow cytometry [77], will permit more efficient characterization of drug effects on cancer cells with CA and will likely continue to evolve as a proven platform for drug discovery [78]. When combined with tissue-on-a-chip [79] and/or 3D cell-culture technologies [80, 81], these high-throughput methods might one day streamline the drug development/approval process by accurately predicting in vivo results.
Highlights.
Cancer cells often have supernumerary centrosomes and rely on centrosomal clustering to divide.
Centrosomal clustering leads to chromosomal instability (CIN) through the formation of merotelic kinetochore attachments.
CIN results in changes to the sequence and copy number of oncogenes, providing a mechanism for cancer cells to adapt.
Inhibiting centrosomal clustering forces cells to undergo multipolar anaphase and often results in cell death.
Acknowledgments
The authors would like to thank Dr. J. Oakey for critically reading the manuscript and his insightful thoughts and comments. Additionally, we would like to acknowledge all authors who have contributed greatly to our understanding of the centrosome and its role in cancer, but whose work was not cited due to space limitations. Lastly, we would like to thank the National Institute for General Medical Sciences (R01GM102428 to J.C. Gatlin and R15GM101636 to J. Oakey), the Marine Biological Laboratories Whitman Center fellows program, and the Pew Charitable Trusts Biomedical Scholars program for funding our research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- 1.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Santaguida S, Amon A. Short- and long-term effects of chromosome mis-segregation and aneuploidy. Nat Rev Mol Cell Biol. 2015;16:473–485. doi: 10.1038/nrm4025. [DOI] [PubMed] [Google Scholar]
- 3.Schvartzman J, Sotillo R, Benezra R. Mitotic chromosomal instability and cancer: mouse modeling of the human disease. Nature Reviews Cancer. 2010;10:102–115. doi: 10.1038/nrc2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paintrand M, Moudjou M, Delacroix H, Bornens M. Centrosome organization and centriole architecture: their sensitivity to divalent cations. J Struct Biol. 1992;108:107–128. doi: 10.1016/1047-8477(92)90011-x. [DOI] [PubMed] [Google Scholar]
- 5.Gonczy P. Towards a molecular architecture of centriole assembly. Nat Rev Mol Cell Biol. 2012;13:425–435. doi: 10.1038/nrm3373. [DOI] [PubMed] [Google Scholar]
- 6.Janke C. The tubulin code: molecular components, readout mechanisms, and functions. Journal of Cell Biology. 2014;206:461–472. doi: 10.1083/jcb.201406055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barisic M, Silva e Sousa R, Tripathy SK, Magiera MM, Zaytsev AV, Pereira AL, Janke C, Grishchuk EL, Maiato H. Mitosis. Microtubule detyrosination guides chromosomes during mitosis. Science. 2015;348:799–803. doi: 10.1126/science.aaa5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song Y, Brady S. Post-translational modifications of tubulin: pathways to functional diversity of microtubules. Trends in Cell Biology. 2015;25:125–136. doi: 10.1016/j.tcb.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *9.Mennella V, Keszthelyi B, McDonald KL, Chhun B, Kan F, Rogers GC, Huang B, Agard DA. Subdiffraction-resolution fluorescence microscopy reveals a domain of the centrosome critical for pericentriolar material organization. Nat Cell Biol. 2012;14:1159–1168. doi: 10.1038/ncb2597. A particularly good illustration of a useful application of superresolution microscopy. This paper is one of the first to characterize the molecular structure of the PCM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prosser SL, Pelletier L. Centrosome biology: the ins and outs of centrosome assembly. Curr Biol. 2015;25:R656–659. doi: 10.1016/j.cub.2015.06.038. [DOI] [PubMed] [Google Scholar]
- 11.Petry S, Vale R. Microtubule nucleation at the centrosome and beyond. Nature Cell Biology. 2015;17:1089–1093. doi: 10.1038/ncb3220. [DOI] [PubMed] [Google Scholar]
- 12.Teixido-Travesa N, Roig J, Luders J. The where, when and how of microtubule nucleation - one ring to rule them all. Journal of Cell Science. 2012;125:4445–4456. doi: 10.1242/jcs.106971. [DOI] [PubMed] [Google Scholar]
- 13.Lin T, Neuner A, Schiebel E. Targeting of y-tubulin complexes to microtubule organizing centers: conservation and divergence. Trends in Cell Biology. 2015;25:296–307. doi: 10.1016/j.tcb.2014.12.002. [DOI] [PubMed] [Google Scholar]
- 14.Meraldi P. Centrosomes in spindle organization and chromosome segregation: a mechanistic view. Chromosome Res. 2015 doi: 10.1007/s10577-015-9508-2. [DOI] [PubMed] [Google Scholar]
- 15.Kotak S, Gonczy P. Mechanisms of spindle positioning: cortical force generators in the limelight. Current Opinion in Cell Biology. 2013;25:741–748. doi: 10.1016/j.ceb.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 16.Weaver BA, Cleveland DW. Aneuploidy: instigator and inhibitor of tumorigenesis. Cancer Res. 2007;67:10103–10105. doi: 10.1158/0008-5472.CAN-07-2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Musacchio A. The molecular biology of spindle assembly checkpoint signaling dynamics. Current Biology. 2015;25:1002–1018. doi: 10.1016/j.cub.2015.08.051. [DOI] [PubMed] [Google Scholar]
- 18.Needleman DJ, Groen A, Ohi R, Maresca T, Mirny L, Mitchison T. Fast microtubule dynamics in meiotic spindles measured by single molecule imaging: evidence that the spindle environment does not stabilize microtubules. Mol Biol Cell. 2010;21:323–333. doi: 10.1091/mbc.E09-09-0816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heald R, Khodjakov A. Thirty years of search and capture: the complex spimplicity of mitotic spindle assembly. Journal of Cell Biology. 2015;211:1103–1111. doi: 10.1083/jcb.201510015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reber S, Hyman AA. Emergent Properties of the Metaphase Spindle. Cold Spring Harb Perspect Biol. 2015;7 doi: 10.1101/cshperspect.a015784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gatlin JC, Bloom K. Microtubule motors in eukaryotic spindle assembly and maintenance. Semin Cell Dev Biol. 2010;21:248–254. doi: 10.1016/j.semcdb.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khodjakov A, Cole RW, Oakley BR, Rieder CL. Centrosome-independent mitotic spindle formation in vertebrates. Curr Biol. 2000;10:59–67. doi: 10.1016/s0960-9822(99)00276-6. [DOI] [PubMed] [Google Scholar]
- 23.Meunier S, Vernos I. Acentrosomal microtubule assembly in mitosis: the where, when, and how. Trends in Cell Biology. 2015;15:S0962–8924. doi: 10.1016/j.tcb.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 24.Gregan J, Polakova S, Zhang L, Tolic-Norrelykke IM, Cimini D. Merotelic kinetochore attachment: causes and effects. Trends Cell Biol. 2011;21:374–381. doi: 10.1016/j.tcb.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hassold T, Hunt PA. To err (meiotically) is human: the genesis of human aneuploidy. Nature Reviews Genetics. 2001;2:280–291. doi: 10.1038/35066065. [DOI] [PubMed] [Google Scholar]
- **26.Ganem NJ, Godinho SA, Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature. 2009;460:278–282. doi: 10.1038/nature08136. This paper describes work that directly addresses Boveri’s original hypothesis: that multipolar mitosis leads to aneuploidy and cancer. Using long-term live cell imaging, the authors followed the fates of daughter cells after multipolar and pseudo-bipolar mitosis and found that multipolar divisions were not only rare, but that the progeny did not survive. This led them to assess other mechanisms that lead to CIN. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **27.Quintyne NJ, Reing JE, Hoffelder DR, Gollin SM, Saunders WS. Spindle multipolarity is prevented by centrosomal clustering. Science. 2005;307:127–129. doi: 10.1126/science.1104905. This pioneering work was one of the first to address the molecular mechanisms of centrosomal clustering. [DOI] [PubMed] [Google Scholar]
- 28.Kwon M, Godinho SA, Chandhok NS, Ganem NJ, Azioune A, Thery M, Pellman D. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev. 2008;22:2189–2203. doi: 10.1101/gad.1700908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Conduit PT, Wainman A, Raff JW. Centrosome function and assembly in animal cells. Nat Rev Mol Cell Biol. 2015;16:611–624. doi: 10.1038/nrm4062. [DOI] [PubMed] [Google Scholar]
- 30.Fu J, Hagan IM, Glover DM. The centrosome and its duplication cycle. Cold Spring Harb Perspect Biol. 2015;7:a015800. doi: 10.1101/cshperspect.a015800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lacey K, Jackson P, Stearns T. Cyclin-dependent kinase control of centrosome duplication. Proc Natl Acad Sci. 1999;96:2817–2822. doi: 10.1073/pnas.96.6.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hunt T, Nasmyth K, Novak B. The cell cycle. Philos Trans R Soc Lond B Biol Sci. 2011;366:3494–3497. doi: 10.1098/rstb.2011.0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Norbury C, Nurse P. Animal cell cycles and their control. Annu Rev Biochem. 1992;61:443–470. doi: 10.1146/annurev.bi.61.070192.002301. [DOI] [PubMed] [Google Scholar]
- 34.Stearns T, Firat-Karalar E. The centriole duplication cycle. Philos Trans R Soc Lond B Biol Sci. 2014;369 doi: 10.1098/rstb.2013.0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hinchcliffe E, Li C, Thompson E, Maller J, Sluder G. Requirement of Cdk2-Cyclin E activity for repeated centrosome reproduction in Xenopus egg extracts. Science. 1999;283:851–854. doi: 10.1126/science.283.5403.851. [DOI] [PubMed] [Google Scholar]
- 36.Fisher D. Control of DNA replication by clclin-dependent kinases in development. Results Probl Cell Differ. 2011;53:201–217. doi: 10.1007/978-3-642-19065-0_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mardin BSE. Breaking the ties that bind: new advances in centrosome biology. Journal of Cell Biology. 2012;197:11–18. doi: 10.1083/jcb.201108006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang G, Jiang Q, Zhang C. The role of mitotic kinases in coupling the centrosome cycle with the assembly of the mitotic spindle. Journal of Cell Science. 2014;127:4111–4122. doi: 10.1242/jcs.151753. [DOI] [PubMed] [Google Scholar]
- 39.Martin CA, Ahmad I, Klingseisen A, Hussain MS, Bicknell LS, Leitch A, Nurnberg G, Toliat MR, Murray JE, Hunt D, et al. Mutations in PLK4, encoding a master regulator of centriole biogenesis, cause microcephaly, growth failure and retinopathy. Nat Genet. 2014;46:1283–1292. doi: 10.1038/ng.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *40.Vitre B, Holland AJ, Kulukian A, Shoshani O, Hirai M, Wang Y, Maldonado M, Cho T, Boubaker J, Swing DA, et al. Chronic centrosome amplification without tumorigenesis. Proc Natl Acad Sci U S A. 2015;112:E6321–6330. doi: 10.1073/pnas.1519388112. The authors generate a Cre-inducible Plk4 mouse and examine the effects of Plk4 overexpression on tumorigenesis. They found that increased levels of Plk4 lead to CA and expected mitotic errors, but did not confer a proliferative advantage to cells or induce increased tumor formation in mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Coelho PA, Bury L, Shahbazi MN, Liakath-Ali K, Tate PH, Wormald S, Hindley CJ, Huch M, Archer J, Skarnes WC, et al. Over-expression of Plk4 induces centrosome amplification, loss of primary cilia and associated tissue hyperplasia in the mouse. Open Biol. 2015;5 doi: 10.1098/rsob.150209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holland A, Fachinette D, Zhu Q, Bauer M, Verma I, Nigg E, Cleveland D. The autoregulated instability of polo-like kinase 4 limits centrosome duplication to once per cell cycle. Genes Dev. 2012;26:2684–2689. doi: 10.1101/gad.207027.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nigg E. The centrosome duplication cycle in health and disease. FEBS Letters. 2014;588:2366–2372. doi: 10.1016/j.febslet.2014.06.030. [DOI] [PubMed] [Google Scholar]
- 44.Gonczy P. Centrosomes and cancer: revisiting a long-standing relationship. Nat Rev Cancer. 2015;15:639–652. doi: 10.1038/nrc3995. [DOI] [PubMed] [Google Scholar]
- *45.Chan JY. A clinical overview of centrosome amplification in human cancers. Int J Biol Sci. 2011;7:1122–1144. doi: 10.7150/ijbs.7.1122. This review provides one the most complete pictures to date of the prevalence of CA in different cancer types. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wong YL, Anzola JV, Davis RL, Yoon M, Motamedi A, Kroll A, Seo CP, Hsia JE, Kim SK, Mitchell JW, et al. Cell biology. Reversible centriole depletion with an inhibitor of Polo-like kinase 4. Science. 2015;348:1155–1160. doi: 10.1126/science.aaa5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kollman J, Merdes A, Mourey L, Agard D. Microtubule nucleation by y-tubulin complexes. Nature Reviews Molecular Cell Biology. 2011;12:709–721. doi: 10.1038/nrm3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nigg EA. Centrosome aberrations: cause or consequence of cancer progression? Nat Rev Cancer. 2002;2:815–825. doi: 10.1038/nrc924. [DOI] [PubMed] [Google Scholar]
- 49.Boveri T. Ueber den Antheil des Spermatozoon an der Teilung des Eies. Sitzungsber. Ges Morph Physiol München. 1887;3:151–164. [Google Scholar]
- 50.Boveri T. Concerning the origin of malignant tumuors by Theodor Boveri. Translated and annotated by Henry Harris. Journal of Cell Science. 2008;121:1–84. doi: 10.1242/jcs.025742. [DOI] [PubMed] [Google Scholar]
- 51.Godinho SA, Kwon M, Pellman D. Centrosomes and cancer: how cancer cells divide with too many centrosomes. Cancer Metastasis Rev. 2009;28:85–98. doi: 10.1007/s10555-008-9163-6. [DOI] [PubMed] [Google Scholar]
- 52.Vitre BD, Cleveland DW. Centrosomes, chromosome instability (CIN) and aneuploidy. Curr Opin Cell Biol. 2012;24:809–815. doi: 10.1016/j.ceb.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marthiens V, Piel M, Basto R. Never tear us apart--the importance of centrosome clustering. J Cell Sci. 2012;125:3281–3292. doi: 10.1242/jcs.094797. [DOI] [PubMed] [Google Scholar]
- 54.Yang Z, Loncarek J, Khodjakov A, Rieder CL. Extra centrosomes and/or chromosomes prolong mitosis in human cells. Nat Cell Biol. 2008;10:748–751. doi: 10.1038/ncb1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leber B, Maier B, Fuchs F, Chi J, Riffel P, Anderhub S, Wagner L, Ho AD, Salisbury JL, Boutros M, et al. Proteins required for centrosome clustering in cancer cells. Sci Transl Med. 2010;2:33ra38. doi: 10.1126/scitranslmed.3000915. [DOI] [PubMed] [Google Scholar]
- **56.Silk AD, Zasadil LM, Holland AJ, Vitre B, Cleveland DW, Weaver BA. Chromosome missegregation rate predicts whether aneuploidy will promote or suppress tumors. Proc Natl Acad Sci U S A. 2013;110:E4134–4141. doi: 10.1073/pnas.1317042110. In this paper, the authors test the hypothesis that there is a tolerable level of chromosome missegregation below which CIN and tumorigenesis is supported and above which it is not. Using mice heterzygous for the molecular motor CENP-E and already prone to mis-segregation defects and tumorigenesis, they found that increased levels of chromosome missegregation actually reduce tumor formation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kulukian A, Holland AJ, Vitre B, Naik S, Cleveland DW, Fuchs E. Epidermal development, growth control, and homeostasis in the face of centrosome amplification. Proc Natl Acad Sci U S A. 2015;112:E6311–6320. doi: 10.1073/pnas.1518376112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bhakta-Guha D, Saeed M, Greten H, Efferth T. Dis-organizing centrosomal clusters: specific cancer therapy for a generic spread? Curr Med Chem. 2015;22:685–694. doi: 10.2174/0929867322666141212114529. [DOI] [PubMed] [Google Scholar]
- 59.Korzeniewski N, Hohenfellner M, Duensing S. The centrosome as potential target for cancer therapy and prevention. Expert opinion on therapeutic targets. 2013;17:43–52. doi: 10.1517/14728222.2013.731396. [DOI] [PubMed] [Google Scholar]
- 60.Kawamura E, Fielding AB, Kannan N, Balgi A, Eaves CJ, Roberge M, Dedhar S. Identification of novel small molecule inhibitors of centrosome clustering in cancer cells. Oncotarget. 2013;4:1763–1776. doi: 10.18632/oncotarget.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hara Y, Merten CA. Dynein-Based Accumulation of Membranes Regulates Nuclear Expansion in Xenopus laevis Egg Extracts. Dev Cell. 2015;33:562–575. doi: 10.1016/j.devcel.2015.04.016. [DOI] [PubMed] [Google Scholar]
- 62.Ramdas NSG. Cytoskeletal control of nuclear morphology and chromatin organization. Journal of Molecular Biology. 2015;427:695–706. doi: 10.1016/j.jmb.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 63.Maizels Y, Gerlitz G. Shaping of interphase chromosomes by the microtubule network. FEBS Journal. 2015;282:3500–3524. doi: 10.1111/febs.13334. [DOI] [PubMed] [Google Scholar]
- 64.Starr D. A nuclear-envelope bridge positions nuclei and moves chromosomes. Journal of Cell Science. 2009;122:577–586. doi: 10.1242/jcs.037622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kimura A, Onami S. Computer simulations and image processing reveal length-dependent pulling force as the primary mechanism for C. elegans male pronuclear migration. Dev Cell. 2005;8:765–775. doi: 10.1016/j.devcel.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 66.Wuhr M, Dumont S, Groen AC, Needleman DJ, Mitchison TJ. How does a millimeter-sized cell find its center? Cell Cycle. 2009;8:1115–1121. doi: 10.4161/cc.8.8.8150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vertii A, Bright A, Delaval B, Hehnly H, Doxsey SJ. New frontiers: discovering cilia-independent functions of cilia proteins. EMBO reports. 2015;16:1233–1408. doi: 10.15252/embr.201540632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Watts CA, Richards FM, Bender A, Bond PJ, Korb O, Kern O, Riddick M, Owen P, Myers RM, Raff J, et al. Design, synthesis, and biological evaluation of an allosteric inhibitor of HSET that targets cancer cells with supernumerary centrosomes. Chem Biol. 2013;20:1399–1410. doi: 10.1016/j.chembiol.2013.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rebacz B, Larsen TO, Clausen MH, Ronnest MH, Loffler H, Ho AD, Kramer A. Identification of griseofulvin as an inhibitor of centrosomal clustering in a phenotype-based screen. Cancer Res. 2007;67:6342–6350. doi: 10.1158/0008-5472.CAN-07-0663. [DOI] [PubMed] [Google Scholar]
- 70.Ronnest MH, Rebacz B, Markworth L, Terp AH, Larsen TO, Kramer A, Clausen MH. Synthesis and structure-activity relationship of griseofulvin analogues as inhibitors of centrosomal clustering in cancer cells. J Med Chem. 2009;52:3342–3347. doi: 10.1021/jm801517j. [DOI] [PubMed] [Google Scholar]
- 71.Raab MS, Breitkreutz I, Anderhub S, Ronnest MH, Leber B, Larsen TO, Weiz L, Konotop G, Hayden PJ, Podar K, et al. GF-15, a novel inhibitor of centrosomal clustering, suppresses tumor cell growth in vitro and in vivo. Cancer Res. 2012;72:5374–5385. doi: 10.1158/0008-5472.CAN-12-2026. [DOI] [PubMed] [Google Scholar]
- 72.Basto R, Brunk K, Vinadogrova T, Peel N, Franz A, Khodjakov A, Raff JW. Centrosome amplification can initiate tumorigenesis in flies. Cell. 2008;133:1032–1042. doi: 10.1016/j.cell.2008.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *73.Good MC, Vahey MD, Skandarajah A, Fletcher DA, Heald R. Cytoplasmic volume modulates spindle size during embryogenesis. Science. 2013;342:856–860. doi: 10.1126/science.1243147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *74.Hazel J, Krutkramelis K, Mooney P, Tomschik M, Gerow K, Oakey J, Gatlin JC. Changes in cytoplasmic volume are sufficient to drive spindle scaling. Science. 2013;342:853–856. doi: 10.1126/science.1243110. These two complimentary references describe the application of microfluidic-based encapsulation of cell-free extracts to address the basic biological question of how the size of a cell and the spindle it assembles are related. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jimenez AM, Roche M, Pinot M, Panizza P, Courbin L, Gueroui Z. Towards high throughput production of artificial egg oocytes using microfluidics. Lab Chip. 2011;11:429–434. doi: 10.1039/c0lc00046a. [DOI] [PubMed] [Google Scholar]
- 76.Roth S, Laan L, Dogterom M. Reconstitution of cortical Dynein function. Methods Enzymol. 2014;540:205–230. doi: 10.1016/B978-0-12-397924-7.00012-1. [DOI] [PubMed] [Google Scholar]
- 77.Fan B, Li X, Chen D, Peng H, Wang J, Chen J. Development of Microfluidic Systems Enabling High-Throughput Single-Cell Protein Characterization. Sensors. 2016;16 doi: 10.3390/s16020232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Eribol P, Uguz A, Ulgen K. Screening applications in drug discovery based on microfluidic technology. Biomicrofluidics. 2016;10 doi: 10.1063/1.4940886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Esch E, Bahinski A, Huh D. Organs-on-chips at the frontiers of drug discovery. Nature Reviews Drug Discovery. 2015;14:248–260. doi: 10.1038/nrd4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Joshi P, Lee M. High Content Imaging (HCI) on Miniaturized Three-Dimensional (3D) Cell Cultures. Biosensors. 2015;5:768–790. doi: 10.3390/bios5040768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rijal G, Li W. 3D scaffolds in breast cancer research. Biomaterials. 2016:135–156. doi: 10.1016/j.biomaterials.2015.12.016. [DOI] [PubMed] [Google Scholar]